

Abstract
Fcγ-receptor (FcγR) activation by antibody derived soluble immune complexes (sICs) is a major contributor to inflammation in autoimmune diseases such as systemic lupus erythematosus (SLE). A robust and scalable test system allowing for the detection and quantification of sICs with regard to receptor activation is missing. We developed a comprehensive cell-based reporter system capable of measuring the sIC-mediated activation of human and mouse FcγRs individually. We show that compared to human FcγRs IIB and III, human FcγRs I and IIA lack sensitivity to sICs. The assay enables measurement of FcγR activation in response to sIC size and demonstrates a complete translation of the Heidelberger-Kendall precipitation curve to FcγR responsiveness. The assay also proved useful to quantify sICs-mediated FcγR activation using sera from SLE patients and mouse models of lupus and arthritis. Thus, in clinical practice, our assay might be employed as a diagnostic tool to measure FcγR activation as a biomarker for disease activity in immune-complex mediated disease.
Introduction
Immunoglobulin G (IgG) is the dominant immunoglobulin isotype in chronic infections and in antibody-mediated autoimmune diseases. The multi-faced effects of the IgG molecule rely both on the F(ab) regions, which recognize a specific antigen to form immune complexes (ICs), and the constant Fc region (Fcγ), which is detected by Fcγ receptors (FcγRs) found on most cells of the immune system (Lu et al., 2018). When IgG binds to its antigen ICs are formed, which, depending on the respective antigen, are either cell-bound or soluble (sICs). The composition of sICs is dependent on the number of epitopes recognized by IgG on a single antigen molecule and the ability of the antigen to form multimers. Fcγ-FcγR binding is necessary but not sufficient to activate FcγRs with receptor cross-linking generally underlying receptor activation (Bruhns et al., 2009b; Ravetch and Bolland, 2001; van der Poel et al., 2011). Cell bound ICs are readily able to cross-link FcγRs (Bruhns et al., 2009b; Lux et al., 2013). This induces various signaling pathways (Greenberg et al., 1994; Kiefer et al., 1998; Luo et al., 2010) which in turn regulate immune cell effector functions (Bournazos et al., 2017; Nimmerjahn and Ravetch, 2010). It is also suggests that sICs can dynamically tune FcγR activation, meaning that changes in sIC size directly impact FcγR responses (Lux et al., 2013). However, the molecular requirements are largely unknown. Also, a functional reproduction of the paradigmatic Heidelberger-Kendall precipitation curve, describing that the molecular size of sICs determined by the antibody:antigen ratio dynamically tunes FcγR activation(Heidelberger and Kendall, 1929), is missing.
Generally, IC-mediated FcγR cross-linking is indispensable to initiate the full signal cascade following immune cell activation (Duchemin et al., 1994; Getahun and Cambier, 2015; Luo et al., 2010). Human FcγRs are membrane resident receptors recognizing Fcγ. Among all type I FcγRs, FcγRIIB (CD32B) is the only inhibitory one signaling via immunoreceptor tyrosine-based inhibitory motifs (ITIMs) while the activating receptors are associated with immunoreceptor tyrosine-based activation motifs (ITAMs). Another exception is FcγRIIIB (CD16B), which is glycosylphosphatidylinositol (GPI)-anchored (Bruhns, 2012; Bruhns and Jonsson, 2015; Nimmerjahn and Ravetch, 2006; Nimmerjahn and Ravetch, 2008). FcγRI (CD64) is the only receptor with high affinity binding to monomeric IgG not associated with antigen and is primarily tasked with phagocytosis linked to antigen processing and pathogen clearance (Guilliams et al., 2014; Indik et al., 1994). All the other FcγRs only efficiently bind to complexed, meaning antigen-bound IgG (Bruhns, 2012; Bruhns and Jonsson, 2015; Lu et al., 2018). While FcγRI, FcγRIIB and FcγRIIIA are able to recognize sICs, this has not been reported for FcγRIIA (CD32A), rather this receptor has recently been shown to depend on the neonatal Fc receptor (FcRn) to do so (Fossati et al., 2002b; Hubbard et al., 2020).
Activation of FcγRs leads to a variety of cellular effector functions such as antibody-dependent cellular cytotoxicity (ADCC) by natural killer (NK) cells via FcγRIIIA, antibody-dependent cellular phagocytosis (ADCP) by macrophages via FcγRI, cytokine and chemokine secretion by NK cells and macrophages via FcγRIIIA. Furthermore, reactive oxygen species (ROS) production of neutrophils and neutrophil extracellular traps formation (NETosis) via FcγRIIIB, dendritic cell (DC) maturation and antigen presentation via FcγRIIA and B cell selection and differentiation via FcγRIIB (Berger et al., 1996; Bournazos et al., 2017; Granger et al., 2019; Kang et al., 2016; Laborde et al., 2007; Pincetic et al., 2014; Tay et al., 2019; Vidarsson et al., 2014). Consequently, FcγRs regulate and connect both innate and adaptive branches of the immune system. Various factors have been indicated to influence the IC-dependent FcγR activation profiles, including Fcγ-FcγR binding affinity and avidity (Koenderman, 2019), IgG subclass, glycosylation patterns and genetic polymorphisms (Bruhns et al., 2009a; Pincetic et al., 2014; Plomp et al., 2017; Vidarsson et al., 2014), stoichiometric antigen-antibody-ratio (Berger et al., 1996; Lux et al., 2013; Pierson et al., 2007) and FcγR clustering patterns (Patel et al., 2019). For example, glycosylation patterns of the IgG Fc domain initiate either pro- or anti-inflammatory effector pathways by tuning the binding affinity to activating or inhibitory FcγRs, respectively (Bohm et al., 2014). However, despite being explored individually, the functional consequences of these features when acting in combination on a single receptor are still not fully understood. Therefore, an assay allowing for the systematic functional assessment of IC-mediated FcγR activation is strongly required. sICs and immobilized ICs represent intrinsically different stimuli for the immune system (Fossati et al., 2002a; Granger et al., 2019). Soluble circulating ICs are commonly associated with chronic viral or bacterial infections (Wang and Ravetch, 2015; Yamada et al., 2015) and some autoimmune diseases, such as systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) (Antes et al., 1991; Koffler et al., 1971; Zubler et al., 1976). Typically, sICs related disorders are characterized by systemic cytokine secretion (Mathsson et al., 2007; Vogelpoel et al., 2015) as well as immune cell exhaustion and senescence (Bano et al., 2019; Tahir et al., 2015). In order to study sIC-dependent activation of FcγRs in detail, we employed a cell-based assay which has been previously utilized to study immobilized ICs (Corrales-Aguilar et al., 2013) and adapted it into a sIC sensitive reporter system capable of distinguishing the activation of individual FcγRs and their responses to varying complex size. This allowed for the first time a complete reproduction of the Heidelberger-Kendall precipitation curve measuring actual FcγR activation. The assay also enables a quantification of clinically relevant sICs in sera from SLE patients and autoimmune-prone mice with immune-complex-mediated arthritis and lupus using reporter cells expressing chimeric mouse FcγRs.
