

Abstract
The ultimate goal of HIV-1 is integration into the host chromatin to optimize the release of high levels of viral progeny and discretely coexist with the host. To uncover the HIV-1 DNA fate in the nuclear landscape we directly tracked the viral DNA (vDNA) and the viral RNA (vRNA) by coupling HIV-1 ANCHOR technology with RNA FISH or MCP-MS2 RNA-tagging bacterial system. Our computational imaging analysis revealed that proviral forms are early located in proximity of the nuclear periphery of mitotic and non-mitotic cells. We also observed that HIV-1 infection prompts clustering formation of the host factor CPSF6 restructuring membraneless organelles enriched in both viral proteins and speckle factors. Interestingly, we observed that integrase proteins are retained in CPSF6 clusters, while the late retrotranscribed DNA was excluded from HIV-induced membranelless organelles (HIV-1 MLOs), indicating that those structures are not proviral sites, but orchestrate viral events prior to the integration step. HIV-1 MLOs are in the vicinity of pre-existing LEDGF clusters. Importantly, we identified actively transcribing proviruses localize, outside HIV-1 MLOs, in LEDGF-abundant regions, known to be active chromatin sites. This study highlights single functional host-proviral complexes in their nuclear landscape, which is markedly restructured by HIV-1 to favor viral replication.
Introduction
Immediately after fusion at the plasma membrane, HIV-1 cores are released into the cytoplasm and move towards the nucleus, while the viral RNA genome begins the process of reverse transcription into double-stranded DNA (dsDNA) (Campbell and Hope, 2015; Di Nunzio, 2013). Once imported in the nucleus, the interplay between the HIV-1 DNA genome and the host chromatin compartment is crucial for the fate of the virus-host coexistence. HIV-1 can adopt either an episomal or a proviral form (Butler et al., 2001). The choice between these two possibilities and their surrounding chromatin landscape dictates the evolution of HIV infection (Maldarelli, 2016; Sharkey et al., 2011; Sharkey et al., 2000). The mechanisms determining the fate of the proviral DNA in the nucleus are still under investigation (Liu et al., 2020; Olson et al., 2019) also because of the limits imposed by available technologies. Real-time imaging approaches provide new insight into unprecedented spatial information of individual HIV-1 infected cells. Several strategies have enabled the visualization of the viral components during nuclear entry: Integrase (IN)-TC/FlAsH, APOBEC 3F (A3F)-YFP, IN-YFP, Gag-iGFP, Cyclophilin A (CypA)-DsRed/ Capsid (CA), INsfGFP, and CA-GFP (Burdick et al., 2017; Hubner et al., 2009; Lelek et al., 2012) (Francis et al., 2016; Francis and Melikyan, 2018) (Mamede et al., 2017) (Albanese et al., 2008; Francis et al., 2014). Previously, biochemistry studies supported only a limited and secondary role of the viral CA in the early steps of infection (Farnet and Haseltine, 1991). State-of-the-art imaging technologies developed in recent years have shed light on the central role of the viral CA in nuclear import(Blanco-Rodriguez et al., 2020; Burdick et al., 2020) dictated by the CA interplay with host factors, such as CPSF6 (Buffone et al., 2018; Francis et al., 2020; Price et al., 2014) and Nup153 (Di Nunzio et al., 2013; Lelek et al., 2015). Although these techniques improved our knowledge in the HIV field, the labeling of viral proteins only allows the visualization of steps prior to the establishment of proviral or episomal forms. Mechanisms underlying viral post-nuclear entry steps and the role of nuclear structures induced by HIV-1 during viral replication could be revealed only by the direct co-labeling of viral DNA (vDNA) forms and host factors. Thus far, the vDNA can be visualized either by supplying 5-Ethynyl-2’-deoxyuridine (EdU) during infection (Peng et al., 2014), which is limited to fixed non-dividing cells (De Wit et al., 2019) or by DNA-FISH (Marini et al., 2015), which is characterized by harsh conditions of fixed sample preparation that limits its coupling with other techniques, impeding the study of the interplay between the vDNA and the host factors. Here, we were able to live track episomal and proviral forms, showing their divergent intranuclear behavior. In parallel, we detect the location of functional proviruses in the nuclear space of the main target cells of the virus, such as CD4+T cells and macrophages, by using HIV-1 ANCHOR methodology (Blanco-Rodriguez et al., 2020) to label the vDNA coupled to RNA FISH or to the live bacterial approach MCP-MS2 (Tantale et al., 2016) to identify the vRNA foci. Next, we deepened the study of the nuclear landscape that surrounds proviruses. We observed that viral infection reprogramed the nuclear location of a viral partner, CPSF6. CPSF6 is a paraspeckle factor (Fox et al., 2002; Naganuma and Hirose, 2013) involved in viral nuclear entry though its interaction with the viral CA (Buffone et al., 2018; Francis et al., 2020; Lee et al., 2010; Price et al., 2014). Here we found that CPSF6 relocates in HIV-1 induced membraneless organelles (HIV-1 MLOs), enriched in speckle factors, SC35 (Spector and Lamond, 2011) and SON (Sharma et al., 2010), near LEDGF clusters. Previous studies reported the importance of LEDGF in HIV-1 integration sites distribution (Ciuffi et al., 2005), however a complex between the transcribing proviral DNA and LEDGF has never been visualized. In this study, we determined the nuclear position of several viral forms, including actively transcribing proviruses, with respect to HIV-1-MLOs or the nuclear envelope (NE). In addition, we found that the late retro-transcribed DNA co-exists, but remains separated, from viral INs in the host nucleus. We observed that the viral INs are sequestered in HIV-1 MLOs, suggesting the role of these nuclear structures in the Pre-Integration Complex (PIC) maturation. On the other hand, the late reverse transcripts are fully excluded from HIV-1 MLOs, in fact we found that HIV-1 MLOs are not the sites of viral integration. Finally, we identified functional host-viral complexes, formed by vDNA/vRNA associated to LEDGF, excluded from the inner cavity of HIV-1 MLOs (avg distances from HIV-1 MLOs < 0.6 μm) and located in the vicinity of the NE (avg of distances from NE < 2 μm). In this study we show that HIV-1 DNA localization is key to identifying partners of HIV-1 that aid the virus to hijack cellular mechanisms to persist in the host cells and to highlight the nuclear landscape surrounding single viral genomes. Our results provide new insights into how HIV reprograms and markedly restructures the nuclear environment to orchestrate viral replication steps.
Results
Live tracking of HIV-1 DNA forms in the nucleus of target cells
