

Abstract
The novel betacoranavirus SARS-CoV-2 causes a form of severe pneumonia disease, termed COVID-19 (coronavirus disease 2019). Recombinant human antibodies are proven potent neutralizers of viruses and can block the interaction of viral surface proteins with their host receptors. To develop neutralizing anti-SARS-CoV-2 antibodies, antibody gene libraries from convalescent COVID-19 patients were constructed and recombinant antibody fragments (scFv) against the receptor binding domain (RBD) of the S1 subunit of the viral spike (S) protein were selected by phage display. The selected antibodies were produced in the scFv-Fc format and 30 showed more than 80% inhibition of spike (S1-S2) binding to cells expressing ACE2, assessed by flow cytometry screening assay. The majority of these inhibiting antibodies are derived from the VH3-66 V-gene. The antibody STE90-C11 showed an IC50 of 0.56 nM in a plaque-based live SARS-CoV-2 neutralization assay. The crystal structure of STE90-C11 in complex with SARS-CoV-2-RBD was solved at 2.0 Å resolution showing that the antibody binds at the same region as ACE2 to RBD. In contrast to other published anti-SARS-CoV-2 antibodies, the binding of STE90-C11 is not blocked by known RBD mutations, endowing our antibody with higher intrinsic resistance to those possible escape mutants.
Introduction
Antibody phage display is a widely used in vitro technology to select human antibody fragments for the development of therapeutic antibodies (Frenzel et al., 2016). Currently, 14 EMA or FDA approved antibodies were generated by antibody phage display (Alfaleh et al., 2020). Human antibodies can be selected from two kind of antibody gene libraries: universal libraries and immune libraries. Universal libraries which have a naïve, semi-synthetic or synthetic antibody gene repertoire, allow – in theory - the selection of antibodies against any kind of molecule, while immune libraries derived from immunized donors typically facilitate the antibody generation against the pathogen that affected the donors (Bradbury et al., 2011; Frenzel et al., 2017; Kretzschmar and von Rüden, 2002; Kügler et al., 2018; Kuhn et al., 2016). Immune phage display libraries from convalescent patients or immunized human donors were used in the past for the successful generation of antibodies against infectious diseases (Duan et al., 2009; Rahumatullah et al., 2017; Trott et al., 2014; Wenzel et al., 2020a; Zhang et al., 2003).
The severe pneumonia COVID-19 (coronavirus disease 2019) is a disease caused by the novel coronavirus SARS-CoV-2 and was described at the end of 2019 in Wuhan China (Lu et al., 2020; Zhou et al., 2020). This new human pathogenic coronavirus is closely related to the bat coronavirus RATG13 indicating an animal-to-human transition (Shang et al., 2020a). The spike (S) protein of SARS-CoV-2 binds to the human zinc peptidase angiotensin-converting enzyme 2 (ACE2) as main receptor for cell entry. The S protein has two subunits, the N-terminal S1 harbouring the receptor binding domain (RBD) and the viral membrane-anchored C-terminal S2 subunit, which is required for trimerization and fusion of the virus and host membrane for viral entry (Shang et al., 2020b; Starr et al., 2020; Walls et al., 2020; Wang et al., 2020b; Wrapp et al., 2020; Yan et al., 2020). Blocking of the RBD-ACE2 interaction by therapeutic antibodies as a strategy to treat COVID-19 (Zhou and Zhao, 2020) is a promising approach since the successful generation of neutralizing antibodies against S1 and RBD was demonstrated already for SARS-CoV and MERS (Coughlin and Prabhakar, 2012; Widjaja et al., 2019; Zhu et al., 2007). Some of the anti-SARS-CoV antibodies showed also cross-reaction and cross-neutralization of SARS-CoV-2 (Huo et al., 2020; Tian et al., 2020; Wang et al., 2020a). Human antibodies were also selected directly against SARS-CoV-2 using different approaches including single B-cell cloning or antibody phage display (Bertoglio et al., 2020; Cao et al., 2020; Hansen et al., 2020; Kreer et al., 2020; Li et al., 2020; Liu et al., 2020a, 2020b; Shi et al., 2020; Zeng et al., 2020).