Results
Experimental assay setup
The assay used in this study was adapted from a previously described cell-based FcγR activation assay designed to measure receptor activation in response to opsonized virus infected cells (Corrales-Aguilar et al., 2013) or therapeutic Fc-fusion proteins (Lagasse et al., 2019). We changed the assay setup to enable measurement of sICs when directly incubated with reporter cells stably expressing the ectodomains of the human FcγR fused to the signaling module of the mouse CD3-ζ chain (FcγRI: Acc# LT744984; FcγRIIA: Acc# M28697; FcγRIIB/C: Acc# LT737639; FcγRIIIA/B: Acc# LT737365). Ectodomains of FcγRIIIA and FcγRIIIB as well as ectodomains of FcγRIIB and FcγRIIC are identical. Second generation reporter cells were generated to stably express chimeric FcγRs compared to the stable transfectants used in the original assay (Corrales-Aguilar et al., 2013). To this end, BW5147 cells were transduced as described previously via lentiviral transduction (Corrales-Aguilar et al., 2013; Van den Hoecke et al., 2017). Human FcγR expression on transduced cells after puromycin selection is shown in Fig. 1A. Activation of the reporter cells is measured by quantification of mouse IL-2 (mIL-2) secretion into the cell culture supernatant using an anti-IL-2 sandwich ELISA as described previously (Corrales-Aguilar et al., 2013). In order to employ the original assay, designed to measure ICs on adherent infected cells, for the detection of soluble ICs we first determined the suspension of IgG achieved by pre-blocking a 96 well ELISA microtiter plate using PBS/10%FCS. To this end, we compared different concentrations of FCS in the blocking reagent and measured the threshold at which IgG (rituximab, Rtx) no longer binds to the plate and stays in solution. Fig. 1B clearly shows that FCS supplementation to 1% (v/v) or higher is sufficient to keep antibodies in solution and prevents IgG binding to the plastic surface. Using this adapted protocol, the assay allows for the characterization of FcγR interaction with immobilized ICs versus sICs as shown schematically in Fig. 1C. Next, we set out to test if immobilized IgG is an appropriate substitute for opsonized cells or immobilized ICs with regard to FcγR activation as suggested before (Tanaka et al., 2009).

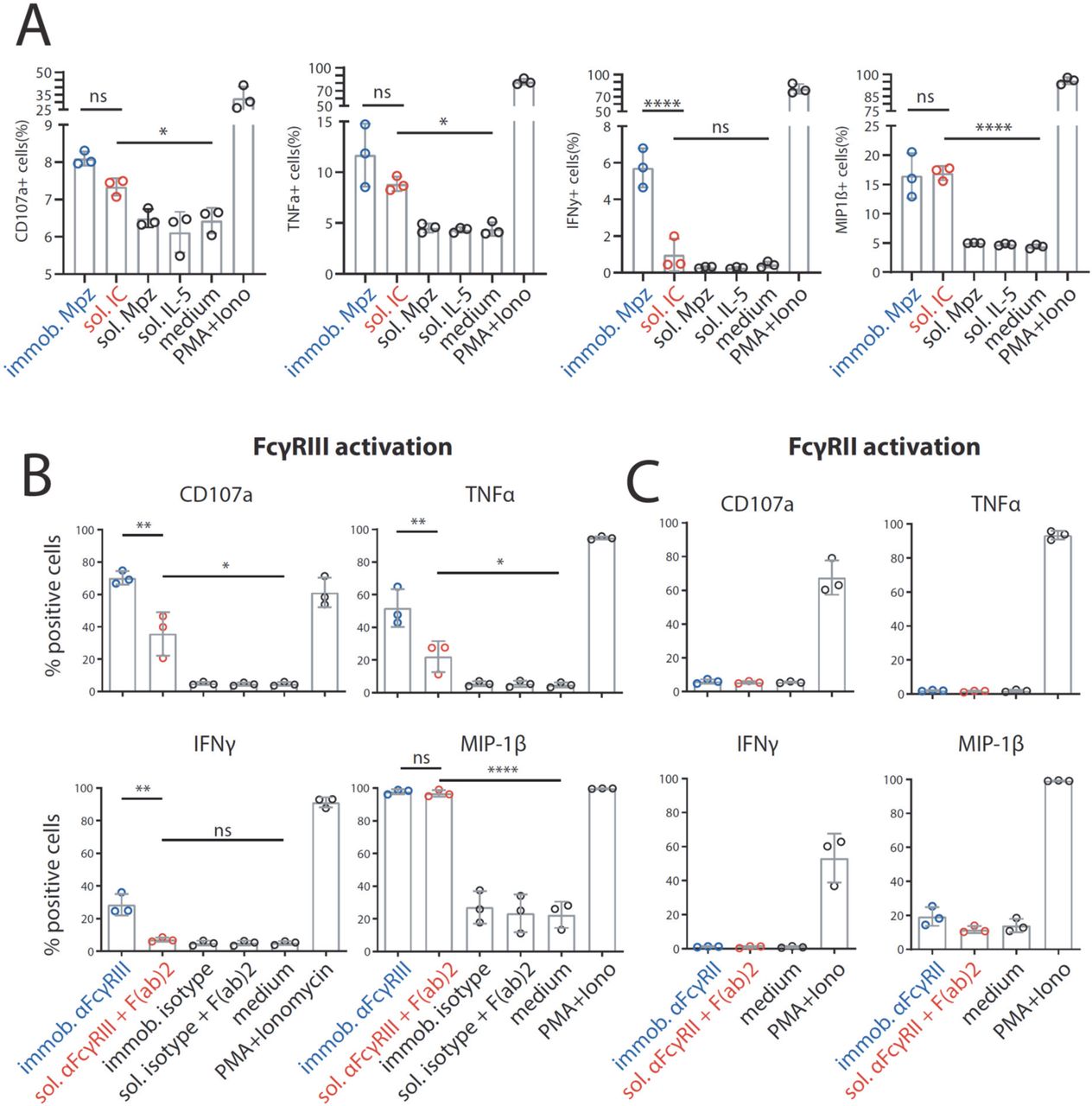
A) BW5147 reporter cells stably expressing human FcγRs or BW5147 parental cells were stained with FcγR specific conjugated mAbs as indicated and measured for surface expression of FcγRs via flow cytometry. Fcγ binding was determined using a PE-TexasRed-conjugated human IgG-Fc fragment. B) FCS coating of an ELISA plate allows for suspension of subsequently added IgG. Plate bound IgG was quantified via ELISA. PBS supplemented with >1% FCS (v/v) avoids adhesion of IgG (rituximab, Rtx) to the ELISA plate. C) Schematic of an immobilized IC or soluble IC setup. BW5147 reporter cells expressing chimeric human FcγR receptors secrete IL-2 in response to FcγR activation by clustered IgG. Soluble ICs are generated using mAbs and multivalent antigens (blue). Solubility is achieved by pre-blocking an ELISA plate using PBS supplemented with 10% FCS (orange).
There was no qualitative difference in FcγR activation between immobilized Rtx, immobilized ICs (Rtx + CD20) or Rtx-opsonized 293-CD20 cells, showing that FcγR cross-linking by clustered IgG alone is sufficient for receptor activation (Fig. 2). As sICs formed by monomeric CD20 peptide (aa 141-188) and Rtx completely failed to activate FcγRs, we hypothesized that, in order to generate sICs able to activate FcγRs, antigens have to be multivalent. Of note, to reliably and accurately differentiate between soluble and immobilized triggers using this assay, reagents for the generation of ICs need to be of high purity and consistent stability. Only combinations of therapy grade ultra-pure mAbs and ultra-pure antigens (size exclusion chromatography) showed reproducible, dose-dependent and specific activation of the reporter assay (data not shown).

