

ABSTRACTS
Ostracods are one of the oldest crustacean groups with an excellent fossil record and high importance for phylogenetic analyses but genome resources for this class are still lacking. We have successfully assembled and annotated the first reference genomes for three species of non-marine ostracods; two with obligate sexual reproduction (Cyprideis torosa and Notodromas monacha) and the putative ancient asexual Darwinula stevensoni. This kind of genomic research has so far been impeded by the small size of most ostracods and the absence of genetic resources such as linkage maps or BAC libraries that were available for other crustaceans. For genome assembly, we used an Illumina-based sequencing technology, resulting in assemblies of similar sizes for the three species (335-382Mb) and with scaffold numbers and their N50 (19-56 kb) in the same orders of magnitude. Gene annotations were guided by transcriptome data from each species. The three assemblies are relatively complete with BUSCO scores of 92-96%, and thus exceed the quality of several other published crustacean genomes obtained with similar techniques. The number of predicted genes (13,771-17,776) is in the same range as Branchiopoda genomes but lower than in most malacostracan genomes. These three reference genomes from non-marine ostracods provide the urgently needed basis to further develop ostracods as models for evolutionary and ecological research.
BACKGROUND
Relevance of ostracods
Ostracoda are small, bivalved crustaceans, widely occurring in almost all aquatic habitats as part of the meiobenthos and periphyton. There are 2,330 formally described species of extant non-marine ostracods (Meisch et al. 2019) and at least another 7,000 described species of extant marine ostracod species (see Schön and Martens 2016 for an estimate by S. Brandao). Their calcified valves are preserved as microfossils, making them the extant arthropod group with the most extensive fossil record. The group has an estimated (Cambrian) age of c 500 myr (millions of years) according to a molecular clock (Oakley et al. 2013), and c. 450 myr (Ordovician; Maddocks 1982) to 509 myr (Wolfe et al. 2016) according to the fossil record. This makes them one of the oldest extant pancrustacean groups (Figure 1). Because of their excellent fossil data, evolutionary events can be dated with real time estimates making ostracods ideal models for evolutionary research (Butlin and Menozzi 2000; Oakley and Cunningham 2002; Oakley et al. 2013; Schön and Martens 2016;).

Modified from Oakley et al. (2013). Different pancrustaceans are indicated by branches in different colours. The Ostracoda include the Podocopida, Platycopida and Myodocopida. Here, three representatives of the Platycopida (indicated in purple) have been sequenced. The phylogenetic clade to which Darwinula stevensoni belongs, is indicated by D, the clade to which Cyprideis torosa and Notodromas monacha belong, is indicated by *. Black horizontal bars represent the range of age estimates in myr from Bayesian analyses by Oakley et al. (2013). The letters A-C in the black boxes indicated fossils that were used for calibrations of age estimates.
Contrary to the extensive focus on this group for palaeontological research, there is a total lack of published ostracod genomes, and even isolated genomic data from ostracods in open access databases are still rare. Thus, the only resources available beyond individual gene sequences are four mitogenomes (the marine ostracods Vargula hilgendorfii (Ogoh and Ohmiya 2004; GenBank accession number NC_005306) and Cypridina dentata (Wang et al. 2019; NC_042792); and two unpublished mitogenomes from V. tsujii (NC_039175) and Cyprideis torosa (PRJNA302529)). Also, raw Illumina DNA sequencing reads of the podocopid ostracod Eucypris virens have been generated as part of a study testing DNA extraction methods for high throughput sequencing in zooplankton (SRX8021019; Beninde et al. 2020) but these have neither been assembled nor annotated. In studies on crustacean phylogenies and gene expression (see Table S1 for details), raw RNA-sequencing reads have been generated for a total of 12 species coming from the three major ostracod lineages (Mydocopida, Halocyprida and Podocopida), but the number of assembled and annotated ostracod genes in these studies remains very limited, ranging between 4 and 822 genes.
Choice of model species
Extant non-marine ostracods show a high prevalence of asexual reproduction (Chaplin et al. 1994; Butlin et al. 1998; Martens et al. 1998), which has evolved several times independently in different ostracod lineages and is most frequent in the Cyprididae and the Darwinulidae. Ostracods are thus an ideal group to further study the paradox of sex, which remains one of the most puzzling questions in evolutionary biology (Bell 1982; Otto and Lenormand 2002; Schön et al. 2009a; Neiman et al. 2018). The most important sets of hypotheses explaining why sex is advantageous despite its direct costs are based on the fact that physical linkage among loci generates different forms of selective interference (recently reviewed in Otto 2020). Genome-wide data are very valuable to test if asexuals indeed are affected by these predictions (e.g., Glémin et al. 2019; Jaron et al. 2020) and to develop insights into mechanisms such as gene conversion (Omilian et al. 2006), DNA repair (Schön and Martens, 1998; Hecox-Lea and Mark Welch 2018) or horizontal gene transfer (Gladyshev et al. 2008; Danchin et al. 2010; Boschetti et al. 2012; Flot et al. 2013; Paganini et al. 2012). Such data are also needed to further test for general consequences of asexuality beyond lineage-specific effects (Jaron et al. 2020). For many animal groups in which asexuality is frequent, genomic data are limited to a few representatives only (Tvedte et al. 2019) or are totally absent like in the Ostracoda.