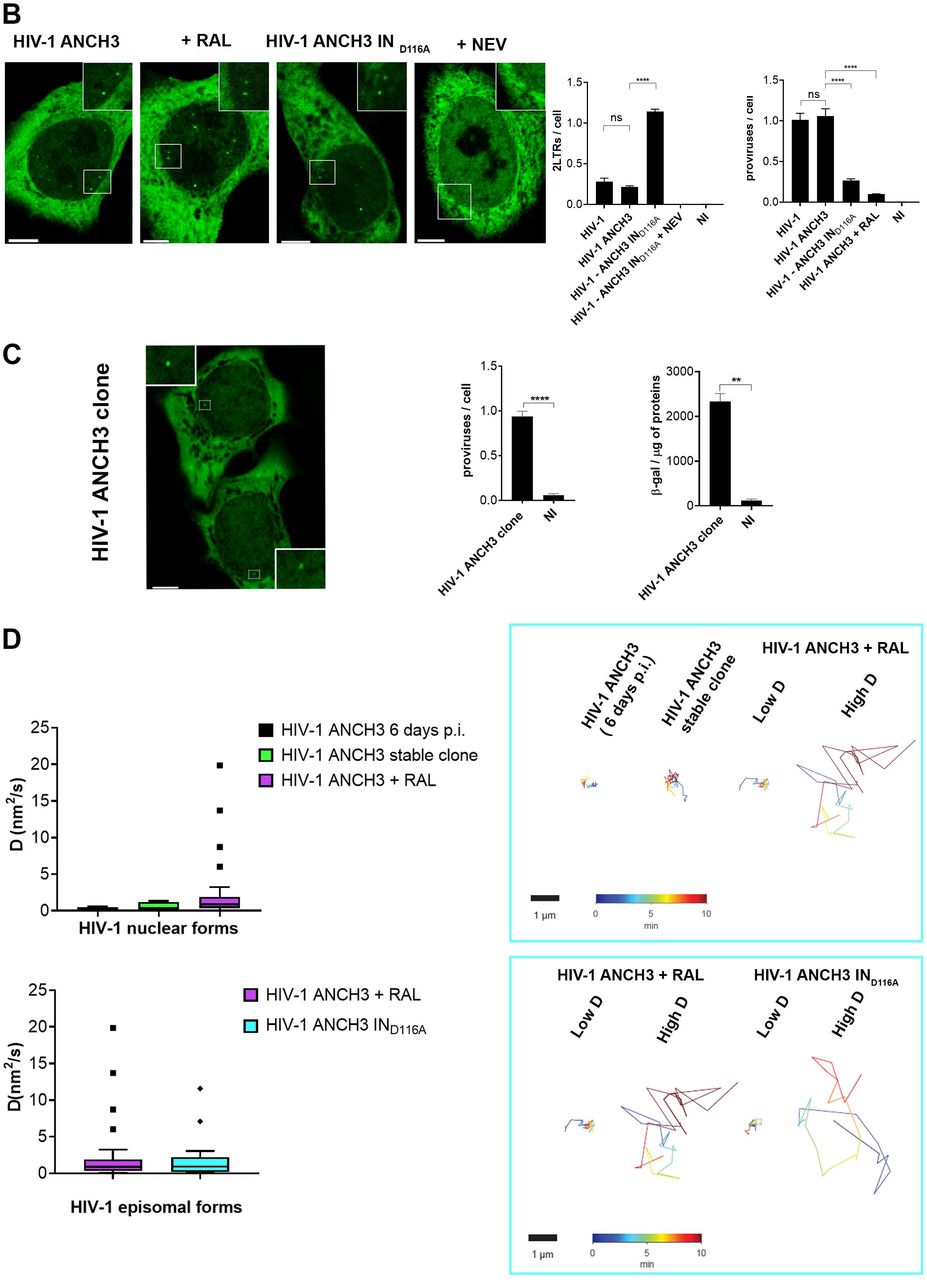
With the aim of studying HIV-1 DNA nuclear fate in different cell types, we benefited of the HIV-1 ANCHOR DNA labeling system (Fig. 1A) (Blanco-Rodriguez et al., 2020). Once fully retrotranscribed, the HIV-1 genome carrying the bacterial sequence ANCH3 is specifically recognized by OR-GFP protein, which is expressed in the target cells, genetically modified by the lentiviral vector (LV) OR-GFP (Blanco-Rodriguez et al., 2020). The accumulation of OR-GFP, which is a modified version of the bacterial ParB protein (Graham et al., 2014; Sanchez et al., 2015), on the ANCH3 sequence generates bright nuclear signals (Fig.1A). Importantly, OR protein binds exclusively double stranded DNA (Saad et al., 2014). We cloned ANCH3 in the nef gene, thus, HIV-1 ANCHOR system exclusively detects late reverse transcripts, because only late retrotranscripts contain ANCH3 sequence as double stranded DNA (Fig. 1A). Therefore, HIV-1 ANCHOR system allows only the visualization of potential functional vDNA. We optimized and set up our imaging approach in HeLa P4R5 cells (Charneau et al., 1994), a widely used cellular model for HIV-1 infection. Previous studies showed that HeLa cells enable single particle tracking of HIV-1 proteins (Burdick et al., 2020; Francis and Melikyan, 2018). However, since HIV-1 genome dissociates from viral proteins to become a proviral DNA or an extrachromosomal DNA, the track of these viral forms is the only direct measurement to investigate the fate of the viral genome into the host nuclear space. Unlike previous studies, we are now able to directly visualize by live imaging the nuclear vDNA as GFP puncta appearing in the host nucleus once retrotranscribed (Fig. 1A). On the contrary, bright individual spots are rarely visualized in the cytoplasm, where the vDNA is only partially accessible to OR-GFP, as we previously demonstrated (Blanco-Rodriguez et al., 2020). HIV-1 ANCHOR is able to specifically label the vDNA as shown by the correlation between the nuclear GFP puncta and the MOI (multiplicity of infection) used (Fig. S1A). This specificity was corroborated using the reverse transcription inhibitor, Nevirapine (NEV), which completely prevents the detection of vDNA (Fig. 1B, S1B). We then investigated whether HIV-1 ANCHOR allows the detection of different intranuclear vDNAs, such as episomal and integrated forms. To generate episomal forms, either we infected cells with HIV-1 ANCH3 in the presence of the integration inhibitor Raltegravir (RAL), or using an integration-deficient virus, which is mutated at the catalytic site of the integrase (IN) enzyme (HIV-1 ANCH3 IND116A). We detected intranuclear GFP puncta under both conditions (Fig. 1B), meaning that HIV-1 ANCHOR permits the detection of nuclear episomal vDNA forms. The experiments were validated by qPCR, which confirmed the extrachromosomal forms generated during infection when the integration step is impeded (Fig. 1B). We next assessed whether HIV-1 ANCHOR is sufficiently sensitive to allow the detection of integrated vDNA at the level of a single provirus. We selected single HeLa P4R5 clones carrying the HIV-1 ANCH3 genome. The imaging data correlates with results obtained by ALU PCR (Fig. 1C), demonstrating that this technology is powerful enough to detect a single provirus. Thus, we used this system to live track proviruses and episomal forms to investigate their behaviour in the nucleus (movies S1, S2, and S3). We performed live tracking of vDNAs in cells at six days post infection (p.i.). (movie S1), which contain a negligible amount of episomal forms, likely lost during cell division (Fig. S1C), in the single-cell clone (movie S2) or in cells containing only the episomal vDNA (movie S3). Then, we compared vDNA trajectories in the aforementioned experimental conditions (Fig. 1D). The stable clone and the cells at six days p.i. show vDNA with similar trajectories and diffusion coefficient (D (nm2/s) < 5). Instead, infected cells carrying unintegrated viruses harbored two populations composed of episomal forms with divergent behaviors, one with a low D (D (nm2/s) < 5) and the other with a high D (5 < D (nm2/s) < 20) (Fig. 1D). Our data hint that most, but not all, episomal forms are retained in particular nuclear regions, most likely interacting with host factors or chromatin compartments, consistent with the findings of other studies (Geis and Goff, 2019; Zhu et al., 2018), whereas some episomal forms are free to move in the nuclear space. On the other hand, the integrated forms showed a uniform, low-diffusive behavior.
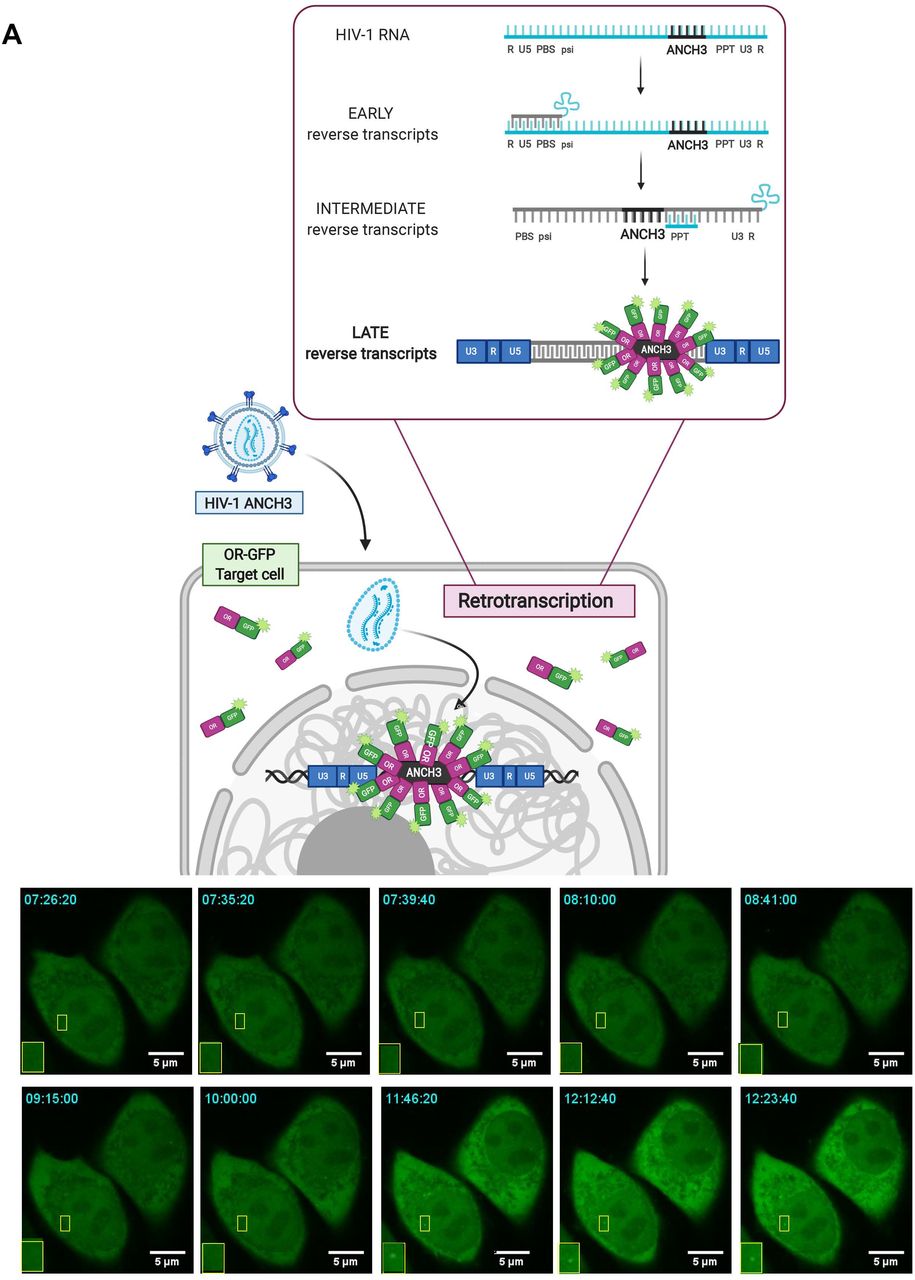
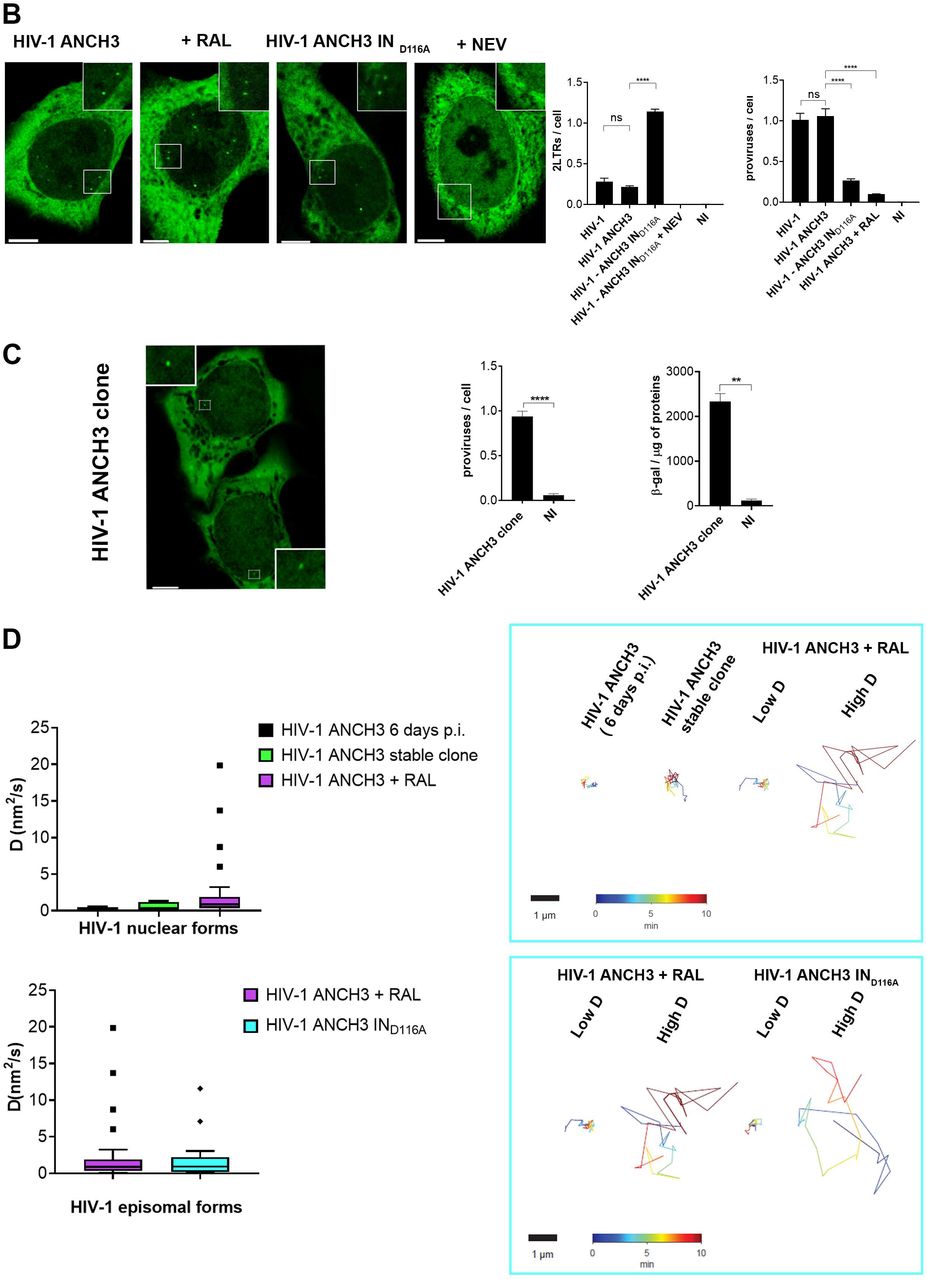
A) Scheme of vDNA labeled by the HIV-1 ANCHOR system. ANCH3 is the target sequence of the OR protein. ANCH3 has been cloned in the HIV-1 genome in place of Nef gene (HIV-1 ANCH3). The late retrotranscribed DNA of HIV-1 ANCH3 can be visualized in cells stably expressing the OR-GFP protein, thanks to the accumulation of OR protein to the double-stranded ANCH3 sequence. Viral DNA detection occurs mostly in the nucleus, where late reverse transcripts are abundant and are more likely to be exposed to the binding of OR-GFP. Starting from early time points of infection, it is possible to follow HIV-1 ANCH3 in target cells. The labelling results in the appearance of a bright fluorescent spot that can be tracked over time (time post-infection is indicated in the upper-left corner). The time-lapse images are representative of three independent experiments. The cartoon model was created with BioRender.com. B) Confocal images of HeLa P4R5 cells expressing OR-GFP infected at MOI 30 with HIV-1 ANCH3, HIV-1 ANCH3 + RAL (20 μM), HIV-1 ANCH3 IND116A and HIV-1 ANCH3 + NEV (10 μM), 24 h p.i. On the right, histogram plots of 2-LTR circles and ALU-PCR, comparing the different infection conditions, represent the nuclear import and integration rates, respectively. All graphs contain ± SD. C) Confocal image of HIV-1 ANCH3 provirus in a single-cell clone. The clone was transduced with OR-GFP LV for the detection of HIV-1 integrated DNA. On the right, histogram plot of ALU-PCR and β-galactosidase expression of the HIV-1 ANCH3 single-cell clone compared to non-infected cells (NI). All graphs contain ± SD. C) On the left, Tukey’s box plots of the diffusion coefficient (D (nm2/s)) of episomal and integrated HIV-1 DNA forms tracked in 3D time lapses (movies S1, S2, S3). On the right, an example of live track for each experimental group is shown. Low D corresponds to D (nm2/s) < 5 and high D to 5 < D (nm2/s) < 20. The color range indicates the time up to 10 min. Statistics: Student’s t test for two comparisons, one-way ANOVA for more than two comparisons. ns=non-significant, *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001. Scale bars: 5 μm.