Dose-dependent activation of FcγR-bearing reporter cells by immobilized IC can be mimicked by immobilized IgG. Response curves of human FcγRIIB/C and FcγRIIIA/B are similar between opsonized cells (293T cells stably expressing CD20 + Rtx), immobilized IC (rec. soluble CD20 + Rtx) and immobilized IgG (Rtx). SIC formed by monovalent antigen (rec. soluble CD20) do not activate human FcγRs. X-Axis shows sample concentration determined by antibody molarity. Y-Axis shows FcγR activation determined by reporter cell IL-2 production.
Quantification of human FcγR responsiveness to multimeric sICs
There are only few commercially available antibody-antigen pairs that meet both the above mentioned high grade purity requirements while also allowing for multimeric sIC formation. We focused on three pairs of multivalent antigens and their respective mAbs that were available in required amounts enabling large-scale titration experiments; trimeric rhTNFα:IgG1 infliximab (TNFα:Ifx), dimeric rhVEGFA: IgG1 bevacizumab (VEGFA/Bvz) and dimeric rhIL-5: IgG1 mepolizumab (IL-5/Mpz). As lymphocytes express TNFα-receptors I and II while not expressing receptors for IL-5 or VEGFA, we tested whether our mouse lymphocyte derived BW5147 thymoma reporter cell line is sensitive to high concentrations of rhTNFα. Toxicity testing revealed that even high concentrations up to 76.75 nM rhTNFα did not affect viability of reporter cells (Fig. S1). Next, we measured the dose-dependent activation of human FcγRs comparing immobilized IgG to soluble ICs using the FcγR reporter cell panel (Fig. 3).

Three different multivalent ultra-pure antigens (Ag) mixed with respective therapy-grade mAbs were used to form sICs as indicated for each set of graphs (top to bottom). IC pairs: infliximab (Ifx) and rhTNFα; mepolizumab (Mpz) and rhIL-5; bevacizumab (Bvz) and rhVEGFA. X-Axis: concentrations of stimulant expressed as molarity of either mAb or Ag monomer and IC (expressed as mAb molarity) at a mAb:Ag ratio of 1:2. Soluble antigen or soluble antibody alone served as negative controls and were not sufficient to activate human FcγRs. FcγR responses were normalized to immobilized rituximab (Rtx) at 1μg/well (set to 1) and a medium control (set to 0). All FcγRs show dose-dependent activation towards immobilized IgG. FcγRIIA shows low activation at high sIC concentrations compared to immobilized IgG activation. FcγRI shows no activation towards sICs. FcγRIIIA/B and FcγRIIB/C are dose-dependently activated by sICs with responses comparable in strength to immobilized IgG stimulant. Experiments performed in technical replicates. Error bars = SD. Error bars smaller than symbols are not shown.
Soluble antigen or mAb alone served as negative controls showing no background activation even at high concentrations. Immobilized rituximab served as a positive control for inter-experimental reference. We observed that all FcγRs are strongly activated in a dose-dependent manner when incubated with immobilized IgG. Incubating the FcγR reporter cells with sICs at identical molarities showed FcγRIIB/C and FcγRIIIA/B to be efficiently activated by sICs, while in contrast, FcγRIIA and FcγRI did not respond to sICs. We furthermore observed FcγRIIIA/B to be efficiently activated by sICs with responses even surpassing those achieved with immobilized IgG for TNFα/Ifx and IL-5/Mpz ICs. FcγRIIB/C showed a generally weaker reactivity towards sICs compared to immobilized ICs, especially at high concentrations whereas an inversion of this order was seen for TNFα/Ifx and VEGFA/Bvz ICs at lower concentrations. IL-5/Mpz, FcγRIIB/C and FcγRIIIA/B showed similar responsiveness towards immobilized or sICs with a generally stronger activation on immobilized ICs. These experiments demonstrate that sICs of different composition vary in the resulting FcγR activation pattern, most likely due to the antigens being either dimeric, trimeric or different in size.
Next, to validate the measured differences in response to sICs vs. immobilized IC for primary cells, we determined FcγRIIIA activation using primary human NK cells isolated from PBMCs of healthy donors. Measuring a panel of activation markers and cytokine responses by flow cytometry, we observed a differential activation pattern depending on ICs being soluble or immobilized at equal molarity (Fig. 4A). We chose IL-5/Mpz sICs as NK cells do not express the IL-5 receptor. While MIP1-β responses were comparable between the two triggers, degranulation (CD107a) and TNFα responses showed a trend towards lower activation by sICs compared to immobilized IgG (Mpz). Strikingly, IFNγ responses were significantly weaker when NK cells were incubated with sICs compared to immobilized IgG. In order to confirm this hierarchy of responses and to enhance the overall low activation by Fcγ compared to the PMA control, we changed the IC setup by generating reverse-orientation sICs consisting of human FcγR-specific mAbs and goat-anti-mouse IgG F(ab)2 fragments. NK cell activation by reverse sICs was compared to NK cell activation by immobilized FcγR specific mAbs (Fig. 4B). Here, we not only confirm our previous observations regarding MIP-1β and IFNγ, but we also confirm significantly lower TNFα and CD107a responses towards soluble complexes compared to immobilized mAbs. Importantly, these experiments validate that sICs readily activate primary NK cells and induce immunological effector functions. As in 10% of the population NK cells express FcγRIIC (Breunis et al., 2008; Metes et al., 1998), we also tested if this receptor plays a role in our measurements. Using the same three donors and an FcγRII specific mAb as described above, we did not observe an FcγRII-mediated response. Accordingly, we conclude that FcγRIIC expression did not play a role in our experiments (Fig. 4C). Taken together, we show that multivalent but not dimeric soluble immune complexes govern primary NK cell response and FcγRIIIA/B activation (Fig. 2A).

Primary NK cells purified from PBMCs of three different donors were tested for NK cell activation markers. Error bars = SD. Two-way ANOVA (Turkey). A) NK cells were incubated with immobilized IgG (mepolizumab, Mpz), soluble IC (Mpz:IL-5 = 1:1), soluble Mpz or soluble IL-5 (all at 200 nM, 106 cells). Incubation with PMA and Ionomycin (Iono) served as a positive control. Incubation with medium alone served as a negative control. B) NK cells were incubated with immobilized FcγRIII-specific mAb, soluble mouse-anti-human IgG F(ab)2 complexed FcγRIII-specific mAb (reverse sICs), immobilized IgG of non-FcγRIII-specificity (isotype control) or soluble F(ab)2 complexed isotype control (all at 1μg, 106 cells). Incubation with PMA and Ionomycin served as a positive control. Incubation with medium alone served as a negative control. C) As in B using an FcγRII-specific mAb. NK cells from the tested donors in this study do not react to FcγRII activation.