Of all extant non-marine ostracods, the Cyprididae (cyprids) are most speciose, comprising 42% of all known species (Meisch et al. 2019). They would thus be an obvious choice for genomic studies, also because in this ostracod family, mixed reproduction with sexual and asexual females and geographic parthenogenesis is very common (Horne et al. 1998). Asexual cyprids, however, are often polyploid (Symonova et al. 2018; Adolfsson et al. 2010), probably because of hybridization between males and asexual females through accidental mating (Schmit et al. 2013). Consequently, genome sizes are relatively large (Jeffery et al. 2017; Gregory 2020) up to 3.13 pg which equals more than 3 Gb. These features are likely to seriously complicate genomic assemblies and annotations in the absence of any genomic resources for ostracods, which is why we did not choose any asexual cypridid ostracods for this genome project. Instead, we have selected three other species of nonmarine ostracods, one putative ancient asexual ostracod and two species with obligate sexual reproduction.
The ostracod family Darwinulidae is one of the two last remaining animal groups which are most likely true ancient asexuals (Heethoff et al. 2009; Schön et al. 2009b; Schwander 2016) and comprises about 35 morphospecies (Meisch et al. 2019). All darwinulids are brooders with valve dimorphisms between males and females that are detectable in the fossil record. Martens et al. (2003) showed that males have been absent in this family for at least 200 myr. One study reported a few males in a single darwinulid species (Smith et al. 2006) but proof of the functionality of these males for successful mating and meaningful genetic exchange could not been provided. Such (potential) atavistic males have also been reported in other putative ancient asexuals (Heethoff et al. 2009). The type species of the Darwinulidae, Darwinula stevensoni, has been asexual since c 20 myr (Straub 1952), occurs on all continents except Antarctica (Schön et al. 2012) and in a wide range of habitats (Schön et al. 2009b). Darwinula stevensoni is the best investigated darwinulid ostracod so far and has been the subject of ecological (Van Doninck et al. 2002, 2003a & b; Van den Broecke et al. 2013) and molecular research using DNA sequence data from single genes (Schön et al. 1998; Schön et al. 2003; Martens et al. 2005; Schön et al. 2012). These studies revealed that D. stevensoni is most likely apomictic or functionally mitotic (following the definition of apomixis in animals as in Schön et al. 2009a). The species also has low mutation rates. as there appears to be no (Schön et al. 1998) or low (Schön and Martens 2003; Schön et al. 2009b) allelic divergence within individuals, and genetic differences between populations from different continents can be attributed to ancient vicariant processes (Schön et al. 2012). It has also been suggested that gene conversion is common in this species (Schön and Martens 1998, 2003). These results, however, were based on a limited number of genes and require further confirmation with genome-wide data. Darwinula stevensoni has a life cycle of 1 year in Belgium (Van Doninck et al. 2003b) and up to 4 years in more northern regions (McGregor 1969 in Northern America; Ranta 1979 in Finland), which is exceptionally long for a non-marine ostracod. It can survive a wide range of temperatures, salinities (Van Doninck et al. 2002) and oxygen concentrations (Rossi et al. 2002). The total genome size of D. stevensoni has been estimated as 0.86-0.93 pg with flow cytometry (Paczesniak, unpublished), approximating 900 Mb. There is no information on the ploidy level of D. stevensoni, except for the study by Tétart (1979) showing 22 dot-like chromosomes. Because of its putative ancient asexuality, no close sexual relatives of D. stevensoni are available for comparative, genomic analyses. We have chosen two fully sexual non-marine ostracod species from the Cytherideidae and the Notodromadidae with high population densities in Belgium as comparisons to the putative ancient asexual: Cyprideis torosa and Notodromas monacha respectively. Cyprideis torosa inhabits brackish waters and is the only extant species of this genus in Europe (Meisch 2000). It has been the subject of various biological and especially palaeontological and geochemical studies (see for example: Heip, 1976a, b; De Deckker et al. 1999; Keyser 2005). Frogley and Whittacker (2016) suggested that C. torosa is at least of Pleistocene origin (c 2.5 myr) but might be older. There are only two molecular studies of this species based on single genes (Schön and Martens 2003; Schön et al. 2017). No information on the genome size or the karyotype of C. torosa is currently available.
The second sexual ostracod species analysed here, Notodromas monacha, occurs throughout the Northern hemisphere and is a non-marine ostracod with a most peculiar behaviour: it is partially hyponeustonic, hanging upside down attached to the water surface (Meisch 2000). The fossil record of N. monacha goes back to the Miocene (max 23 myr - Janz 1997), and its genome size is at 0.87pg (Jeffery et al. 2017; Gregory 2020) very similar to that of D. stevensoni. This species has not yet been the subject of any molecular studies.
Our aim here is to provide the first reference genome data of non-marine ostracods from three different species with varying reproductive modes: the putative ancient asexual D. stevensoni and the two obligate sexuals, C. torosa and N. monacha. We also generate transcriptomes of these species to facilitate genome annotations.
MATERIAL AND METHODS
Sample collection for genome and transcriptome sequencing
All three non-marine ostracod species were sampled in Belgian lakes where previous research had shown that these species occurred (Schön and Martens 2003; Merckx et al. 2018). Living ostracods were sampled using a hand net with a mesh size of 150 μm. The hand net was swept in between the vegetation and forcefully right above the surface of the sediment for collecting Darwinula stevensoni and Cyprideis torosa. Notodromas monacha was sampled by moving the net on the water surface. Non-marine ostracods were kept in habitat water. Their taxonomic identity was confirmed, and they were sorted alive under a binocular microscope as described by Martens and Horne (2016). Individual ostracods were picked with a pipette and transferred into sterilized EPA water in which they were maintained until DNA and RNA was extracted. More details on the origin of biological samples are provided in Table S2.