Detection of actively transcribing viral DNA at single-cell level in HIV-1 target cells
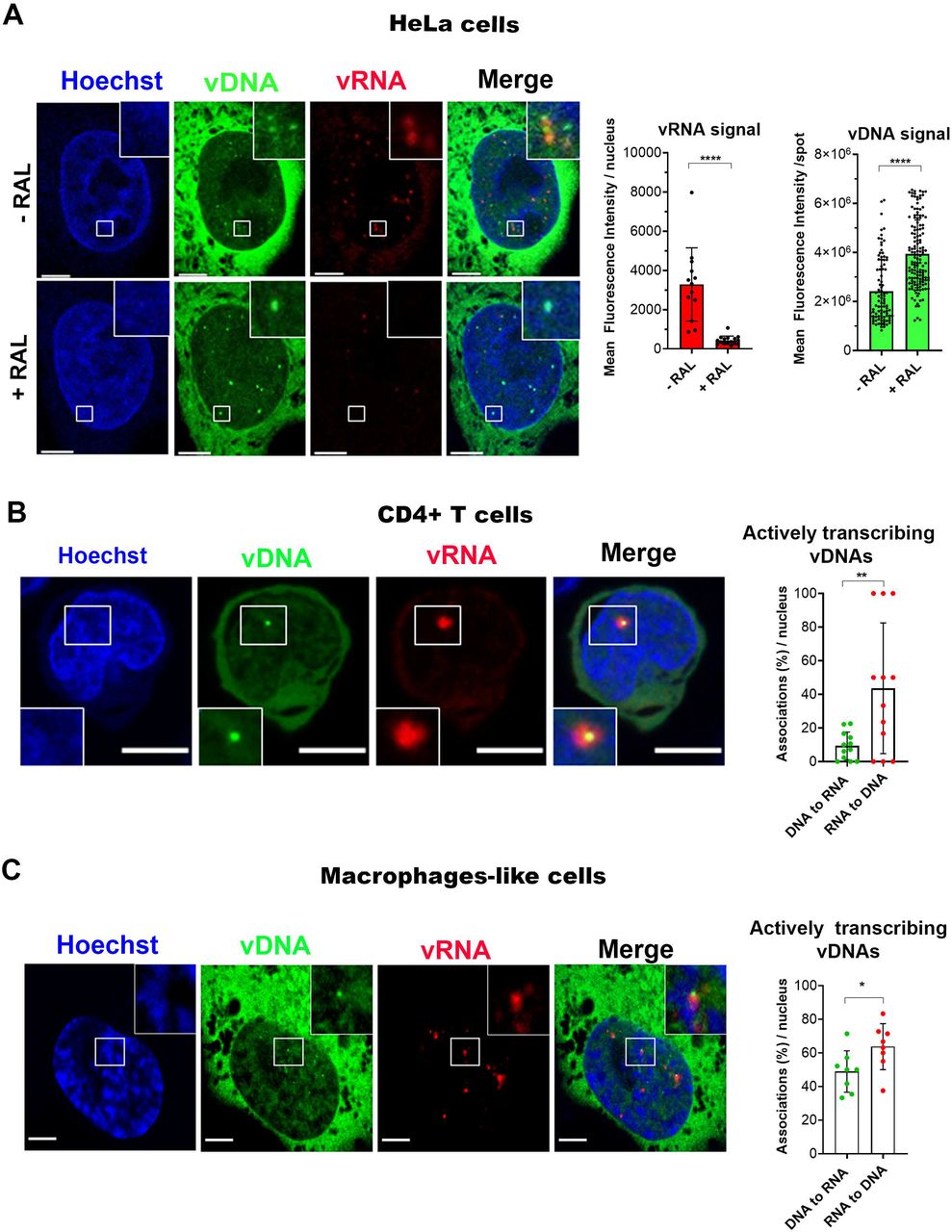
The identification of functional vDNA in the nucleus of infected cells has been one of our goals. For this purpose, we combined HIV-1 ANCHOR technology with RNA FISH (single molecule inexpensive FISH, smiFISH) against HIV-1 polymerase transcripts, to detect vRNA foci of transcription (Tsanov et al., 2016). As expected, in cells treated with RAL, where the integration step is blocked, vRNA signal is highly depleted. In untreated cells it is possible to observe vDNA-vRNA associations, highlighting the proviruses location (Fig. 2A), in fact proviruses are the only transcribing forms. We strengthen our results on the ability to visualize proviruses by showing the vDNA-vRNA association by live imaging. HIV-1 transcriptional activity was tracked by MCP-GFP system (kind gift from E. Bertrand) to label vRNA (Tantale et al., 2016) coupled with HIV-1 ANCHOR technology (Fig. S2A, B). Next, we compared the intensity of ANCHOR signals of the episomal forms (RAL) with those of the integrated forms (untreated cells) and observed that the signal of unintegrated DNA was two-fold brighter than the proviral DNA spot (Fig. 2A). Interestingly, we also remarked, during live imaging, that the vDNA of the integration-deficient virus (HIV-1 ANCH3 IND116A) aggregated to form “flower-shaped structures” in the nucleus (Fig. S3, movie S4). Aggregation of extrachromosomal forms could prompt a brighter appearance of viral episomal DNA compared to a dimmer signal of the individual proviral DNA (Fig. 2A). Next, we tested the HIV-1 ANCHOR system in the context of the main target cell of HIV-1, primary CD4+T cells, derived from healthy donors. CD4+T cells challenged with various MOIs of HIV-1 ANCH3 showed that the number of nuclear spots increased with the increasing of the MOI (Fig. S4), meaning that HIV-1 ANCHOR specifically detects the vDNA in the nucleus of primary lymphocytes. The correlation between different MOIs used and the number of proviruses (r=0.9412), analysed by qPCR, indicates that OR-GFP does not impede the viral integration step (Fig. S4). We tested, as well, the co-detection of viral transcriptional foci and vDNA in T lymphocytes (primary CD4+T and Jurkat cells) through RNA FISH (Fig. 2B, Fig. S5). We observed that not all vDNAs associated to a transcriptional focus at 48 h p.i., most likely because of the presence of silent episomal forms. On the other hand, the majority of cells analyzed contain vRNA foci colocalized with vDNA (Fig. 2B), meaning that we are able to detect single transcribing proviruses. We also performed similar experiments in non-dividing cells, THP-1, which are macrophage-like cells. Thus, we imaged and analyzed THP-1 cells at 3 days p.i. and we observed that also in these cells we could distinguish individual transcribing proviruses from silent viral forms (Fig. 2C). Of note, the RNA signal considered in our images was exclusively due to the detection of viral transcription foci, since samples treated with NEV did not show any significant intra-nuclear signal (Fig.S6). Taken together, our results suggest that we are able to distinguish episomal forms (mainly silent) from integrated forms that are the only ones able to replicate.

A) Labeling of vDNA and vRNA in HeLa cells expressing OR-GFP obtained by coupling of HIV-1 ANCHOR (vDNA) with RNA FISH (vRNA). On the left, confocal images of HIV-1 RNA FISH with or without RAL (20 μM), 24 h p.i. On the right, scatter plot with bars showing the mean fluorescence intensity of the vRNA signal (red) per nucleus and of vDNA spots detection (green) (for vRNA: n = 13 cells (-RAL), 16 cells (+RAL); for vDNA: 95 (-RAL) and 141 (+RAL) spots, n = 13 cells (-RAL), 16 cells (+RAL)). The analysis was performed in 2D. B) Labeling of vDNA and vRNA in CD4+T cells expressing OR-GFP. On the left, confocal image of HIV-1 RNA FISH in primary activated CD4+T cells, 3 days p.i‥ On the right, scatter plot with bars showing the percentage of vDNA associated to vRNA signal and vice versa, per nucleus (n= 262 (vDNA spots); 64 (vRNA foci) in infected Jurkat cells, 48 h p.i. (some examples of cells are shown in Fig. S5). The analysis was performed in 3D. C) The same techniques have been used to label vDNA and vRNA in macrophage-like cells, THP-1, expressing OR-GFP. On the left, confocal image of infected THP-1, 3 days p.i. On the right, scatter plot with bars showing the percentage of vDNA associated to vRNA signal and vice versa, per nucleus (n=130 (vDNA spots), 99 (vRNA foci). The analysis was performed in 3D. All graphs contain ± SD. Statistics: Student’s t test. ns=non-significant, *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001. Scale bars: 5 μm.