Measurement of FcγR activation in response to changing sIC size
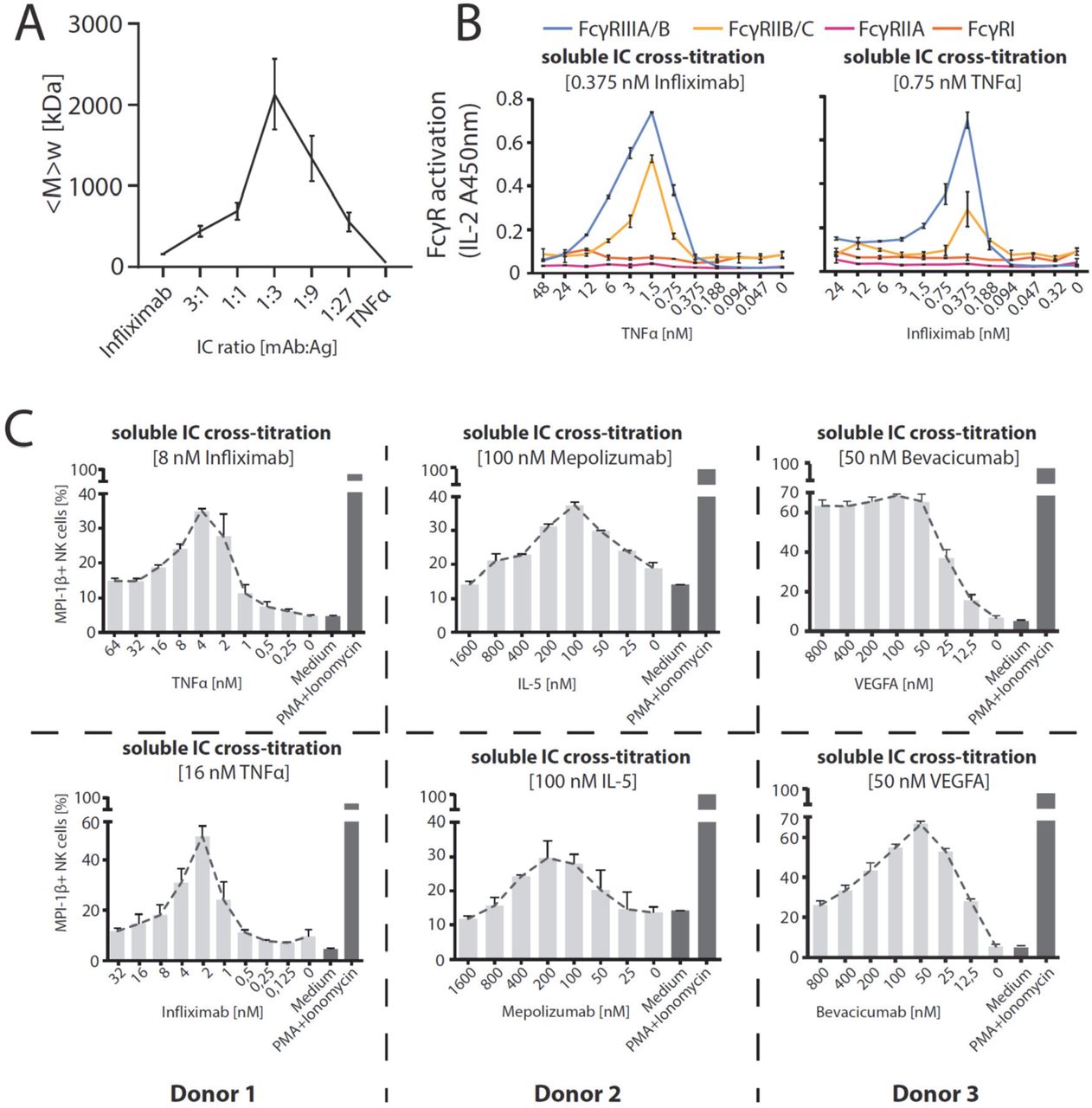
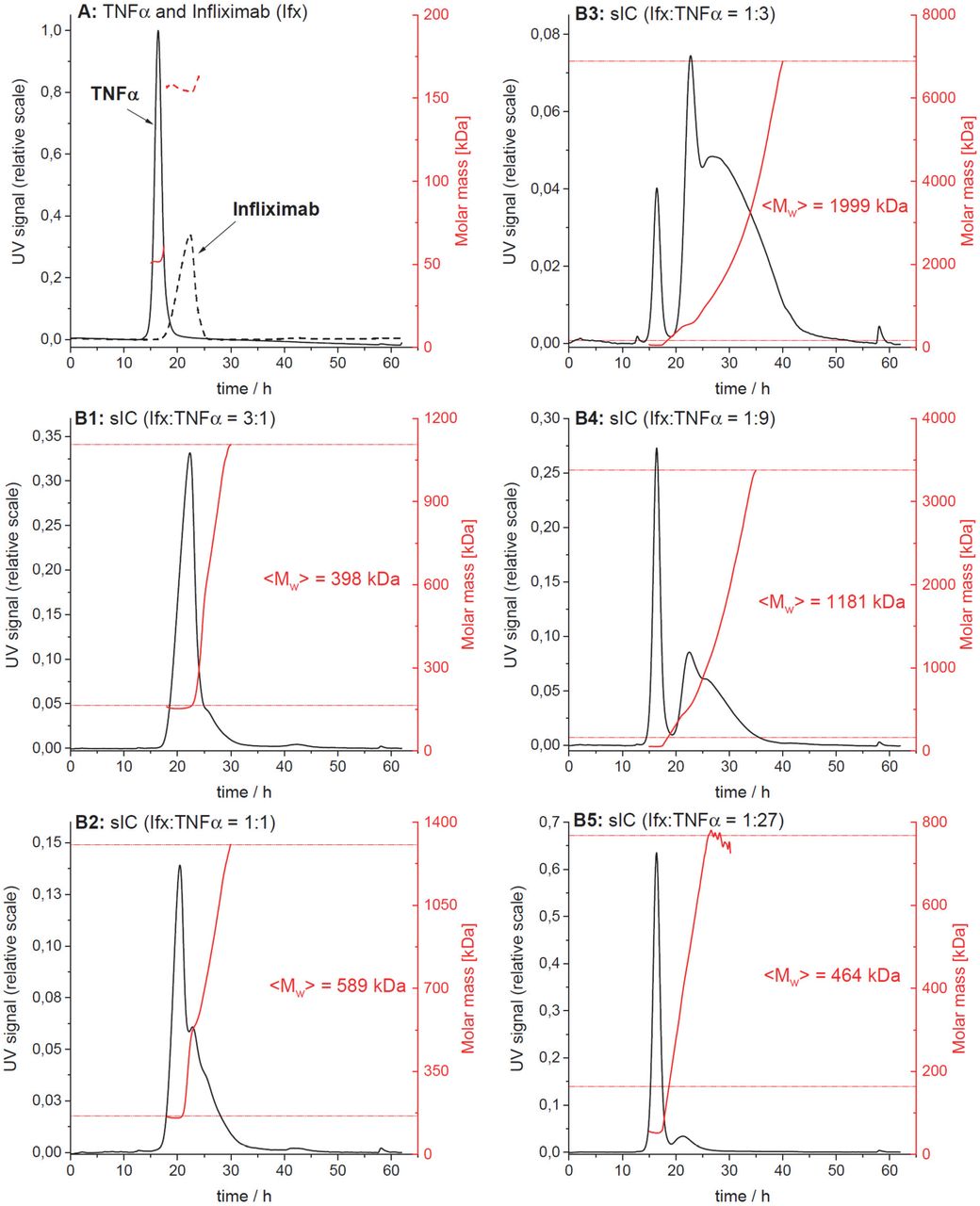
We observed that the dimeric sIC CD20:Rtx completely failed to activate our reporter cells, while potentially larger sICs based on multimeric antigens showed an efficient dose-dependent FcγR activation. In order to determine whether the assay is able to respond to changes in sIC size, we tested cross-titrated amounts of antibody (mAb, infliximab, Ifx) and antigen (Ag, rhTNFα). To this end, the reporter cells were incubated with sIC of different mAb:Ag ratio by fixing one parameter and titrating the other. According to the Heidelberger-Kendall precipitation curve (Heidelberger and Kendall, 1929) describing sIC size as being dependent on the mAb:Ag ratio, this should result in varying sIC sizes as an excess of either antigen or antibody results in the formation of smaller complexes compared to the large molecular complexes formed at around equal molarity. Changes in sIC size due to a varying mAb:Ag ratio were quantified using asymmetrical flow-field flow fractionation (AF4) (Fig. 5A and Table S1). Fig. S2 shows an exemplary complete run of such an analysis. AF4 analysis identifies the highest sIC mean molecular being approximately 2130 kDa at a 1:3 ratio (mAb:Ag) with sICs getting smaller with increasing excess of either antigen or antibody, recapitulating a Heidelberger-Kendall-like curve. Incubation of the FcγR reporter cells with ICs of varying size indeed shows that the assay responds to changes in sIC size (Fig. 5B). Accordingly, both FcγR types showed the strongest responses at mAb:Ag ratios of approximately 1:3. We then set out to test the accuracy of our reporter cell assay as a surrogate marker for primary human immune cells expressing FcγRs. To this end, we isolated primary NK cells from three individual donors and measured NK cell MIP1-β upregulation in response to synthetic sICs of varying size and composition again using a similar assay setup optimized for NK cell activation. We chose MIP-1β upregulation as a cell surface marker to measure NK cell activation as it showed the highest responsiveness in previous experiments (Fig. 4). We could observe that primary immune cells expressing FcγRIIIA respond to IC size, confirming our assay to be an accurate surrogate for primary immune cell responses to soluble ICs (Fig. 5C). Convincingly, NK cell responses to sICs generated from trimeric antigen (rhTNFα) peaked at a different mAb:Ag ratio compared to NK cell responses to sICs generated from dimeric antigens (rhIL-5 and rhVEGFA). Of note, TNFα and VEGFA activate resting NK cells thus leading to higher MIP1-β positivity when NK cells are incubated in the presence of excess antigen. As NK cells do not express IL-5 receptor, this is not observed in the presence of excess IL-5. Regarding TNFα, NK cells still show a stronger activation by sICs generated under optimal mAb:Ag ratios compared to conditions where excess antigen is used. This shows a clear correlation between IC size and effector response. Conversely, when changing antibody concentrations using fixed amounts of antigen, a consistent reduction of NK cell activation is observed in the presence of excess IgG for all three mAb/Ag pairs.

A) infliximab (mAb) and rhTNFα (Ag) were mixed at different ratios (17 μg total protein, calculated from monomer molarity) and analysed via AF4. sIC size is maximal at a 1:3 ratio of mAb:Ag and reduced when either mAb or Ag are given in excess. <M>w = mass-weighted mean of the molar mass distribution. Three independent experiments. Error bars = SD. Data taken from Table S1. One complete run analysis is shown in Fig. S2. B) FcγR BW5147 reporter cell activation is sensitive to sIC size. sICs of different size were generated by cross-titration according to the AF4 determination. Reporter cells were incubated with fixed amounts of either mAb (infliximab, left) or Ag (rhTNFα, right) and titrated amounts of antigen or antibody, respectively. X-Axis shows titration of either antigen or antibody, respectively (TNFα calculated as monomer). IL-2 production of reporter cells shows a peak for FcγRIIB/C and FcγRIIIA/B activation at an antibody:antigen ratio between 1:2 and 1:4. FcγRs I and IIA show no activation towards sICs in line with previous observations. Two independent experiments. Error bars = SD. C) Primary NK cells purified by negative selection magnetic bead separation from three different donors were incubated with cross-titrated sICs as in A. NK cells were measured for MIP-1β expression (% positivity). Incubation with PMA and Ionomycin served as a positive control. Incubation with medium alone served as a negative control. Measured in technical replicates. Error bars = SD.