For generating reference genomes, DNA was extracted from a single female of each species using the QIAamp DNA Micro kit according to manufacturer’s instructions. The extracted DNA from single females was amplified in two independent reactions using the SYNGIS TruePrime WGA kit and then pooled, to generate sufficient DNA for preparing different libraries. To generate transcriptomes for annotation of reference genomes, RNA was extracted from 40 pooled individuals per species from the same collection batch. For this, individuals were frozen in liquid nitrogen and, after addition of Trizol (Life Technologies), mechanically crushed with beads (Sigmund Lindner). Next, chloroform and ethanol-treatment was applied to the homogenized tissue and the aqueous layer transferred to RNeasy MinElute Columns (Qiagen). Subsequent steps of RNA extraction were done following the RNeasy Mini Kit protocol, including DNase digestion. Finally, RNA was eluted into water and stored at −80°C. RNA quantity and quality were estimated with the NanoDrop (Thermo Scientific) and Bioanalyzer (Agilent).
Genome assembly
We prepared five genomic DNA libraries for each reference genome (three 2×125bp paired-end libraries with average insert sizes of 250-300, 550 and 700bp, and two mate-pair libraries with average insert sizes of 3000 and 5000bp; see Table S3 for more details) with the Illumina TruSeq DNA Library Prep Kit. Reads were generated with the Illumina HiSeq 3000 system for a total coverage between 351X and 386X (Table S3).