Nuclear distance of actively transcribing proviruses from the nuclear envelope at single-cell level in dividing and non-dividing cells
The location of actively transcribing proviruses in the nuclear space of dividing cells is still a subject of debate (Achuthan et al., 2018; Burdick et al., 2017; Chin et al., 2015; Di Primio et al., 2013; Lelek et al., 2015; Marini et al., 2015), whereas their nuclear location in terminally differentiated cells, as far as we know, has never been studied before. In dividing cells, the 3D analysis of the nuclear location of the vDNA showed the vDNA signals at 48 h p.i. to be more randomly distributed throughout the nucleus compared to the vDNA spots at seven days p.i. (Avg distances from the NE: ~2.5 μm at 48 h p.i. vs ~1.4 μm at 7 days p.i.) (Fig. 3A). The difference in the average distance of the vDNA spots from the NE between the early and late time points p.i. may be due to the co-existence of heterogeneous viral forms at 48 h p.i. (episomal and proviral forms), whereas the vDNA population is more homogeneous at seven days p.i. (Fig. 1D, 3A). In fact, at seven days p.i., almost all remaining vDNAs in the nucleus consisted of integrated forms, as suggested by the imaging of dividing cells following prolonged treatment with RAL (Fig. S1C). Interestingly, we did not detect differences between the distances of the vRNA foci from the NE (~ 1.5 μm) at both time points (48h p.i. and 7d p.i.) and the vDNA spots at seven days p.i. (~ 1.4 μm), indicating that in dividing cells the integration of the proviral DNA occurs early during infection near to the NE (Fig. 3A). We performed similar 3D analysis to calculate the distances of vDNAs and vRNAs from the NE in non-dividing cells at three days p.i‥ The lack of mitosis hinders the loss of the episomal forms in non-dividing cells, so we could not discriminate between integrated and unintegrated vDNA forms. However, when we compared vDNAs (episomal and proviral forms) versus vRNA foci (actively transcribing proviral forms) their analysis of distances from the NE revealed a major vicinity of the proviruses to the NE compared to the total vDNA (~2.2 μm for the vDNA vs ~1.8 μm for the vRNA) (Fig.3B). Overall, our data show a similar nuclear spatial distance of actively transcribing proviral forms from the NE at single-cell level in both, dividing and non-dividing cells (distance of vRNA foci from the NE: ~1.7 μm in HeLa cells and ~1.8 μm in THP-1 cells) (Fig.3C).
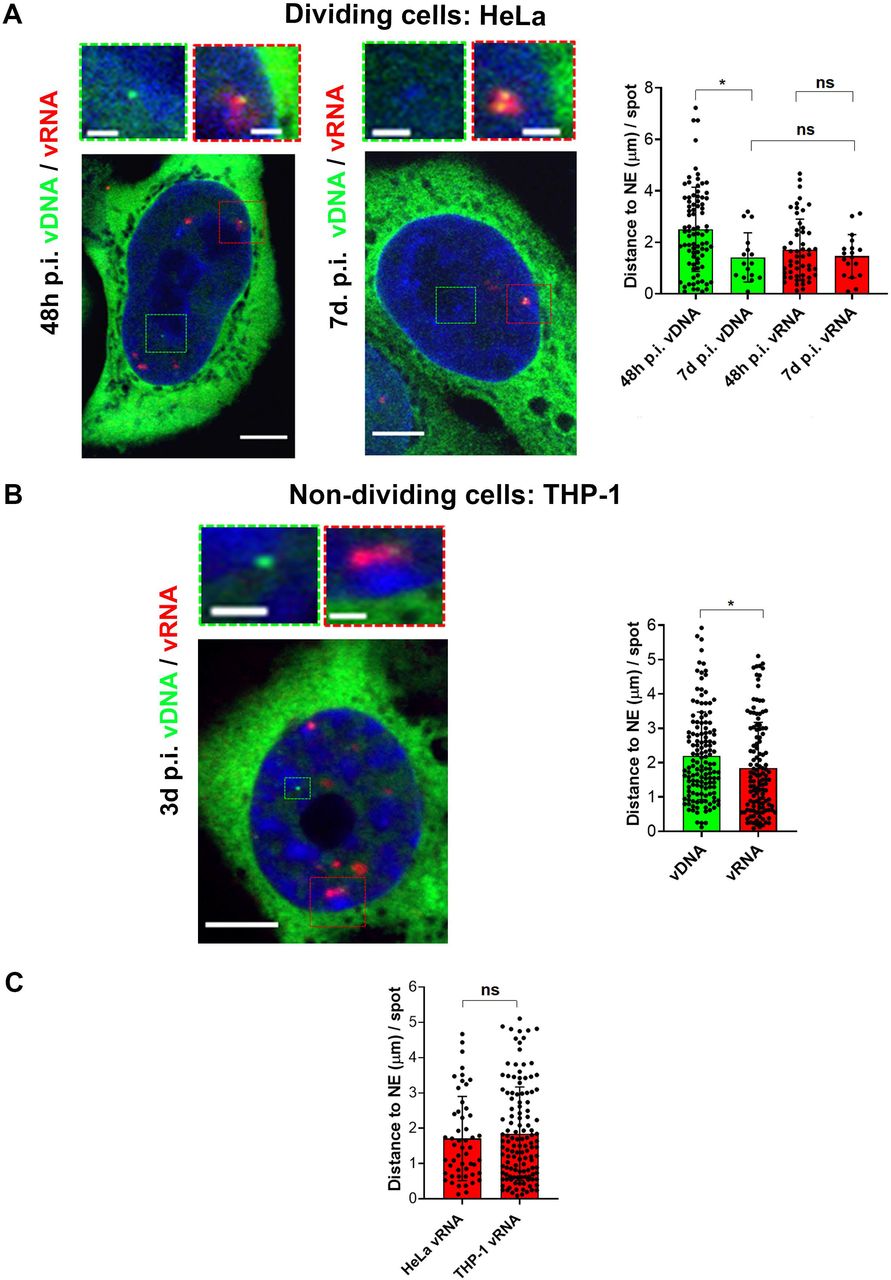
A) On the left, confocal images of HIV-1 DNA labeling by HIV-1 ANCHOR system and HIV-1 RNA labeling by RNA FISH in HeLa cells expressing OR-GFP infected with HIV-1 ANCH3 at 48 h p.i. compared to 7 days p.i‥ On the right, scatter plot with bars indicating the distance from the nuclear envelope (NE) per detected vDNA spot (green) and vRNA focus (red) (n=vDNA spots: 83 (48 h p.i.) and 16 (7 d p.i.); n=vRNA foci: 52 (48h p.i.) and 18 (7d p.i.)). B) On the left, confocal images to detect vDNA and vRNA in THP-1 cells expressing OR-GFP infected with HIV-1 ANCH3, 3 days p.i‥ On the right, scatter plot with bars indicating the distance from the nuclear envelope (NE) per detected vDNA spot (green) and vRNA focus (red) (n=139 vDNA spots; n= 126 vRNA foci). C) Scatter plot with bars comparing the distances from the nuclear envelope (NE) per detected vRNA focus in HeLa P4R5, 48h p.i. and THP-1, 3 days p.i. (vRNA foci: 52 (HeLa), 126 (THP-1)). All the analyses were performed in 3D. All graphs contain ± SD. Statistics: Student’s t test for two comparisons, one-way ANOVA for more than two comparisons. ns=non-significant, *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001. Scale bars: 5 μm, inset 1 μm.
The majority of IN proteins are retained in CPSF6 clusters and separate from the late retrotranscribed DNA
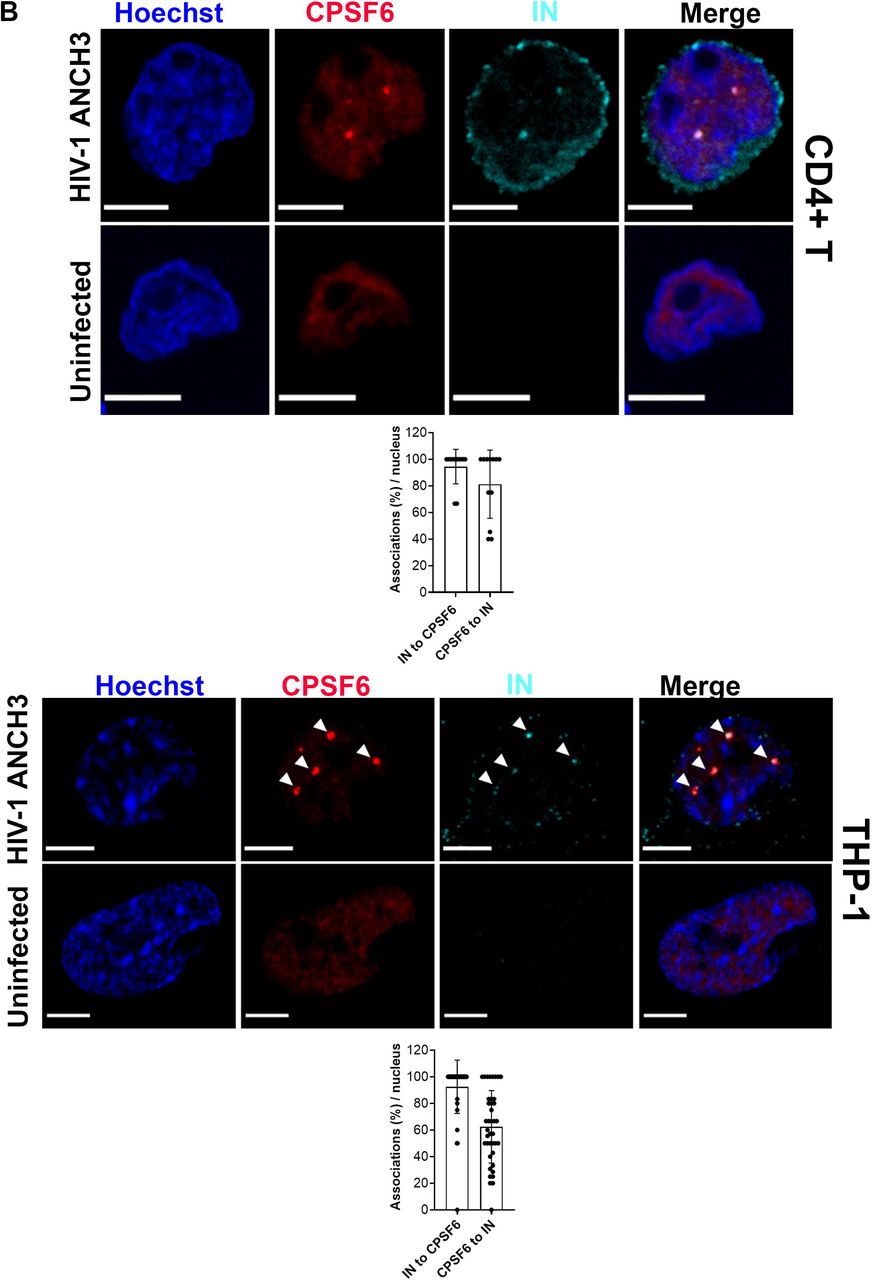
A long-standing question in the early steps of HIV life cycle is whether viral IN proteins remain all associated with late reverse transcribed DNA for long time or only for a short time after the nuclear entrance. This association may not be detectable because of the short-time nature of the event or due to the technical limitations of vDNA and IN visualization. Thus, we exploited our vDNA tracking system to investigate about IN proteins localization in primary CD4+T cells in comparison to terminally differentiated cells, THP-1. We challenged both cell types with HIV-1 ANCH3 and we analysed them at 20h and 30h p.i., respectively. We found that in both cells the vDNA/IN association event was very rare (only one single event in 1 cell for THP-1 and in 3 cells for CD4+T cells) (Fig. 4A). Nuclear IN protein clusters were found in the nucleus, but not at the site of the late reverse transcripts (Fig. 4A). To confirm our data we performed live-imaging, asking whether we could pinpoint the dynamic evolution of the association between the vDNA and the IN in the nucleus of infected cells by coupling the HIV-1 ANCHOR system with the GIR virus (generous gift from Edward Campbell)(Dharan et al., 2016; Hulme et al., 2015). We produced the GIR virus using a viral genome carrying the IN with a mutation in the catalytic site (HIV-1 ANCH3 IND116A), as consequence, the only active IN in these viral particles was the IN-Ruby (Fig. S7). Analysis of the dynamics of the vDNA / IN association shows the two signals to be separated (~ 0.6 to 0.8 μm) within the nucleus for several hours (Fig. S7, movie S5). Our results suggest that, after nuclear entry, the main localization of IN is not at the exact site of the viral DNA forms, even though we can sometimes spot them in proximity. Next, we investigated whether the viral IN was retained in particular nuclear regions by some nuclear host factors. We first found that the viral IN occupies the same positions as the viral CA and its host nuclear partner, CPSF6 (Fig. S8A). We confirmed that HIV-1 reprograms the nuclear localization of CPSF6 from being randomly dispersed in the nucleoplasm to form particular aggregates in both primary lymphocytes and macrophages (Francis et al., 2020) (Rensen et al., BioRxiv doi.org/10.1101/2020.04.12.038067) (Fig. 4B). We also observed that CPSF6-IN-CA clustering is independent of DNA synthesis (Fig. S8B). Then, we quantified the percentage of association per nucleus between the viral IN and CPSF6. We found an average of association of IN to CPSF6 ~ 94.4% in CD4+T cells and ~92.5% in THP-1 cells (Fig. 4B). Taken together our results show that in dividing and in non-dividing cells there is an intra-nuclear separation of the viral DNA and of the IN proteins. Interestingly, the viral IN proteins persist after viral nuclear entry associated to CPSF6 clusters. This observation suggests that the late reverse transcripts are released from the majority of IN proteins which instead are retained in a separated nuclear location occupied by CPSF6. Thus, CPSF6 clusters could serve as sites to release a mature PIC that could contain just few IN proteins necessary for the integration step (Ballandras-Colas et al., 2017; Hare et al., 2009).