Quantification of reactive sICs in sera of SLE patients
In order to apply the assay to a clinically relevant condition we measured circulating sICs present in the serum of SLE patients with variable disease activity. Sera from 4 healthy donors and 25 SLE patients were investigated for FcγRIIIA/B and FcγRIIB/C activation. Reporter cells produced IL-2 in response to patient sera in a dose-dependent manner (shown exemplarily for one group of SLE patients in Fig. 6A), which was not the case when sera from healthy controls were tested. Consistent with the observation that FcγRI and FcγRIIA do not respond to synthetic sICs, reporter cells expressing these receptors did also not respond to the tested serum samples. While this reaction pattern already indicated that sICs are the reactive component measured in SLE patients’ sera, we further demonstrate that FcγRIIIA/B and FcγRIIB/C activation depends on the presence of serum ICs by analyzing patient serum before and after polyethylene glycol (PEG) precipitation of sICs (Fig. 6B). Next, we calculated the area under the curve (AUC) values for all 25 SLE patient titrations and normalized them to the AUC values measured for healthy individuals. The resulting index values were then correlated with established biomarkers of SLE disease activity, such as anti-dsDNA titers (α-dsDNA) and concentrations of the complement cleavage product C3d (Fig. 6C). Both biomarkers are believed to indirectly indicate the presence of sICs. We observed a significant correlation between our FcγRIIIA/B activation index values and the determined disease activity markers, anti-dsDNA titers and C3d (p=0.0078 and p=0.0118, respectively). FcγRIIB/C on the other hand showed no significant correlation with either biomarker. We assume this may be due to the influence of IgG sialyation found to be reduced in active SLE (Vuckovic et al., 2015). Generally, de-sialylation of IgG leads to stronger binding by the activating receptors FcγRI, FcγRIIA and FcγRIII while it reduces the binding affinity of the inhibitory FcγRIIB (Kaneko et al., 2006). In sum, our assay allows the indirect quantification of clinically relevant sICs in sera of SLE patients.

Serum derived sIC from systemic lupus erythematosus (SLE) patients activate human FcγR reporter cells. 25 patients and 4 healthy control individuals were separated into three groups for measurement. A) Experiments shown for an exemplary group of 8 SLE patients and two healthy individuals. Dose-dependent reactivity of FcγRs IIIA/B and IIB/C was observed only for SLE patient sera and not for sera from healthy individuals. FcγRs I and IIA show no reactivity towards clinical IC in line with previous observations. B) Activation of FcγRs IIB/C and IIIA/B by patient serum is mediated by serum derived sICs. Patient serum was depleted of sICs by PEG precipitation and the supernatant was compared to untreated serum regarding FcγR activation (left). IgG concentration in the precipitate (SN), supernatant (SN) and untreated serum is shown in the bar graph (right). IC precipitation did not remove IgG from the supernatant. C) FcγRIIIA/B activation, but not FcγRIIB/C activation, significantly correlates with known SLE disease markers. FcγR activation data from A was correlated to established SLE disease markers (α-dsDNA levels indicated as IU/ml or C3d concentrations indicated as mg/ml). FcγR activation from a dose-response curve as in A was calculated as area under curve (AUC) for each SLE patient (n=25) or healthy individual (n=4) and expressed as fold change compared to the healthy control mean. SLE patients with α-dsDNA levels below 50 IU/ml and C3d values below 9 mg/ml were excluded. Two-tailed Pearson correlation.