Reads were filtered with Trimmomatic v0.36 (Bolger et al. 2014) and NxTrim v0.4.1 (O’Connell et al. 2015). We employed non-standard methods for de novo genome assemblies owing to uneven coverage produced by PCR-based whole-genome amplification (Chen et al. 2013; Oyola et al. 2014)._Filtered reads were normalized using BBMap v36.59 (Bushnell 2014) and then assembled into contigs with SPAdes v3.10.1 (Bankevich et al. 2012). Scaffolding was performed using SSPACE v3.0 (Boetzer et al. 2012). Scaffolds identified as contaminants were filtered out using Blobtools v1.0 (Laetsch and Blaxter 2017). The completeness of genomes assemblies was assessed with BUSCO v3.0.2 (Seppey et al. 2019) against the arthropoda_odb9 lineage. More details of the assembly pipelines and the applied parameters can be found in supplementary materials (SM1).
Protein coding gene annotation
Libraries were prepared using the Illumina TruSeq Stranded RNA, following the manufacturer’s instructions. RNA reads were generated with the Illumina HiSeq 2500 system (Table S4). Reads were filtered with Trimmomatic v0.36. All trimmed reads were mapped against the genomes with STAR v2.5.3a (Dobin et al. 2013) and further assembled with Trinity v2.5.1 (Haas et al. 2013) under the “genome guided” mode to produce transcriptome assemblies.
The obtained transcriptomes and protein evidence were used to train and predict protein coding genes using MAKER v2.31.8 (Holt and Yandell 2011). Predicted protein coding genes were functionally annotated with Blast2GO v5.5.1 (Conesa et al. 2005; Götz et al. 2008) against the NCBI non-redundant arthropods protein database (v 2018-10). More details of the annotation pipelines and the applied parameters can be found in supplementary materials (SM2).
GenomeScope analyses
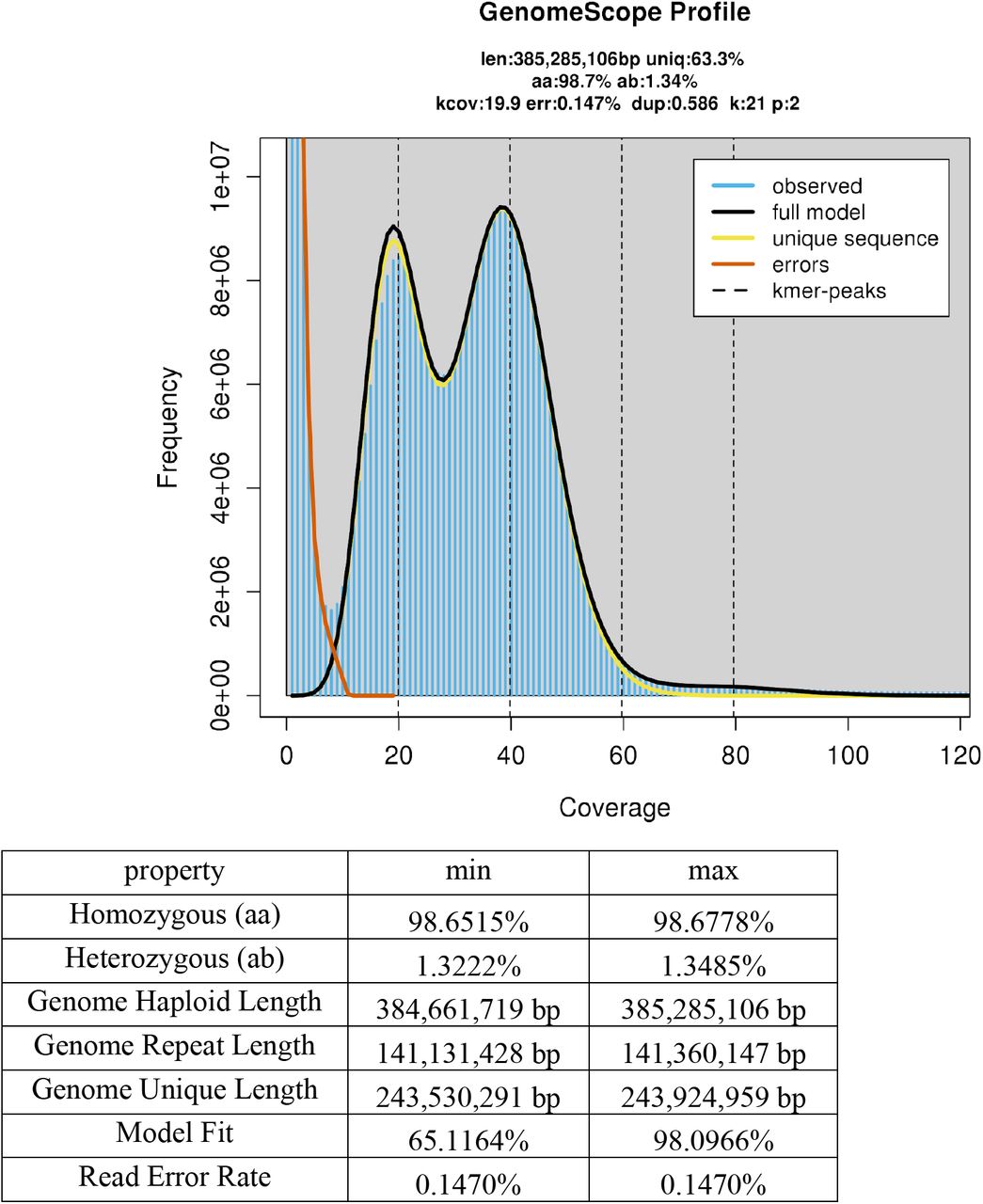
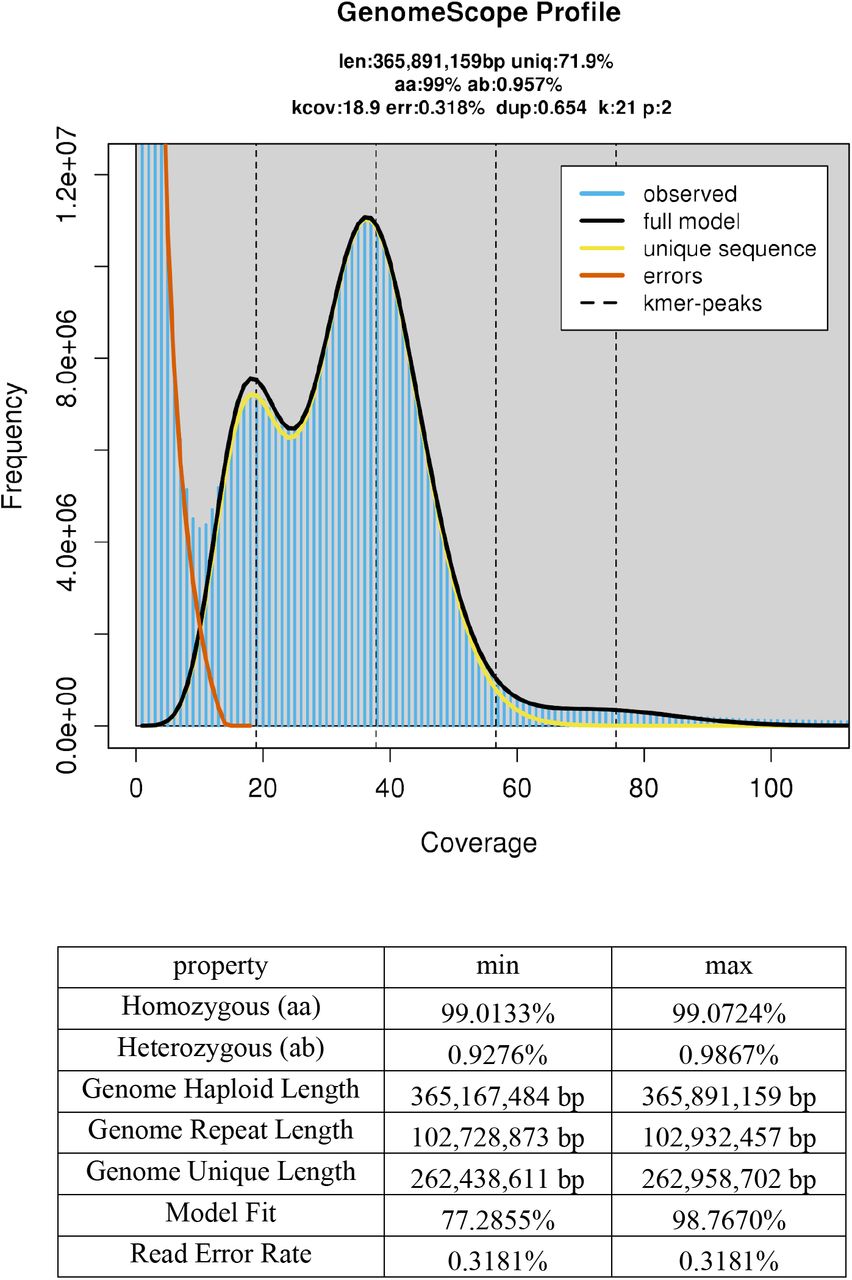
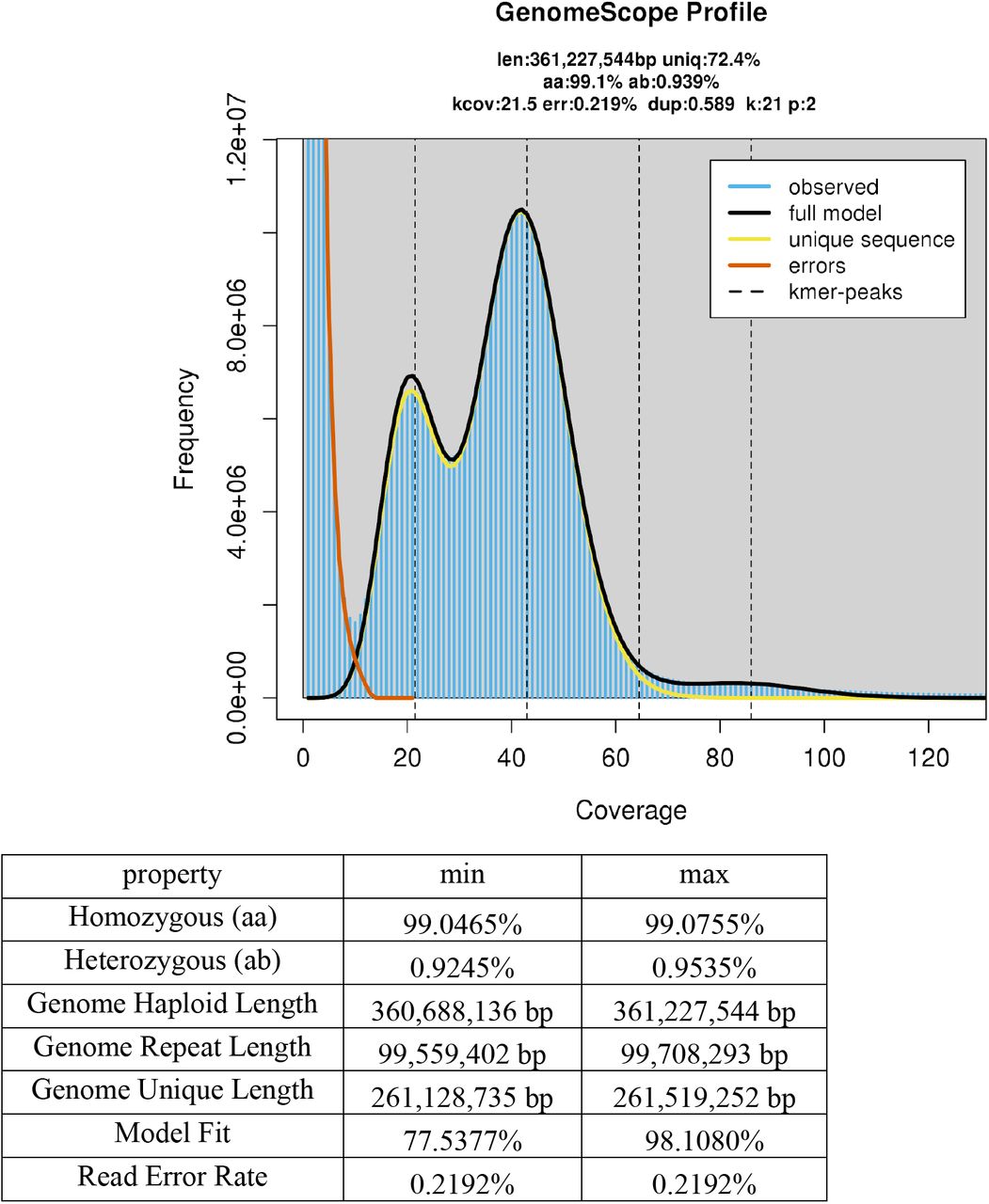