A) Confocal images of primary activated CD4+T and THP-1 cells expressing OR-GFP infected with HIV-1 ANCH3, 20h and 30h p.i, respectively, compared to uninfected cells. Co-visualization of vDNA (green) and viral IN (magenta). On the bottom scatter plot with bars comparing the percentages of vDNAs associated to IN per nucleus (n=vDNA: 54 (CD4+T), 100 (THP-1)). The analysis was performed in 3D. B) Confocal images of primary activated CD4+T and THP-1 cells infected with HIV-1 ANCH3, 20h and 30h p.i, respectively, compared to uninfected cells. Co-visualization of CPSF6 (red) and viral IN (cyan). The graphs show the percentages of IN-CPSF6 association events and vice versa, per nucleus (n= for CD4+T: 46 (CPSF6), 33 (IN); for THP-1: 206 (CPSF6), 132 (IN)). All graphs contain ± SD. Statistics: Student’s t test. ns=non-significant, *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001. Scale bars: 5 μm.
Proviruses are excluded from HIV-1-induced membraneless organelles
CPSF6 participates to the viral nuclear translocation and post-nuclear entry steps by interacting with the viral CA (Bejarano et al., 2019; Burdick et al., 2017; Lee et al., 2010; Price et al., 2014) (Francis et al., 2020). Recent studies reported a high level of co-localization between the paraspeckle factor, CPSF6, or the speckle factor and splicing regulator, SC35, (Spector and Lamond, 2011) and the vDNA labeled with EdU (Francis et al., 2020) (Bejarano et al., 2019)(Rensen et al., BioRxiv doi.org/10.1101/2020.04.12.038067). Importantly, it has been shown that EdU labels nuclear clusters composed of multiple viral episomal forms (Francis et al., 2020) (Rensen et al.,BioRxiv doi.org/10.1101/2020.04.12.038067). Therefore, EdU labeling can detect multiple nascent vDNAs but it is likely not sufficiently sensitive to detect an individual viral genome. Contrary to EdU labeling, the HIV-1 ANCHOR system allows the visualization of a single provirus (Fig.1C) and does not interfere with viral transcription (Blanco-Rodriguez et al., 2020) (Rensen et al.,BioRxiv doi.org/10.1101/2020.04.12.038067). Thus, we used HIV-1 ANCHOR technology to visualize the late retrotranscripts in infected macrophage-like cells at 3 days p.i‥ We observed that late retrotranscripts do not colocalize with CPSF6 (Fig. 5A). However, upon viral infection, CPSF6 relocates and fully colocalizes with speckle-simile organelles labeled with SC35 (Fig. 5A). SC35 is strongly associated to another speckle factor, SON, independently of viral infection (r pearson ~0.995). More specifically, we found that the nuclear reorganization of CPSF6 is not required for the integration step, as the nuclear organization of CPSF6 was similar in cells treated or not with RAL (Fig. S9). Then, we investigated whether the vDNA colocalizes with SC35. We computed vDNA distances in 3D space from the boundary of SC35 bodies in cells treated with RAL (avg ~ 0.4 μm (Fig. S9)) or in absence of RAL (avg ~ 0.6 μm (Fig. 5A)). Therefore, similarly to CPSF6 also SC35 clusters exclude late reverse transcripts (Fig. 5A). In addition, we found that viral transcriptional foci where also located outside CPSF6/SC35 organelles (Fig. 5B). These results are independent of the presence of vpx (Fig. S10) that we used to increase viral infectivity in THP-1 (Hrecka et al., 2011; Laguette et al., 2011). Taken together analysis of the nuclear positions of vDNA (episomal and proviral forms) and vRNA foci (proviruses) showed that these viral nuclear forms are excluded from HIV-1 MLOs (Fig. 5C), indicating that those organelles cannot be sites for viral integration or replication.
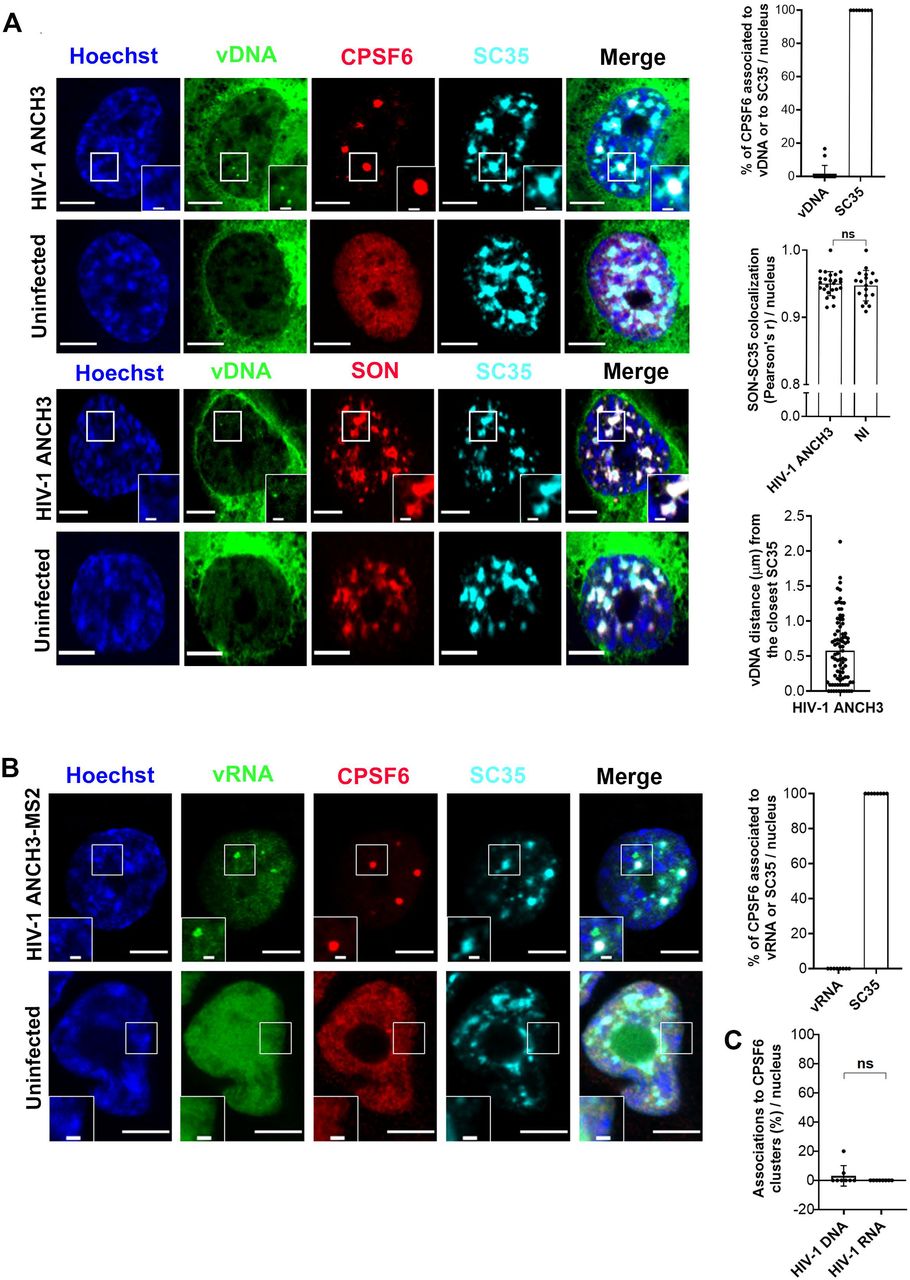
A) Confocal images of THP-1 cells expressing OR-GFP infected with HIV-1 ANCH3, 3 days p.i., to co-visualize vDNA (green), CPSF6 (red), SC35 (cyan) or to co-visualize vDNA (green), SON (red), SC35 (cyan), compared to uninfected cells. On the right, the graphs show: the percentages of CPSF6 clusters associated to vDNA or to SC35, per nucleus (n= CPSF6 clusters ˃ 50); the comparison of Pearson’s r coefficients of SON-SC35 colocalization between infected and uninfected cells, per nucleus (n = 25 cells (HIV-1 ANCH3), 19 cells (NI)); the average distance of each vDNA from the closest SC35-speckle border (n= vDNAs: 83). B) Confocal images of THP-1 cells expressing MCP-GFP infected with HIV-1 ANCH3 MS2, 3 days p.i., to co-visualize vRNA (green), CPSF6 (red), SC35 (cyan), compared to uninfected cells. The graph shows the percentages of CPSF6 clusters associated to vRNA foci or to SC35 (n= CPSF6 clusters: 42). C) Scatter plot with bars indicating the percentage of vDNA or vRNA associated to CPSF6, per nucleus (n= 69 (vDNA), 30 (vRNA)). All the analyses were performed in 3D. All graphs contain ± SD. Statistics: Student’s t test. ns=non-significant, *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001. Scale bars: 5 μm, inset 1 μm.