Translation to clinically relevant in vivo lupus and arthritis mouse models
BW5147 reporter cells stably expressing chimeric mouse as well as rhesus macaque FcγRs have already been generated using the here described method and were successfully used to measure FcγR activation by opsonized adherent cells in previous studies (Kolb et al., 2019; Van den Hoecke et al., 2017) (mFcγR reporter cells). As the human FcγR reporter cells described here are sensitive to sICs, we next aimed to translate the assay to clinically relevant animal models. To this end, we incubated previously described FcγR reporter cells expressing chimeric mouse FcγRs (Van den Hoecke et al., 2017) with sera from lupus (NZB/WF1) or arthritis (K/BxN) mice with active disease. The reporter assay was performed as described above. We chose to measure the stimulation of the activating receptors, mFcγRIII and mFcγRIV, that correspond to human FcγRIII and show a comparable cellular distribution and immune function (Bruhns and Jonsson, 2015). Incubation with synthetic sICs generated from rhTNFα and mouse-anti-hTNFα IgG1 showed both of the mFcγR reporter cells to be responsive to sICs (Fig. 7A). Parental BW5147 cells expressing no FcγRs served as a negative control. The sera of three mice per group were analysed and compared to sera from wildtype C57BL/6 mice, which served as a negative control. We consistently detected mFcγR activation by sera from K/BxN or NZB/WF1 compared to BL6 mice (Fig. 7B). While the mFcγRIII responses were generally high and similar between K/BxN and NZB/WF1 mice, we found a lower and more variable mFcγRIV responsiveness. We assume this might be explained by differences in mFcγR affinity to mouse IgG subclasses. While mFcRIII binds IgG1, IgG2a and IgG2b with comparable affinity, IgG1 is not detected by mFcRIV (Bruhns and Jonsson, 2015). Further, we found that the response of mFcγRIV-expressing reporter cells shows no strict dose-dependency. This effect was more pronounced for NZB/WF1 compared to K/BxN mice. In addition, in this case we assume an influence of factors such as glycosylation patterns or subclass composition, not further addressed here. Nevertheless, the assay enables the reliable detection of sICs in sera of mice with immune-complex mediated diseases making it a promising novel tool to monitor sICs as a biomarker of disease activity.

A) Reporter cells expressing mFcγRIII, mFcγRIV or parental BW5147 cells were incubated with titrated amounts of synthetic sICs generated from rhTNFα and mouse-anti-hTNFα at a 1:1 ratio by mass. One experiment in technical replicates. Error bars = SD. B) Titrations of 3 mouse sera per group (BL/6, K/BxN or NZB/WF1) were incubated with mFcγR reporter cells and FcγR activation was assessed as described above. Sera from BL/6 mice served as negative control. Performed in technical replicates. Error bars = SD.
Discussion
A novel assay for the quantification of synthetic as well as disease-associated sICs
We developed a cell-based reporter system capable of quantifying the IgG-IC mediated activation of individual human and mouse FcγRs. We show that the assay is exquisitely specific for immobilized ICs as well as soluble ICs. This assay presents a meaningful advancement in methodology as it enables the direct detection of receptor-activating ICs. This is of great value for autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), where circulating ICs crucially contribute to disease manifestations (Koffler et al., 1971; Zubler et al., 1976) and for certain infectious diseases such as COVID-19 caused by SARS-CoV2 (Vuitton et al., 2020) or chronic hepatitis B virus (HBV) infection, in which circulating sICs are generated (Madalinski et al., 1991; Wang and Ravetch, 2015). We demonstrated that the here described assay enables quantification of serum-derived circulating sICs from SLE patients with regard to FcγR reactivity. Further, the sIC-mediated FcγR activation correlated with SLE disease markers. Thus, the assay may serve as promising new tool to measure sIC as disease biomarker in autoimmunity or infection. We also show the potential of this assay to be translated to clinically relevant animal models. As the assay also enables the measurement of dose-dependent FcγR responses this allows the (semi-) quantitative detection of sICs in various samples such as mAb preparations for clinical use. The presence of ICs in therapeutic preparations or their formation following patient treatment is unwanted due to the dangers of side effects such as lupus-like syndrome which has been linked to mAb treatment in patients receiving infliximab (Wetter and Davis, 2009). As this assay is sensitive to any aggregation of IgG, it also represents a tool to control the purity and quality of mAb preparations developed for therapeutic use in patients. In addition, the assessment of sIC-mediated FcγR activation allows for optimization of mAb preparations targeting cytokines and soluble factors, which would result in sIC formation, designed for reduced or enhanced FcγR activation such as glyco-engineered mAbs or LALA-mutant mAbs (Li et al., 2017; Saunders, 2019). Notably, the scalability of this cell-based test system does allow for large-scale screening of samples.
FcγR activation profiles differ dependent on the solubility and size of clustered IgG
We observed a difference in the response patterns for FcγRIIIA/B and FcγRII/B/C depending on the solubility of clustered IgG (immobilized versus soluble ICs) which we validated for FcγRIIIA using primary human NK cells. It should be emphasized that multimeric, but not dimeric sICs can trigger FcγR activation. This highlights the fundamental influence of the antigen and subsequent sIC size on FcγR-dependent signal transduction. The ability of the here described assay to quantify the activation of individual FcγR by sICs is not achievable or directly comparable using primary cells due to non-uniform immune responses upon FcγR activation and as different immune cells express FcγRs in different combinations as well as variable densities. Finally, and in contrast to primary human cells, our murine reporter cells are largely inert to human cytokines, which provides a key advantage to measure FcγR activation selectively in response to sIC.
Human FcγRIIIA mediates ADCC elicited by NK cells and the induction of a pro-inflammatory cytokine profile by CD16+ monocytes, while FcγRIIIB is a GPI-anchored receptor on neutrophils. FcγRIIB is an inhibitory receptor expressed by B cells and dendritic cells (DCs) regulating B cell activation, antibody production by plasma cells and the activation state of DCs, while the activating FcγRIIC is found on NK cells mediating ADCC. However, as FcγRIIC is only expressed by less than 20% of the human population, its role is still poorly understood (Anania et al., 2019; Lisi et al., 2011). Given the here shown difference in FcγR reactivity towards multimeric sICs versus immobilized IgG it is tempting to speculate that FcγRIIIA/B- and FcγRIIB/C-positive immune cells might have adapted to differentially perceive the different FcγR ligands (sICs versus membrane bound insoluble ICs) and translate them into distinct reaction patterns. This could be achieved by these cells via differences in receptor density, signal transduction or regulation of receptor expression. Consulting the literature indeed supports our hypothesis with neutrophils, B cells and NK cells being efficiently activated by sICs via essentially the same receptor ectodomains (Goodier et al., 2016; Kang et al., 2016; Mayadas et al., 2009; Nimmerjahn and Ravetch, 2008), while the immunological outcome of their reaction very much differs.
Revisiting the Heidelberger-Kendall curve: Dynamic sIC size measurement and monitoring of activity in sIC-associated diseases
We provide for the first time a simultaneous functional and biophysical assessment of the paradigmatic Heidelberger-Kendall precipitation curve (Heidelberger and Kendall, 1929). While previous work revealed that large and small sICs show differential impact on IL-6 production in PBMCs (Lux et al., 2013), the dynamics of FcγR activation resulting from constant changes in sIC size have not been explored in great detail yet. This was achieved in this study by directly analyzing native molecules via AF4 (Fig. 5A, Fig. S2, Table S1). Our data reveal that sIC size is indeed governed by antibody:antigen ratios covering a wide range of sizes up to several megadaltons. In the presence of increasing amounts of antibody or antigen deviating from an optimal antibody:ratio, sIC size steadily decreases. Further, by the measurement of FcγR activation of we now translate sIC size directly to an immunologically meaningful read-out. In doing so, we show that sIC size essentially tunes FcγR activation and thereby immune cell responses. Thus, our new test system can not only contribute to the functional detection and quantification of clinically relevant sICs but also provides a starting point on how to avoid pathological consequences by influencing the sIC size, for example by administering therapeutic antibodies or recombinant antigens in optimized concentrations, thus becoming relevant in clinical pharmacokinetics.