The whole genome amplification approach, which we used in the present study because of the small body size of individual ostracods, generated unequal read coverage of ostracod genomes and prevented us from directly estimating genome sizes and levels of heterozygosity from the assemblies. To overcome this problem, we re-sequenced two individual ostracods each of D. stevensoni and N. monacha without whole genome amplification, preparing libraries with the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina. Reads were filtered with Trimmomatic v0.36 and analyzed using GenomeScope v2.0 (Ranallo-Benavidez et al. 2020) to correctly estimate genome size and heterozygosity. More details on the analyses are provided in the supplementary material (SM3).
RESULTS AND DISCUSSION
First ostracod reference genomes and their features
We successfully produced the first de novo reference genomes of non-marine ostracods, namely of the three species Darwinula stevensoni, Cyprideis torosa and Notodromas monacha with different reproductive modes (see SM1 and Tables S3–S4 for more details on the assemblies). Given the small size of individual non-marine ostracods and the limited amount of soft tissue suitable for DNA and RNA extractions (Schön and Martens 2016), this was not a trivial task. We used a whole genome amplification approach (WGA), because the TruSeq DNA Nano library prep kit for Illumina sequencing or low input protocols for PacBio (Duncan et al. 2019) were not yet available when these assemblies were generated. We would not recommend WGA for future studies because this PCR-based method generated uneven coverage, and consequently, problems for applying routine genome assembly methods and estimates of genome size and heterozygosity. Despite these limitations, our approach produced genome assemblies that are useful for future research as will be outlined below.