Late reverse transcripts DNA and viral RNA foci form complexes with LEDGF, spatially independent of CPSF6 clusters
We extended our study on THP-1 cells at 3 days p.i. to investigate whether Lens Epithelium-Derived Growth Factor (LEDGF), which is a partner of the viral IN and leads HIV-1 integration sites selection (Cherepanov et al., 2003; Ciuffi et al., 2005; Emiliani et al., 2005; Ferris et al., 2010; Hare et al., 2009; Lelek et al., 2015; Llano et al., 2006; Shun et al., 2008; Shun et al., 2007; Wang et al., 2014), is associated to the proviruses forming a complex visible by imaging. We observed that CPSF6 proteins cluster in proximity of LEDGF sites (~ 43%), without co-localizing (Fig. 6A). There is a similar number of LEDGF clusters in both infected and uninfected cells (Fig. 6A), while CPSF6 clusters are mainly formed upon infection, suggesting that CSPF6 is prompted by the virus to localize near the pre-existing LEDGF regions (Fig.6A). Of note, the PWWP domain of LEDGF has been shown to interact with H3K36me3 (Pradeepa et al., 2014; Pradeepa et al., 2012), which is a marker of euchromatin. In particular, this post-translational modification is enriched in HIV-1 integration sites (Ciuffi et al., 2005; Lelek et al., 2015; Vansant et al., 2020; Wang et al., 2009). LEDGF also interacts with splicing factors (Singh et al., 2015). Therefore, we analysed the location of proviral foci of transcription in relation to LEDGF and CPSF6. Our previous data shows that CPSF6 co-localizes with SC35, however the distance between the vDNA and SC35 has been estimated to be ~ 0.6 μm (Fig. 5A), therefore, the late retrotranscribed DNA is too distant from those HIV-1 MLOs to be considered part of the same complex. While, the vDNA of the late retrotranscripts is mostly excluded from CPSF6 clusters, we found that ~64% of the vDNA and ~73% of the vRNA foci associate with LEDGF (Fig. 6B). Next we have characterized in more detail the viral genome associations with LEDGF and CPSF6. We calculated that ~ 87.9% of the vDNA/LEDGF complexes and ~80% of vRNA foci/LEDGF complexes are uncoupled to CPSF6 (Figure 6B). We found only few vDNA (~12.1%) and vRNA (~20%) foci associated to LEDGF clusters coupled to CPSF6 (Fig. 6B). Our results suggest that the two cellular factors, CPSF6 and LEDGF, orchestrate two sequential steps of viral replication, the maturation of the PIC and its integration. Our data indicate that the functional vDNAs are not located inside CPSF6/SC35 clusters, but in external active chromatin domains, enriched in LEDGF, likely to benefit of a favorable environment for viral replication.

A) Confocal images of THP-1 cells expressing OR-GFP infected with HIV-1 ANCH3, 3 days p.i., to co-visualize CPSF6 (red) and LEDGF (white), compared to uninfected cells. On the left scatter plot of the percentages of CPSF6 found associated to LEDGF clusters per nucleus; on the right, scatter plot of the LEDGF and CPSF6 count per nucleus (n = 16 cells). Scale bar: 5 μm, inset 2 μm. B) Confocal images of THP-1 cells expressing OR-GFP infected with HIV-1 ANCH3 and expressing MCP-GFP infected with HIV-1 ANCH3 MS2, to co-visualize vDNA or vRNA (green), CPSF6 (red) and LEDGF (white). The scatter plot shows the percentage of vDNA or vRNA associated to LEDGF (n= 33 (vDNA), 15 (vRNA)). The pie charts show the percentages of the 2 populations of LEDGF clusters associated to vDNA or vRNA: LEDGF alone or LEDGF in proximity of CPSF6. Scale bar: 5 μm, inset 1 μm. All the analyses were performed in 3D. Statistics: Student’s t test. ns=non-significant, *=p ≤ 0.05, **=p ≤ 0.01, ***=p ≤ 0.001, ****=p ≤ 0.0001.
Discussion
Studies of parasites that permanently integrate into the host genome show that they have evolved to target chromatin in such a way as to optimize their coexistence with the host and favor the release of new viral progeny (Bushman, 2003; Craig and Marszalek, 2002). HIV-1 needs to rapidly release high levels of newly generated viruses after infection, intuitively explaining the preference of active genes as integration targets (Ciuffi et al., 2005; Di Nunzio et al., 2013; Lelek et al., 2015; Maldarelli, 2016; Schroder et al., 2002). However, the nuclear location of individual proviruses actively transcribing at the dividing or non-dividing single cell level has never been visualized before. Thus far, the direct detection of HIV-1 genomes after reverse transcription and the distribution of proviral transcriptional foci has been technically challenging. Recent studies have shown the possibility to follow HIV infection in live using surrogate viruses (IN GFP virus (Albanese et al., 2008), a mix of viruses carrying fluorescent and non-fluorescent IN proteins (Hulme et al., 2011), or indirectly follow the viral journey by labeling host viral partners, such as CypA (Francis and Melikyan, 2018) or APOBEC 3F (Burdick et al., 2017)). All these approaches allow to follow viral or host proteins but not viral replication complexes. HIV-1 ANCHOR system allows the live tracking of different nuclear late retrotranscribed DNA forms (Fig.1A), which makes possible to probe their behavior via the measurement of the mean-squared displacement (MSD). We observed a homogenous population formed by proviral DNA with low diffusion coefficient. Episomal forms showed a heterogeneous behavior, characterized by two different populations, implying that these two viral forms can interact with different factors in the nuclear space (Fig. 1D). Moreover, imaging analysis showed that viral episomal forms identified by the accumulation of OR-GFP on ANCH3 sequences gave rise to brighter signals than proviral forms (Fig. 2A). This phenomenon could account for the differences in behavior between integrated and unintegrated forms. Thus, unintegrated forms can be brighter for the following reasons: a) the absence of transcriptional machinery associated with the vDNA may reduce the steric hindrance that can interfere with the accumulation of OR-GFP on the HIV-1 tag sequence (ANCH3) (Fig. 2A), b) episomal forms can aggregate in a flower shape, generating a brighter signal (Fig. S3, movie S4), c) episomal forms are differently chromatinized than the integrated vDNAs, becoming more accessible to OR-GFP (Geis and Goff, 2019). Next, to track the nuclear location of functional proviruses we co-labeled vDNA and vRNA by combining HIV-1 ANCHOR technology with RNA FISH in the main target cells of HIV-1, CD4+T cells and macrophages (Fig.2A,B,C). We also corroborated results on the visualization of functional proviruses using a live track method, MCP-MS2 system (Tantale et al., 2016), coupled to HIV-1 ANCHOR (Fig.S2). Once the performance of our approach for labeling actively transcriptional proviruses was validated, we investigated the spatial location of proviruses in dividing and non-dividing cells. Three-dimensional analysis of the distribution of vDNAs during early (48 h p.i.) infection in mitotic cells displays that they are more randomly distributed than the proviral DNA from the single clone or from cells at 7days p.i. (Fig. 3A). We also observed that the location of viral transcription sites (vRNA) is highly similar between early and late time points of infection, indicating that proviral DNAs, the only transcribing forms (Fig. 2A), immediately localize to near the NE (~1.5 μm) early after infection (Fig. 3A) in dividing and non-dividing cells (Fig.3B,C). We next investigated the interplay among viral components and their association with host factors during post-nuclear entry steps. We corroborated our published data (Rensen et al.,BioRxiv doi.org/10.1101/2020.04.12.038067) showing that IN proteins strongly co-localize with CPSF6 clusters in macrophage-like cells and in this study we observed a similar association between IN and CPSF6 also in CD4+T lymphocytes at early time of infection (Fig. 4B). Contrary to CPSF6, the late vDNA products of retrotranscription rarely co-localize with IN in both types of cells (Fig. 4A). These results are in line with the dynamic coexistence of the IN and vDNA, which remain separated but in the proximity for several hours, as it has been shown by real-time imaging in HeLa cells (Fig. S7, movie S5). This result can appear in contrast with recent data from other groups and from us obtained with another DNA labeling technology, EdU (Francis et al., 2020) (Rensen et al., BioRxiv, 2020), showing the vDNA and the IN strongly colocalized with CPSF6. This apparent discrepancy can be explained by the intrinsic features of vDNA labeling tools. EdU system is capable of detecting nascent episomal forms of vDNA abundant in CPSF6 clusters, sites of nuclear reverse transcription (Rensen et al.,BioRxiv doi.org/10.1101/2020.04.12.038067). Of note, EdU labeling cannot provide direct information on viral replication, as EdU reduces viral transcription (Rensen et al., BioRxiv doi.org/10.1101/2020.04.12.038067). Contrariwise, HIV-1 ANCHOR system allows the detection of late reverse transcripts (Fig.1A) that can generate functional proviral DNA, as we observed in HeLa cells, human lymphocytes, and macrophage-like cells (Fig. 2A,B,C, Fig.S2A,B, Fig. S5). Thus, we combined HIV-1 DNA and HIV-1 RNA labeling to detect the nuclear landscape where functional viral forms are located. Interestingly, we observed that HIV-1 induces the remodeling of speckles characterized by the presence of SC35 and SON, in novel MLOs enriched in viral proteins, such as IN and CA, as well in cellular factors, like the paraspeckle member, CPSF6, (Fig. 4B, 5A,B, S8). Of note, we observed that CPSF6 is fully included inside organelles marked by SC35 during viral infection, otherwise CPSF6 is usually randomly dispersed in the host nucleus of uninfected cells (Fig.4B, Fig.5A). Likely, CPSF6 relocates to speckles prompted by the virus to usurp functions linked to these MLOs. We show that the formation of CPSF6 cluster is independent of viral DNA integration or viral DNA synthesis (Fig. S8B, S9) and that late reverse transcribed products are excluded from them (Fig. 5A). It is possible that once the vRNA is fully retrotranscribed in vDNA (Rensen et al., BioRxiv doi.org/10.1101/2020.04.12.038067), it will quickly leave the HIV-1 MLOs. In fact, vDNAs were located on average at a distance of ~ 600 nm from the boundary of SC35 (Fig. 5A). MCP-labelled vRNA foci shows that actively transcribing proviruses are also located outside HIV-1 MLOs, indicating that those are not sites of viral integration and replication (Fig.5B,C). We also observed that CPSF6 relocates to the vicinity of LEDGF during HIV infection (Fig. 6A). LEDGF is required for the targeting of highly-spliced transcriptional units through its direct interaction with several splicing factors (Singh et al., 2015). LEDGF is also known to determine HIV-1 integration sites distribution in active chromatin sites (Ciuffi et al., 2005; Ferris et al., 2010; Shun et al., 2007), however the visualization of a complex formed by the transcribing provirus and LEDGF remained utopian for a long time. Here, we were able to visualize by fluorescence microscopy a complex formed by the vDNA or vRNA and LEDGF. More than 80% of those complexes are formed by LEDGF uncoupled to CPSF6 (Fig.6B). However, we observed that less than 20% of them can include CPSF6 associated to LEDGF, suggesting that few proviruses need to be near to HIV-1 MLOs without co-localizing, against the majority of them that can replicate farther (Fig. 6B). Whether there is a functional difference between these two complexes it can be further investigated in future studies. Overall, our study highlights the proviral location in the nuclear landscape remodelled by the viral infection (Fig.7). We found that HIV-1-MLOs retain the viral IN but exclude the late retrotranscribed DNA, indicating a role for HIV-1 MLOs prior to the integration step. In particular, we found foci of viral transcription outside HIV-1 MLOs, located in the nuclear periphery, in complex with LEDGF, indicating that HIV-1 MLOs cannot be sites of viral integration and viral transcription. Furthermore, the visualization of the formation of an actively transcribing provirus in complex of a pre-existing cluster of LEDGF, which is known to lay on active chromatin sites (Ciuffi et al., 2005; Singh et al., 2015) (Fig.7), indicates that LEDGF is not part of the viral complex during nuclear entry (Fig.6A). Taken together our results show that functional proviruses establish near to the NE, excluded from CPSF6 clusters, but locate in a favorable transcriptional environment ~ 600nm from HIV-1 MLOs (Fig.7). Our study supports how new single-cell level approaches are pivotal in the study of functional viral nuclear dynamics, to discriminate the cells susceptible to fuel viremia or that concur to viral persistence.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Once retrotranscribed and imported in the nucleus, HIV-1 genome integrates in open chromatin regions enriched in LEDGF proteins, in less than 2 micrometers form the nuclear envelope. In addition, upon infection, CPSF6 clusters in SC35 nuclear speckles with the viral IN restructuring the nuclear landscape and inducing HIV-1 MLOs formation. Late retrotranscribed DNA and proviruses are excluded from CPSF6 clusters. HIV-1 transcription is favored by the nuclear environment surrounding proviral sites that may profit from the proximity to SC35 and other splicing factors for post-transcriptional processes. Created with BioRender.com.