Limitations of the reporter system and conclusions
We found that FcγRI is not activated by sICs in our assay. We assume that FcγRI activation by sICs might require a native cellular environment given that a major function is the uptake of ICs even in the absence of a signaling motif (Indik et al., 1994). However, we find that FcγRI ectodomains alone are not responsive to sICs indicating a different cross-linking threshold for FcγRI possibly linked to it being the only high-affinity FcγR with three extracellular Ig domains compared to the two domains found in other FcγRs. This reflects a general consideration regarding the reporter system. While providing a robust and unified read-out using a scalable cell-based approach, the assay is not able to reflect native immune cell functions governed by cell specific signalling cascades. The major advancements of this reporter system include i) a higher accuracy regarding FcγR activation compared to strict affinity measurement, ii) an sIC size dependent quantification of FcγR responsiveness and iii) the identification of FcγR activating sICs in autoimmunity but also infection. Finally, this scalable, sensitive and robust system to detect FcγR activating sICs in clinical samples might enable their identification diseases that have not been linked to sIC-mediated pathology, yet.
Materials and Methods
Cell culture
All cells were cultured in a 5% CO2 atmosphere at 37°C. BW5147 mouse thymoma cells (BW, obtained from ATCC: TIB-47) were maintained at 3×105 to 9×105 cells/ml in Roswell Park Memorial Institute medium (RPMI GlutaMAX, Gibco) supplemented with 10% (vol/vol) fetal calf serum (FCS, Biochrom), sodium pyruvate (1x, Gibco) and β-mercaptoethanol (0.1 mM, Gibco). 293T-CD20 (kindly provided by Irvin Chen, UCLA (Morizono et al., 2010)) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% (vol/vol) FCS.
FcγR receptor activation assay
FcγR activation was measured adapting a previously described cell-based assay (Corrales-Aguilar et al., 2014; Corrales-Aguilar et al., 2013). The assay was modified to measure FcγR activation in solution. Briefly, 2×105 BW-FcγR (BW5147) reporter cells were incubated with synthetic sIC in a total volume of 100 μl for 16 h at 37°C and 5% CO2. Incubation was performed in a 96-well ELISA plate (Nunc maxisorp) pre-treated with PBS/10% FCS (v/v) for 1 h at 4°C. Immobilized IgG was incubated in PBS on the plates prior to PBS/10% FCS treatment. Reporter cell mIL-2 secretion was quantified via ELISA as described previously (Corrales-Aguilar et al., 2013).
Recombinant antigens and monoclonal antibodies to form sICs
Recombinant human (rh) cytokines TNF, IL-5, and VEGFA were obtained from Stem Cell technologies. Recombinant CD20 was obtained as a peptide (aa141-188) containing the binding region of rituximab (Creative Biolabs). FcγR-specific mAbs were obtained from Stem Cell technologies (CD16: clone 3G8; CD32: IV.3). Reverse sICs were generated from these receptor-specific antibodies using goat-anti-mouse IgG F(ab)2 fragments (Invitrogen) in a 1:1 ratio. Pharmaceutically produced humanized monoclonal IgG1 antibodies infliximab (Ifx), bevacizumab (Bvz), mepolizumab (Mpz) and rituximab (Rtx) were obtained from the University Hospital Pharmacy Freiburg. Mouse anti-hTNFα (IgG2b, R&D Systems, 983003) was used to generate sICs reactive with mouse FcγRs. sICs were generated by incubation of antigens and antibodies in reporter cell medium or PBS for 2 h at 37°C.
Lentiviral transduction
Lentiviral transduction was performed as described previously (Kolb et al., 2019; Van den Hoecke et al., 2017). In brief, chimeric FcγR-CD3ζ constructs (Corrales-Aguilar et al., 2013) were cloned into a pUC2CL6IPwo plasmid backbone. For every construct, one 10-cm dish of packaging cell line at roughly 70% density was transfected with the target construct and two supplementing vectors providing the VSV gag/pol and VSV-G-env proteins (6 μg of DNA each) using polyethylenimine (22.5 μg/ml, Sigma) and Polybrene (4 μg/ml; Merck Millipore) in a total volume of 7 ml (2 ml of a 15-min-preincubated transfection mix in serum-free DMEM added to 5 ml of fresh full DMEM). After a medium change, virus supernatant harvested from the packaging cell line 2 days after transfection was then incubated with target BW cells overnight (3.5 ml of supernatant on 106 target cells), followed by expansion and pool selection using complete medium supplemented with 2 μg/ml of puromycin (Sigma) over a one week culture period.
BW5147 toxicity test
Cell counting was performed using a Countess II (Life Technologies) according to supplier instructions. Cell toxicity was measured as a ratio between live and dead cells judged by trypan blue staining over a 16 h time frame in a 96well format (100 μl volume per well). BW5147 cells were mixed 1:1 with Trypan blue (Invitrogen) and analysed using a Countess II. rhTNFα was diluted in complete medium.
human IgG suspension ELISA
1 μg of IgG1 (rituximab in PBS, 50 μl/well) per well was incubated on a 96well microtiter plate (NUNC Maxisorp) pre-treated (2h at RT) with PBS supplemented with varying percentages (v/v) of FCS (PAN Biotech). IgG1 bound to the plates was detected using an HRP-conjugated mouse-anti-human IgG mAb (Jackson ImmunoResearch).
BW5147 cell flow cytometry
BW5147 cells were harvested by centrifugation at 900 g and RT from the suspension culture. 1×106 cells were stained with PE× or FITC-conjugated anti-human FcγR mAbs (BD) or a PE-TexasRed-conjugated human IgG-Fc fragment (Rockland) for 1h at 4°C in PBS/3%FCS. After 3 washing steps in PBS/3%FCS, the cells were transferred to Flow cytometry tubes (BD) and analysed using BD LSR Fortessa and FlowJo (V10) software.
NK cell activation flow cytometry
PBMC were purified from donor blood using Lymphocyte separation Media (Anprotec). Primary NK cells were separated from donor PBMCs via magnetic bead negative selection (Stem Cell technologies). 96well ELISA plates (Nunc Maxisorp) were pre-treated with PBS/10% FCS (v/v) for 1 h at 4°C. NK cells were stimulated in pre-treated plates and incubated at 37°C and 5% CO2 for 4 h. Golgi Plug and Golgi Stop solutions (BD) were added as suggested by supplier. CD107a (APC, BD, H4A3) specific conjugated mAb was added at the beginning of the incubation period. Following the stimulation period, MIP-1β (PE, BD Pharmigen), IFNγ (BV-510, Biolegends, 4SB3) and TNFα (PE/Cy7, Biolegends, MAB11) production was measured via intracellular staining Cytokines (BD, CytoFix/CytoPerm, Kit as suggested by the supplier). 50 ng/ml PMA (InvivoGen) + 0.5 μM Ionomycin (InvivoGen) were used as a positive stimulation control for NK cell activation. After 3 washing steps in PBS/3%FCS, the cells were transferred to Flow cytometry tubes (BD) and analysed using a BD FACS Fortessa and FlowJo (V10) software. FcγRII or FcγRIII block was performed by addition of receptor specific mAbs (Stem cell technologies, IV.3 and 3G8) at a 1:100 dilution at the beginning of the incubation period. Cells were transferred to Flow cytometry tubes (BD) and analyzed using BD LSR Fortessa and FlowJo (V10) software.