When assessing the quality of the obtained ostracod de novo genome assemblies, the assembly of the putative ancient asexual, D. stevensoni, had the best contiguity, with the largest N50 although the total number of scaffolds was similar to N. monacha (Table 1, Table S6). The genome of the putative ancient asexual is furthermore the most complete as shown by its total BUSCO score of 96% and of 94% for complete single copy genes (Table 1). The quality of the genome from the obligate sexual ostracod Cyprideis torosa is the lowest of the three ostracod species as it has the highest number of scaffolds, and the lowest N50; it is also less complete with a total BUSCO score of 92% (Table S7) and of 87% for complete single copy genes (Table 1). All three species have similar numbers of predicted genes and transcripts (Table S7).
Assembly size is provided in million base pairs, scaffold N50 in kilo base pairs and BUSCO scores in %. Letters behind BUSCO scores indicate the % of complete single copy genes (C) or % of single and fragmented single copy genes (C+F), respectively. Where BUSCO scores lack brackets, no further information on completeness of single copy genes was provided. $ anchoring of scaffolds in existing genome assembly; L = linkage map available; * long read technology; # BAC library available. n. i. = no information available.
Ostracod genome sizes estimated with flow cytometry are somewhat larger than the estimates that we obtained here from GenomeScope analyses of re-sequenced individual ostracods. The haploid genome size of D. stevensoni was estimated at 420-455 Mb with flow cytometry (Paczesniak, unpublished) while we estimated 362 Mb from sequence reads (Figure S1 A-B). Similarly, the size of the haploid genome of N. monacha is estimated at 425 Mb with flow cytometry (Jeffery et al. 2017; Gregory 2020), which is larger than the 385 Mb (Figure S1 CD) that we obtained from sequence reads. It thus seems that some parts of each genome are missing from our sequencing reads. Transposons and repeat-rich genomic regions can contribute to gaps in genomic assemblies (Peona et al. 2020). Some of these missing regions could also be GC-rich, a feature which is known to cause a sequencing-bias with Illumina technology (see for example Chen et al. 2013, Botero-Castro et al. 2017). Acquiring more complete genome assemblies will require the additional application of long read technologies to ostracods.
Genome-wide estimates of heterozygosity are especially interesting for asexual taxa because the absence of recombination is expected to cause accumulation of mutations, resulting in increasing allelic divergences within individuals (Birky 1996). Jaron et al. (2020) identified three factors driving intragenomic heterozygosity in asexuals: how the transition to parthenogenesis occurred, which cytological mechanism underlies parthenogenesis and how long asexual reproduction has been ongoing. Based on sequencing reads from individual ostracods, we estimate heterozygosity of the putative ancient asexual ostracod D. stevensoni to be 0.92-0.99% (Figure S1 A-B) and 1.32-1.43% for the sexual N. monacha (Figure S1 CD). The genome-wide heterozygosity of D. stevensoni matches to some extent an earlier study on intra-individual divergence in three nuclear genes of D. stevensoni (Schön and Martens 2003). The finding of almost 1% heterozygosity in D. stevensoni is remarkable, given that all previous genome-wide estimates for asexual arthropods that did not evolve via hybridization revealed extremely low levels of heterozygosity (Jaron et al. 2020). Yet heterozygosity is clearly less than the estimates for parthenogenetic species with known hybrid origin (1.73-8.5%) or polyploidy (1.84%-33.21%) (Jaron et al. (2020), supporting the view that D. stevensoni is neither a hybrid nor a polyploid. Asexual reproduction in ostracods is thought to be apomictic (Chaplin et al. 1994), implying that observed heterozygosity levels are largely depend on the relative impact of heterozygosity losses from gene conversion and heterozygosity gains from new mutations. Given the apparent absence of sex and recombination for millions of years (Straub 1952), it is perhaps surprising that heterozygosity in this putative ancient asexual ostracod is not larger. This may suggest that genome-wide rates of gene conversion and mutation are comparable in this species.
Genome contiguity of ostracod assemblies as compared to other crustaceans
We here compare the qualities of our ostracod genome assemblies to those of 19 other crustacean species (Table 1) published in the last four years. We only include studies with complete assemblies and sufficient information to assess assembly qualities. We assessed the contiguity of the three de novo ostracod genome assemblies by the number of scaffolds and their N50. On the one hand, both features are comparable to those of the copepod Apocylops royi (Jørgensen et al. 2019) and the amphipod Parhyale hawaiensis (Kao et al. 2016) (Table 1) and better than for crustaceans with larger genomes such as the decapods Cherax quadricarinatus (Tan et al. 2019), Palaeomon carinicauda (Li et al. 2019), Penaeus mondon (Van Quyen et al. 2020), Marsupenaeus japonicus (Yuan et al. 2018) and Procamburus virginalis (Gutekunst et al. 2018; Table 1). On the other hand, genome assemblies of several other crustaceans have smaller scaffold numbers and higher N50 and thus better contiguities than the assemblies obtained here for non-marine ostracods. For the two notostracan Lepidurus species (Savojardo et al. 2018), this can probably be explained by their smaller genome sizes. For other crustaceans, genome assemblies or linkage maps have been available beforehand which have considerably improved assembly qualities (Table 1) as in the examples of the cladocerans Daphnia pulex (Ye et al. 2017), D. magna (Lee et al. 2019) and the copepod Tigriopus japonicus (Jeong et al. 2020). No such genomic resources are currently available for ostracods. Finally, other studies of crustacean genomes with better assembly contiguities (the branchiopod Eulimnadia texana - Baldwin-Brown et al. 2018, and the decapod Erichoir japonica sinensis - Tang et al. 2020), the copepod Tigriopus californicus - Jeong et al. 2020 and the isopod Armadillium vulgare - Chebbi et al. 2019) have used a combination of Illumina and long read technologies (Table 1). Long-read technologies such as PacBio used to require a relatively large amount of high-molecular weight DNA (Solares et al. 2018), which could not be obtained for ostracods with their very low yields of high molecular weight DNA from individual specimens and their small body sizes as compared to many other crustaceans (Schön and Martens 2016). We hope that low input protocols for PacBio (Duncan et al. 2019) and other long read technologies can be successfully applied to ostracods in the future, in which case the genome assemblies obtained here could form the basis for subsequent hybrid assemblies. Optimizing Oxford Nanopore Technology for non-marine ostracods has already commenced (Schön et al. in prep.).