Materials and Methods
Cell lines, primary cells and generation of genetically modified single-cell clones
HEK 293T cells (ATCC) are human embryonic kidney cells used to produce lentiviral vectors and HIV-1 viruses. HeLa P4R5 reporter cells are HeLa CD4+CCR5+CXCR4+, carrying the LacZ gene under the control of the HIV-1 LTR promoter (Charneau et al., 1994). HeLa MCP-GFP is a HeLa clone stably expressing MCP-GFP bacterial fusion protein (kind gift from E. Bertrand) (Tantale et al., 2016). Jurkat cells (ATCC) are human immortalized T lymphocytes derived from acute T cells leukemia. THP-1 cells (ATCC) are human immortalized monocyte cells derived from acute monocytic leukemia, which, once seeded, were differentiated into macrophage-like cells under Phorbol 12-myristate 13-acetate (PMA) treatment (167nM). HEK293T cells and HeLa cells were cultivated in DMEM medium supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin-streptomycin. Jurkat and THP-1 cells were cultivated in RPMI 1640 medium supplemented with 10 % Fetal Bovine Serum (FBS) and 1% penicillin-streptomycin. Primary CD4+T cells were purified from healthy donors’ blood obtained via EFS (Etablissement Français du Sang, Paris), through density gradient centrifugation with Ficoll 400 and isolation of PBMCs buffy coat. Subsequent positive selection was performed with human CD4+Microbeads (Miltenyi Biotec #130-045-101). The day after CD4+T cells were activated with T cell Activation/Expansion kit (Miltenyi Biotec #130-091-441) according to protocol, complete RPMI 1640 medium was supplemented with interleuykin-2 (IL-2) (400 IU/mL). HIV-1 ANCH3 single-cell clone was generated starting from HeLa P4R5 cells which were infected with HIV-1 ANCH3 virus at MOI 1. Cells were diluted to 1 cell per well, in 96-well plates. Cell-clone colonies were tested for β-galactosidase expression to check viral expression (kit Roche #11758241001). Positive clones were transduced with lentiviral vector (LV) CMV-OR-GFP for imaging analyses of HIV-1 ANCH3 provirus. All cells were kept in incubator at 37°C and 5% CO2.
Plasmids and lentiviral vector productions
HIV-1ΔEnvINHAΔNef plasmid encodes for the ΔEnvHIV-1 LAI (BRU) viral genome where the Integrase (IN) protein is fused to the hemagglutinin (HA) tag (kindly provided by F. Mammano)(Petit et al., 1999; Petit et al., 2000). HIV-1ΔEnvINHAΔNef ANCH3 (HIV-1 ANCH3) was obtained through ANCH3 insertion: ANCH3 sequence was cloned by PCR using the template plasmid pANCH3 as we previously described in Blanco et al. (Blanco-Rodriguez et al., 2020). The ANCHOR™technology and sequences are exclusive property of NeoVirTech (Germier et al., 2017; Mariame et al., 2018). HIV-1ΔEnv INHA(D116A) ΔNef ANCH3 was obtained by insertional mutagenesis using the QuikChange II XL Site-Directed Mutagenesis kit (Agilent #200522), for integrase (IN) mutation at D116. The lentiviral vector plasmids CMV OR-GFP and CMV OR-SANTAKA were obtained by cloning OR-GFP/OR-SANTAKA (plamids from NeoVirTech) in a pTripCMV (ΔU3) plasmid through PCR and cloned using restriction site, AgeI and SgrDI. HIV-1ΔEnvINHAΔNef ANCH3 MS2 plasmid was obtained by inserting the MS2×64 sequence amplified by PCR from pMK123-MS2×64 (Tantale et al., 2016) in HIV-1ΔEnvINHAΔNef ANCH3. Lentiviral vectors and HIV-1 viruses were produced by transient transfection of HEK293T cells through calcium chloride coprecipitation. Co-transfection was performed as following, LV carrying OR-GFP/OR-SANTAKA: 10 μg of OR-GFP/OR-SANTAKA LV, 10 μg of NDK plasmid (gag-pol-tat-rev) and 2.5 μg of pHCMV-VSV-G envelope expression plasmid; LV carrying MCP-GFP (kindly provided by E. Bertand): 10 μg of MCP-GFP, 10 μg of NDK plasmid (gag-pol-tat-rev) and 2.5 μg of pHCMV-VSV-G envelope expression plasmid; HIV-1ΔEnv pseudotyped with VSV-G: 10 μg HIV-1ΔEnvINHAΔNef (IN WT or IN(D116A)) plasmid and 2.5 ug of pHCMV-VSV-G plasmid. VSV-G/HIV-1ΔEnv INHA(D116A) ΔNef ANCH3 has been also produced in combination with GIR (Gag-IN-Ruby plasmid, kindly provided by E. Campbell) (Dharan et al., 2016; Hulme et al., 2015). For THP-1 infection, HIV-1 ANCH3 or HIV-1 ANCH3 MS2 viruses and LV OR-GFP or LV MCP-GFP were all produced adding 3 μg of SIVMAC Vpx (Durand et al., 2013) at the moment of HEK293T transfection. After the collection of the supernatant 48h post-transfection, lentiviral particles were concentrated by ultracentrifugation for 1 h at 22000 rpm at 4°C and stored at −80°C. Lentiviral vectors and viruses were tittered through late reverse transcripts (LTR) qPCR in HEK293T cells or HeLa P4R5 cells.
Quantitative PCR and primers
Total DNA of infected cells was extracted at 6 hours post infection for Late Reverse Transcripts (LRTs) qPCR and 24 hours post infection for 2LTR-circles and ALU-PCR, through QIAmp DNA micro kit (QIAGEN #56304). Real-Time PCR of LRTs was performed to assess DNA synthesis and used to normalize qPCR data on viral input. The reactions were carried on in 20 μL, in iTaqUniversal SYBR Green Supermix (Bio-Rad #1725124) using primers for U3 sequence: U3 FX: 5’-TTCCGCTGGGGACTTTCCAGGG-3’, U3 RX: 5’-AGGCTCAGATCTGGTCTAACC-3’. Real-Time PCR of 2LTR-circles was used to asses nuclear import efficiency. Reactions were performed in 20 μL, in Maxima Probe/ROX qPCR Mastermix (ThermoFisher #K0232) using primers for 2LTR-circle junction: 2LTR FX: 5’-AACTAGGGAACCCACTGCTTAAG-3’, 2LTR RX: 5′-TCCACAGATCAAGGATATCTTGTC-3′, 2-LTR probe: 5’-(FAM)-ACACTACTTGAAGCACTCAAG-GCAAGCTTT-(TAMRA)-3’. Real-Time PCR of proviral integrations consisting in a first non-kinetic PCR in 50 μL in Platinum SuperFi DNA Polymerase (ThermoFisher #12351250) and in a second step of qPCR reaction in 20 μL in Maxima Probe/ROX qPCR Mastermix (ThermoFisher #K0232). Primer Alu 166: 5’-TCCCAGCTACTCGGGAGGCTGAGG-3’, Alu 2: 5’-GCCTCCCAAAGTGCTGGGATTACAG-3’, LambdaU3: 5′-ATGCCACGTAAGCGAAACTTTCCGCTGGGGACTTTCCAGGG-3′ for ALU PCR; Lambda: 5’-ATGCCACGTAAGCGAAACT-3’, U5: 5’-CTGACTAAAAGGGTCTGAGG-3’, Probe: 5’-(FAM)-TTAAGCCTCAATAAAGCTTGCCTTGAGTGC-(TAMRA) for ALU qPCR(Di Nunzio et al., 2013). In all experiments β-actin detection was used for normalization. β-actin FX: 5’-AACACCCCAGCCATGTACGT-3’, β-actin RX: 5-CGGTGAGGATCTTCATGAGGTAGT-3’, β-actin probe: (FAM)-CCAGCCAGGTCCAGACGCAGGA-(BHQ1).
Epifluorescence and Immunofluorescence
For imaging studies in adherent cell-lines, the cells were plated on coverslips (12mm diameter in 24 well plates or 18 mm diameter in 12 well plate, #1, ThermoFisher) at least 24h before fixation. For HIV-1 ANCH3 imaging, HeLa P4R5 cells were transduced with OR-GFP LV (MOI 0.2) and then infected within the following 3 days with HIV-1 ANCH3 or HIV-1 IND116A ANCH3 at different MOIs. NEVIRAPINE (NEV) 10 μM or RALTEGRAVIR (RAL) 20 μM were used to block respectively DNA synthesis and integration. For HIV-1 MS2 RNA imaging HeLa-MCP cells were infected with HIV ANCH3 MS2 and fixed 24h post infection. For co-visualization of vDNA and vRNA, HeLa MCP-GFP cells were transduced with OR-SANTAKA LV (MOI 1) and 24h later infected with HIV-1 ANCH3 MS2. All THP-1 cells studies were carried on after 48h of differentiation with PMA (167 nM), then, they were transduced with LV OR-GFP Vpx (MOI 5) or LV MCP-GFP Vpx (MOI 20) and 48 h later they were infected with HIV-1 ANCH3 Vpx (30 h or 3 days p.i) or HIV-1 ANCH3 MS2 ± Vpx (3 days p.i.). The medium was always supplemented with PMA. For HIV-1 ANCH3 imaging in primary lymphocytes, CD4+T cells, 3 days after activation, were transduced with OR-GFP LV (MOI 0.5) for 72 h and then infected with HIV-1 ANCH3 for 20 h. The day of fixation cells in suspension were seeded on poly-L-lysine (Sigma #P4707) coated coverslips and centrifuged to allow attachment. The day of fixation all cells were washed with PBS and fixed with 4% PFA for 15 minutes. For protein staining, cells were treated with glycine 0.15% for 10 min, permeabilized with Triton X‐100 0.5% for 30 min and blocked with 1% bovine serum albumin (BSA) for 30 min. All incubations were carried out at room temperature, in the dark, in humid chamber, 1h with primary antibodies and 45 min with secondary antibodies. Washes between antibody incubations and antibodies dilution were done in 1% BSA. Primary antibodies were diluted as follows: anti-HA 1:500 (Roche #11867423001), anti-HIV-1 p24 (CA) 1:400 (NIH #3537), anti-CPSF6 1:400 (Novus Biologicals #NBP1-85676), anti-SC35 1:200 (Abcam #ab11826), anti-LEDGF 1:200 (BD Bioscience #611715). Secondary antibodies used were the following: Goat anti-Mouse Alexa-647 1:300 (1:100 for CA) (Invitrogen #A21235); Donkey anti-Rat Alexa-488 1:100 (Invitrogen A21208) for IN-HA) or Goat anti-Rat Alexa-647 1:300 (Invitrogen #A21247) (1:100 for IN-HA); Donkey anti-Rabbit Cy3 1:500 (Jackson Lab #711-165-152). Finally, cells were stained with Hoechst 33342 1:5000 (Invitrogen #H3570) for 5 minutes. Coverslips were mounted on glass slides (Star Frost) with Prolong Diamond Antifade Mountant (Life Technologies #P36970). Confocal microscopy was carried out with a Zeiss LSM700 inverted microscope, with a 63X objective (Plan Apochromat, oil immersion, NA=1.4).