Asymmetric flow field flow fractionation (AF4)
The AF4 system consisted of a flow controller (Eclipse AF4, Wyatt), a MALS detector (DAWN Heleos II, Wyatt), a UV detector (1260 Infinity G1314F, Agilent) and the separation channel (SC channel, PES membrane, cut-off 10 kDa, 490 μm spacer, wide type, Wyatt). Elution buffer: 1.15 g/L Na2HPO4 (Merck), 0.20 g/L NaH2PO4 × H2O (Merck), 8.00 g/L NaCl (Sigma) and 0,20 g/L NaN3 (Sigma), adjusted to pH 7.4, filtered through 0.1 μm. AF4 sequence (Vx = cross flow in mL/min): (a) elution (2 min, Vx: 1.0); (b) focus (1 min, Vx: 1.0), focus + inject (1 min, Vx: 1.0, inject flow: 0.2 mL/min), repeated three times; (c) elution (30 min, linear Vx gradient: 1.0 to 0.0); (d) elution (15 min, Vx: 0.0); (e) elution + inject (5 min, Vx: 0.0). A total protein mass of 17±0.3 μg (Ifx, rhTNFα or ICs, respectively) was injected. The eluted sample concentration was calculated from the UV signal at 280 nm using extinction coefficients of 1.240 mL/(mg cm) or 1.450 mL/(mg cm) in the case of TNFα or Ifx, respectively. For the ICs, extinction coefficients were not available and difficult to calculate as the exact stoichiometry is not known. An extinction coefficient of 1.450 mL/(mg cm) was used for calculating the molar masses of all ICs. Especially in the case of ICs rich in TNFα, the true coefficients should be lower, and the molar masses of these complexes are overestimated by not more than 14 %. The determined molar masses for TNFα-rich complexes are therefore biased but the observed variations in molar mass for the different ICs remain valid. The mass-weighted mean of the distribution of molar masses for each sample was calculated using the ASTRA 7 software package (Wyatt).
SLE patient cohort
Sera from patients with SLE were provided by the ImmRheum biobank of the Department of Rheumatology and Clinical Immunology. Biobanking and the project were approved by the local ethical committee of the University of Freiburg (votes 507/16 and 624/14). All patients who provided blood to the biobank had provided written informed consent. Ethical Statement: The study was designed in accordance with the guidelines of the Declaration of Helsinki (revised 2013). Patients with SLE (n = 25) and healthy controls (n = 4) were examined. All patients met the revised ACR classification criteria for SLE. Disease activity was assessed using the SLEDAI-2K score. Serum concentrations of anti–double-stranded DNA (α-dsDNA) and the complement cleavage product Cd3 were monitored and indicated as IU/ml and mg/ml, respectively. Anti-dsDNA concentrations in units and C3d concentrations provide sensitive markers for disease activity in SLE.
Patient serum IC precipitation
For polyethylene glycol (PEG) precipitation human sera were mixed with PEG 6000 (Sigma-Aldrich) in PBS at a final concentration of 10% PEG 6000. After overnight incubation at 4°C, ICs were precipitated by centrifugation at 2000 × g for 30 min at 4 °C, pellets were washed once with PEG 6000 and then centrifugated at 2000 x g for 20 min at 4 °C. Supernatants were harvested and precipitates re-suspended in pre-warmed PBS for 1 h at 37 °C. IgG concentrations of serum, precipitates and supernatants obtained after precipitation were quantified by Nanodrop (Thermo Scientific™) measurement.
Mice and Models
Animal experiments were approved by the local governmental commission for animal protection of Freiburg (Regierungspräsidium Freiburg, approval no. G16/59 and G19/21). Lupus-prone (NZBxNZW)F1 mice (NZB/WF1) were generated by crossing NZB/BlNJ mice with NZW/LacJ mice, purchased from The Jackson Laboratory. KRNtg mice were obtained from F. Nimmerjahn (Universität Erlangen-Nürnberg) with the permission of D. Mathis and C. Benoist (Harvard Medical School, Boston, MA), C57BL/6 mice (BL/6) and NOD/ShiLtJArc (NOD/Lt) mice were obtained from the Charles River Laboratories. K/BxN (KRNtgxNOD)F1 mice (K/BxN) were obtained by crossing KRNtg mice and NOD/Lt mice. All mice were housed in a 12-h light/dark cycle, with food and water ad libitum. Mice were euthanized and blood collected for serum preparation from 16 weeks old BL/6 animals, from 16 weeks old arthritic K/BxN animals and from 26 – 38 weeks old NZB/WF1 mice with established glomerulonephritis.
Statistical analyses
Statistical analyses were performed using Graphpad Prism software (v6) and appropriate tests.
Funding
This work was supported by an intramural junior investigator fund of the Faculty of Medicine to PK (EQUIP - Funding for Medical Scientists, Faculty of Medicine, University of Freiburg), by the German Research foundation (DFG) (FOR2830 HE 2526/9-1) to HH, by the DFG research grant TRR130 to REV and the Ministry of Science, Research, and Arts Baden-Württemberg (Margarete von Wrangell Programm) to NC.
Competing interests
We declare no financial and non-financial competing interests.
Supplemental Data

Cell count and percentage of live cells were unaltered over a 16 h time frame of reporter cell culture in the presence of indicated rhTNFα concentrations and comparable to regular growth in complete medium. Experiments were conducted in 3 replicates. Error bars = SD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The elution profiles from one of three independent runs are shown. Protein concentration in the eluate is shown in black (UV signal at λ = 280 nm, normalized to the highest UV signal found in this experiment), molar masses determined by MALS for a given retention time in red. Horizontal red lines indicate the range of molar masses used to calculate the mass-weighted mean of molar masses <Mw>. A) Overlay of the elution profiles obtained for TNFα and Ifx, respectively; B1 to B5) Elution profiles for sICs formed after incubation of TNFα and Ifx at different molar ratios.
For a given elution time, the AF4 profiles provide the concentration (UV) at which a given molar mass (MALS) of a protein is present in the sample. The molar mass distribution of Ifx, TNFα and their immune complexes (sICs) was obtained by plotting the cumulative frequency as a function of molar mass. For a selected range of molar masses, a mass-weighted mean value (<Mw>) was calculated. All detected molar masses were selected in the case of Ifx and TNFα whereas only molar masses larger than the maximal molar mass found for Ifx were assigned to sICs. The table shows the range of assigned molar masses and the calculated <Mw> for each AF4 run (n = 3).
Acknowledgement
We thank T. Schleyer (ImmRheum Biobank) for providing patient samples.
Footnotes
Summary This study describes a novel cell-based reporter assay enabling the detection and quantification of multimeric soluble IgG immune complexes (sICs). By selective activation of specific FcγRs, the assay shows sensitivity to sIC size and responds to synthetic and clinically relevant sICs in sera from SLE patients and autoimmune-prone mice.
Minor changes to the abstract text and figure resolution.
References