Genome annotations of ostracods and other crustaceans
Because our de novo ostracod genome assemblies are relatively complete (see BUSCO scores in Table 1), we will here also briefly compare some features of predicted protein coding genes with those of other crustaceans (Table S8). We have predicted 13,771 to 17,776 protein coding genes in the three non-marine ostracod genomes, (Tables S7 and S8) with the highest number for the sexual C. torosa and an intermediate estimate for the putative ancient asexual D. stevensoni. The number of annotated protein coding genes in non-marine ostracods is similar to estimates for various branchiopods and the copepods Oithona nana, Tigriopus californicus and T. kingsejongensis but lower than in most malacostracans (Table S8). Not all genome studies of crustaceans cited here contain information on other features of coding genes, such as the average size of genes, introns and exons (Table S7). Comparisons of these features are therefore limited and will not be further discussed here but we provide available data of these features for ostracods and other crustacean genomes for reference.
Gene annotation in general but especially in the crustaceans is challenging; this is for example illustrated by the much lower numbers of protein coding genes (18,440) which are predicted in the novel reference genome of the cladoceran Daphnia pulex by Ye et al. (2017) as compared to the first assembly of D. pulex with more than 30,000 predicted genes (Colbourne et al. 2011). Even more difficult is assigning gene functions to annotated crustacean genomes (Rotlantt et al. 2018). The novel data on predicted genes and transcripts from non-marine ostracods in the current study will significantly contribute to future genome annotations in crustaceans and other arthropods. The genes and transcripts predicted here can also provide the baseline for future gene expression studies of non-marine and marine ostracods.
CONCLUSIONS
We have successfully obtained de novo genome assemblies for three species of non-marine ostracods with different reproductive modes. These represent the first quality reference genomes for ostracods. Given the paucity of genome assemblies from crustaceans as compared to insects or other arthropods, these assemblies are important tools to further develop ostracods as models for evolutionary and ecological research, also including marine species. Even if the de novo genome assemblies are somewhat fragmented and not yet at the chromosome-level, they have a high level of completeness and will thus facilitate future studies of ostracods. The reference genomes of this paper can also provide the first step towards a genomic assessment of the putative ancient asexual status of non-marine darwinulid ostracod species.
DATA AVAILABILITY
Raw sequence reads have been deposited in NCBI’s sequence read archive under the following bioprojects: PRJNA515625 (reference genomes, Table S3) and PRJNA631617 (RNA-seq for annotations and resequenced individuals, Table S4 and Table S5).
Genome assemblies and annotations have been deposited in the European Nucleotide Archive (ENA) under the accession number PRJEB38362 (Table S6 and Table S7). Codes for the analyses are available at: https://github.com/AsexGenomeEvol/Ostracoda_genomes.
ACKNOWLEDGEMENTS, FUNDING, CONFLICT OF INTEREST
This research was funded by Belgian Federal Science Policy (BR/314/PI/LATTECO) and a grant from the Swiss National Science Foundation (CRSII3_160723). Marie Cours, Tijs Van Den Bergen and Jeroen Venderickx are acknowledged for technical support in sampling and sorting ostracod samples. We also thank Kristiaan Hoedemakers and Jeroen Venderickx for their assistance in finalizing the figure.
Supplementary material
SM1: Details of genome assembly pipelines and parameters
The paired-end reads were filtered using Trimmomatic v0.36 (Bolger et al. 2014). Adapters were removed using adapters provided by Trimmomatic. Leading and trailing bases below quality 3 were removed. Reads were scanned using a 4-base sliding window and trimmed when the average quality dropped below 20. Reads were discarded if the size dropped below 60 bp. (parameters: PE ILLUMINACLIP:TruSeq-PE.fa:2:30:10:4 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 AVGQUAL:20 MINLEN:60).
The mate-pair reads were filtered using NxTrim v0.4.1 (O’Connell et al. 2015) (parameters: --preserve-mp --separate). As a result, mate-pair reads flagged as MP and UNKNOWN were concatenated following the authors’ suggestion. Finally, the concatenated mate-pair reads were trimmed using Trimmomatic with the same parameters described previously.