RNA FISH
For RNA FISH studies in adherent cell-lines, the cells were seeded on coverslips. HeLa P4R5 cells expressing OR-GFP were infected with HIV-1 ANCH3 (MOI 30) with or without RAL 20 μM and fixed 24h post infection or infected with HIV-1 ANCH3 (MOI 5) for 48h or 7 days. Jurkat T cells expressing OR-GFP were infected with HIV-1 ANCH3 (MOI 5) and fixed 48h post infection. Primary activated CD4+T cells, 3 days after activation, were together transduced with OR-GFP LV (MOI 10) and infected with HIV-1 ANCH3 (MOI 5) for 3 days,.THP-1 cells were differentiated with PMA for 48h and, after 48h transduction with LV OR-GFP Vpx (MOI 5), the cells were infected with HIV-1 ANCH3 Vpx (MOI 20) for 3 days. The day of fixation Jurkat and CD4+T cells were seeded on poly-L-lysine (Sigma #P4707) coated coverslips and centrifuged to allow attachment. The day of fixation all cells were washed with PBS and fixed with 4% PFA for 15 minutes and incubated in 70% ethanol at −20°C at least for one night. Primary smiFISH probes have a targeting sequence against HIV-1 pol transcript and a shared readout sequence for secondary probe alignment. Twenty-four smiFISH probes (table I) against HIV pol were designed with Oligostan (Tsanov et al., 2016) and purchased from Integrated DNA Technologies (IDT). Primary probes were pre-hybridized with a secondary FLAP probe conjugated to Cy5 fluorophore through pairing with the readout sequence. Washes and hybridization were performed with Stellaris Buffers (WASH buffer A, WASH buffer B, Hybridization Buffer; LGC Biosearch Technologies), following the manufacturer protocol. Hybridization with the probe was carried out at 37°C in a dark humid chamber for 5 hours. Finally, cells were stained with Hoechst 33342 1:5000 (Invitrogen #H3570) for 5 minutes. Coverslips were mounted on glass slides (Star Frost) with Prolong Diamond Antifade Mountant (Life Technologies #P36970). Confocal microscopy was carried out with a Zeiss inverted LSM700 microscope, with a 63X objective (Plan Apochromat, oil immersion, NA=1.4).
Time‐lapse microscopy
For all live imaging studies, the cells were plated on a polymer-coverslip bottom μ-Dish 35 mm (ibidi #81156) or in μ-Dish 35 mm Quad (10,000 cells per chamber, ibidi #80416) and 2D or 3D videos were acquired with an UltraView VOX Spinning Disk Microscope (Perkin-Elmer), based on a CSU-X spinning-disk (Yokogawa), and using a 63X objective (Plan Apochromat, oil immersion, NA=1.4). For integrated provirus imaging, HIV-1 ANCH3 single-cell clone was transduced with OR-GFP LV (MOI 0.2) for 3 days and then plated on the polymer-bottom dish (105cells) the day before acquisition. On the other hand, HeLa P4R5 cells were infected with HIV-1 ANCH3 (MOI 10) and five days post infection cells were plated on the polymer-coverslip bottom dish and transduced with LV OR-GFP for 24h. For non-integrated viral DNA, HeLa P4R5 were firstly transduced with OR-GFP LV (MOI 0.2) and the day after cells were plated on polymer-bottom dish and infected with HIV-1 ANCH3 (MOI 10) with RAL 20 μM or with the mutated HIV-1 INHA (D116A) ANCH3. Cells were imaged in continue for 10 minutes. Experiments of co-live tracking of IN-Ruby (GIR) and vDNA labeled by OR-GFP were performed in HeLa P4R5 cells transduced with LV OR-GFP (MOI 0.2). The following day, cells were infected with HIV-1 INHA(D116A) ANCH3 complemented with the GIR plasmid using an MOI 50. Fluorescence images were taken every 30 seconds for up to 9h post infection. For co-live-imaging of vDNA and vRNA foci, HeLa MCP-GFP cells were transduced with OR-SANTAKA LV (MOI 1) and 24 h later infected with HIV-1 ANCH3 MS2 (MOI 50).
Imaging Analysis and Statistics
All data were analyzed in GraphPad Prism 8 (GraphPad Software, La Jolla California USA, www.graphpad.com), computing Student’s t test for 2 comparisons, One-way ANOVA for more than two comparisons and Pearson’s correlation. P values symbols: ns = non significant, * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001. All images and videos were analyzed in Fiji (Schindelin et al., 2012) and Icy version 2.0.3.0 (de Chaumont et al., 2012) software. All images and video were processed and analyzed in Fiji (Schindelin et al., 2012) or in Icy software version 2.0.3.0 (de Chaumont et al., 2012). Here below the custom-made protocols:
Particle tracking and MSD measurement
3D videos were analyzed in Fiji (Schindelin et al., 2012) and viral nuclear particles were tracked along time in 3D using TrackMate plugin (Tinevez et al., 2017). The resulting tracks were exported in MATLAB (The MathWorks, Natick, USA) and then used for mean-squared displacement (MSD) analysis with @msdanalyzer(Tarantino et al., 2014). MSD curves were fitted by a straight line from which we derived the value of the diffusion coefficient: D = a / (2*nDim), where « a » is the slope of the MSD curve and « nDim » the dimensionality of the image. MSD curves for which the R2 value was lower than 0.8 were discarded from analysis. Only the initial 25% of the MSD curves were used for the linear fit.
vDNA-vRNA association analysis
For the analysis of RNA FISH images ± RAL in HeLa P4R5 cells, 2D confocal images were analysed with Icy software version 2.0.3.0 (de Chaumont et al., 2012) using a custom made protocol (http://icy.bioimageanalysis.org/protocol/batch-spots-detections-and-statistics and doi: 10.5281/zenodo.3925262, example of HeLa confocal image available at doi: 10.5281/zenodo.3925213). vDNA spots were detected extracting the green channel and applying first a median filter of half size = 1 and then 3, the Spot Detector with a scale of 1 (sensitivity to 55) on the nuclear ROI only. Data from the spot detection (e.g. mean fluorescence intensity) were exported for analysis. vRNA mean fluorescence intensity signal was computed from the nuclei ROI. For vDNA-vRNA association studies in Jurkat cells, 3D confocal images were analyzed in Icy version 2.0.3.0 using two successive custom made protocols (first protocol: http://icy.bioimageanalysis.org/protocol/nuclei-segmentation-from-maximum-intensity-projections/ with doi: 10.5281/zenodo.3925181) and second protocol: http://icy.bioimageanalysis.org/protocol/spots-detection-and-colocalisation-analysis-with-soda/ with doi: 10.5281/zenodo.3925191, example of Jurkat confocal image available at doi: 10.5281/zenodo.3925213). Nuclei were segmented on a maximum intensity projection of the Hoechst channel using the Icy block Gaussian filter with a radius of sigma x=5, y=5 and z=5 pixels, the Thresholder block with a manual threshold of 5000 and the Label Extractor block, which uses connected components to separate the nuclei. Cy5 spots were detected with the HK Means block (Dufour et al.,doi.org/10.1109/ICPR.2008.4761748).The logical operation block was used to keep only the spots detected in the nucleus. These spots were added as input parameter “List detection 1” of the SODA block. EGFP spots were detected using the log3D block to enhance the spots, inverting the log transformed image using the Math Operation Expression block with the following expression: A*B with A scalar=-1 and B the log transformed image, detecting the spots with the Local Extrema block and segmenting the spots with a dilation and a connected components operations. The logical operation block was used to keep only the spots detected in the nucleus. These spots were added as input parameter “List detection 2” of the SODA block. The nucleus ROI was kept as input parameter for “ROI of Analysis (Cell’s shape)”. The Cy5 and GFP spots ROIs were then imported in the SODA block (Lagache et al., 2018) to analyse colocalization of spots within the nucleus only with a maximum radius of 10, a step of 1 and no fixed search distance.
vDNA/vRNA nuclear envelope distance analysis
For the analysis of HeLa P4R5 cells 48 h p.i. and 7 days p.i. and in THP-1 3 days p.i. 3D RNA FISH images, the nuclei were segmented on the Hoechst channel using background subtraction followed by a multilevel Otsu thresholding. The resulting nuclei masks were then directly applied on the vDNA and vRNA channels to select the relevant 3D information inside each labelled region. Inside these regions, vRNA spots were identified using 3D non-local means denoising (Buades et al., “Non-local means denoising.” Image Processing On Line 1 (2011): 208-212) and automated segmentation, while vDNA spots were extracted with the image analysis pipeline previously described (Komatsu et al., 2018). For each vDNA and vRNA spot in was computed the distance to the nuclear envelope in the 2D z-plane corresponding to the spot centroid location.
SC35-vDNA distance analysis
The 3D confocal images were processed for multi-channel image splitting, vDNA and SC35 segmentation. The automated 3D segmentation of cell nuclei included a preliminary non-local means denoising step to cope with the strong signal heterogeneity and the segmentation of SC35 was readjusted to low contrast conditions. The 3D boundary-to-boundary distance (rather than centroid-to-centroid which was done for vDNA/vRNA-NE analysis) between each vDNA spot and its closest SC35 speckle was computed.
Competing financial interests
The authors declare no competing financial interests.
Acknowledgements
We wish to thank Guillermo Blanco-Rodriguez, Stella Frabetti, Philippe Souque and Blandine Monel for experimental help. We thank Nicoletta Casartelli for critical reading of the manuscript. We thank Fabrizio Mammano, Edouard Bertrand, Florian Mueller, and Edward Campbell for sharing reagents. We gratefully acknowledge the UtechS Photonic BioImaging (Imagopole), C2RT, Institut Pasteur, supported by the French National Research Agency (France BioImaging; ANR-10–INSB–04; Investments for the Future). We thank the NIH AIDS Reagents program to support us with precious reagents. This work was funded by the ANRS (Agence Nationale de Recherche sur le SIDA) grant ECTZ88162 with a nominative PhD student fellowship ECTZ88177 for V.S., the Sidaction/FRM grant VIH20170718001, the Pasteur Institute.
References