We employed non-standard methods for genome assemblies due to uneven coverage produced by PCR-based whole-genome amplification (Chen et al. 2013; Oyola et al. 2014). Only the overlapped libraries were used for contig assemblies. Filtered paired-end reads were merged into single reads by overlap detection using BBMap v36.59 (Bushnell 2014) and the module BBMerge (parameters: minoverlap=15 mismatches=0 ecct strict). The merged reads were then concatenated with the rest of the single reads and normalization was performed using the module BBNorm with a target average depth of 65x. Normalized data were de novo assembled using SPAdes v3.10.1 (Bankevich et al. 2012) (parameters: --careful -k 21,33,55,77,99,111,127). Contigs were then scaffolded with SSPACE v3.0 (Boetzer et al. 2012) using the remaining libraries and gap-filled with GapCloser v1.12-r6, a module of the SOAP package (Luo et al. 2012) with default parameters. Then, the genome assemblies were decontaminated using BlobTools v1.0 (Laetsch & Blaxter, 2017) under the taxrule “bestsumorder”. Hit files were generated after a blastn v2.7.1+ against the NBCI nt database (v 2016-06), searching for hits with an evalue below 1e-25 (parameters: -max_target_seqs 10 -max_hsps 1 -evalue 1e-25). Scaffolds without hits to metazoans were filtered out from the assemblies.
The completeness of genomes assemblies was assessed with BUSCO v3.0.2 (Seppey et al. 2019) against the arthropoda_odb9 lineage and the --long option.
SM2: Annotation of protein coding genes
Raw paired-end RNA-sequence reads were trimmed according to quality with Trimmomatic v0.36 (Bolger et al. 2014) using the maximum information approach with tolerant parameters.
Any reads with a sequence length of < 80 bp after trimming were discarded (parameters: adapters.fa:2:30:12:1:true LEADING:3 TRAILING:3 MAXINFO:40:0.4 MINLEN:80). All trimmed RNA-sequence reads were then mapped against the genomes using STAR v2.5.3a (Dobin et al. 2013) under the “2-pass mapping” mode and default parameters. Then, the STAR outputs were used to produce transcriptome assemblies using Trinity v2.5.1 (Haas et al. 2013) “genome guided” mode (parameters: -- genome_guided_max_intron 100000 --SS_lib_type RF --jaccard_clip). Finally, the transcriptome assemblies were filtered following Trinity developers recommendations (https://github.com/trinityrnaseq/trinityrnaseq/wiki/Trinity-FAQ): Briefly, filtered RNA-seq reads were mapped back against the transcriptomes using Kallisto v0.43.1 (Bray et al. 2016) with options --bias and --rf-stranded then transcripts with at least 1 TPM in any samples were retained.
Protein coding genes were predicted using MAKER v2.31.8 (Holt & Yandell 2011) in a 2-iterative way described in Campbell et al. (2014) with minor modifications following author recommendations. Only scaffolds above 500bp were annotated. Prior to gene prediction, MAKER used RepeatMasker v4.0.7 (Tarailo-Graovac & Chen 2009) for masking repetitive regions. For the first iteration, genes were predicted using Augustus v3.2.3 (Stanke et al. 2006) trained with the BUSCO v3.0.2 (Seppey et al. 2019) results (SM1). A combination of UniProtKB/Swiss-Prot (release 2018_01) and the BUSCO arthropoda_odb9 proteome were used as protein evidence. The Trinity assembled mRNA-seq reads (described above) were used as transcript evidence. The resulting gene models were then used to retrain Augustus as well as SNAP v2013.11.29 (Korf 2004) and a second iteration was performed. Subsequently, predicted protein coding genes were functionally annotated using Blast2GO v5.5.1 (Conesa et al. 2005; Götz et al. 2008) with default parameters against the NCBI non-redundant arthropods protein database (v 2018-10).
SM3: Estimation of genome size and heterozygosity by genome profiling analysis
Raw paired-end resequencing reads were trimmed using the same strategy as described in SM2. K-mer frequencies were computed using KMC v3.1.1 (Kokot et al. 2017). Genome sizes and heterozygosities were estimated with GenomeScope v2.0 (Ranallo-Benavidez et al. 2020) using parameters recommended by the authors.
No full assemblies or annotations are available from these studies. # of genes = total number of orthologous genes used for phylogenetic or gene expression studies, respectively.
All species were collected in Belgium in 2018. The species printed in blue is a putative ancient asexual, species printed in red are sexually reproducing.
bp = basepairs. G = giga. The coverage is estimated from final assembly sizes. The species printed in blue is a putative ancient asexual, species printed in red are sexually reproducing. Coverage is provided in %.
Data type: paired-end 2x 101bp. bp = basepairs. G = giga. The species printed in blue is a putative ancient asexual, species printed in red reproduce sexually.
Data type: paired-end 2x 101bp. bp = basepairs. G = giga. The coverage is estimated from final assembly sizes. The species in blue is a putative ancient asexual, species printed in red reproduce sexually.
Σ represents the sum of all scaffolds in million basepairs (Mbp). The average N50 was calculated per scaffold in kilo basepairs (Kbp). The BUSCO score is the proportion of conserved single copy ortholog genes among arthropods. N is the proportion of unknown nucleotides (gaps) in the assembly. The species in blue is a putative ancient asexual, species printed in red reproduce sexually.
bp = basepairs. The genome coverage is the proportion of each genome covered by genes. Red species reproduce sexually, while the species indicated in blue is a putative ancient asexual.
Average gene, intron and exon sizes are provided in base pairs.* including all transcripts from different experiments and life stages. n. i. = no information.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Two individuals each of Darwinula stevensoni (Figure S1 A-B) and Notodromas monacha (Figure S1 CD) were resequenced and their reads analyzed to estimate genome sizes and heterozygosities.
Figure S1 A – DS_02
Figure S1 B – DS_05
Figure S1 C – Nm_F03
Figure S1 D – Nm_F07
LITERATURE CITED
References