In this work, immune phage display libraries from six COVID-19 convalescent patients were constructed, RBD-binding antibodies were selected resulting in SARS-CoV-2 inhibiting and neutralizing antibodies. The crystal structure of the best neutralizing candidate STE90-C11 in complex with RBD was elucidated. Together with its unique binding pattern that tolerates a large number of RBD mutations, its properties suggest its use as a therapeutic agent against SARS-CoV-2 infections.
Results
Immune library construction
For the immune library construction, we collected blood samples of 16 q-RT-PCR confirmed COVID-19 convalescent patients. To analyze the immune answer, sera of all patients were titrated on RBD (Supplementary Data 1). The patients 1,2,5,6,9 and 14 showed the highest IgG titers against RBD and were chosen for library construction. PBMCs were isolated and CD19+/CD138+ plasma cells were enriched by FACS from some of these samples (Supplementary Data 2). Both PBMCs and plasma cells were used for library construction. Six individual libraries were constructed from PBMCs of patients 2, 6 and 14, while four additional libraries were made from CD19+/CD138+ plasma cells pools (pool 1: patients 2,6,14; pool 2: patients1,5,9). All libraries were constructed cloning kappa and lambda antibody genes separately, resulting in diversities ranging from 0.7×107 to 1.7×108 estimated independent clones.
Antibody fragments were selected against SARS-CoV-2
Antibodies were selected by panning in microtiter plates against SARS-CoV-2 S1 subunit using S1-S2 in the first panning round, RBD in the second panning round and S1-S2 in the third panning round. This approach focuses on the antibody selection on epitopes located within the RBD while being presented in the correct conformation of the intact spike protein trimer. The two plasma cell-derived immune libraries for kappa, respectively lambda, were combined and the panning was performed separately for lambda and kappa libraries. All kappa and lambda libraries originating from the PBMCs were pooled together. The following single clone screening was performed by antigen ELISA in 96 well MTPs, using soluble monoclonal scFv produced by E. coli. 542 monoclonal hits were identified. DNA sequencing revealed 197 unique antibodies. The V-Gene combinations for kappa (Supplementary Data 3) and lambda (Supplementary Data 4) antibodies were analyzed. The most abundantly used V-genes were VH3-66, VK1D-39 und VL3-21. Antibody sequences that showed potential developabilty liabilities, i.e. glycosylation sites or single cysteines in the CDRs were excluded from further analysis. 135 antibodies were re-cloned into the bivalent scFv-Fc format and produced in Expi293F cell in 5 mL culture scale, with yields ranging from of 70 to 260 mg/L.
Screening for inhibition in a cell-based assay
To screen for SARS-CoV-2 blocking antibodies, an inhibition assay was performed by flow cytometry on ACE2-expressing EXPI293F cells, measuring direct competition between S1-S2 trimer and scFv-Fc antibodies. The entire spike protein ectodomain was used for this inhibition assay for optimal representation of the viral binding. In a first screening, the 135 antibodies were tested at 1500 nM S1-S2 (molar ratio antibody:S1-S2 30:1) (Figure 1). 30 antibodies that inhibited S1-S2 binding more than 80% were selected for further analysis. The germlines genes of these inhibiting scFv-Fc are given in Table 1. VH3-66 was the most abundantly used heavy chain V-gene of the well inhibiting scFv-Fc. All VH3-66 derived antibodies had closely related VH CDRs amino acid sequence while the corresponding light chains were different.

(A) Inhibition prescreen of 135 scFv-Fc antibodies on ACE2 positive cells using 1500 nM antibody and 50 nM spike protein (30:1 ratio). The antibodies selected for detailed analysis are marked in colors. Bars with red background and crossed-striped indicate scFv-Fcs not further analyzed. Data show single measurements.
Antigen binding and cell-based inhibition by IgG
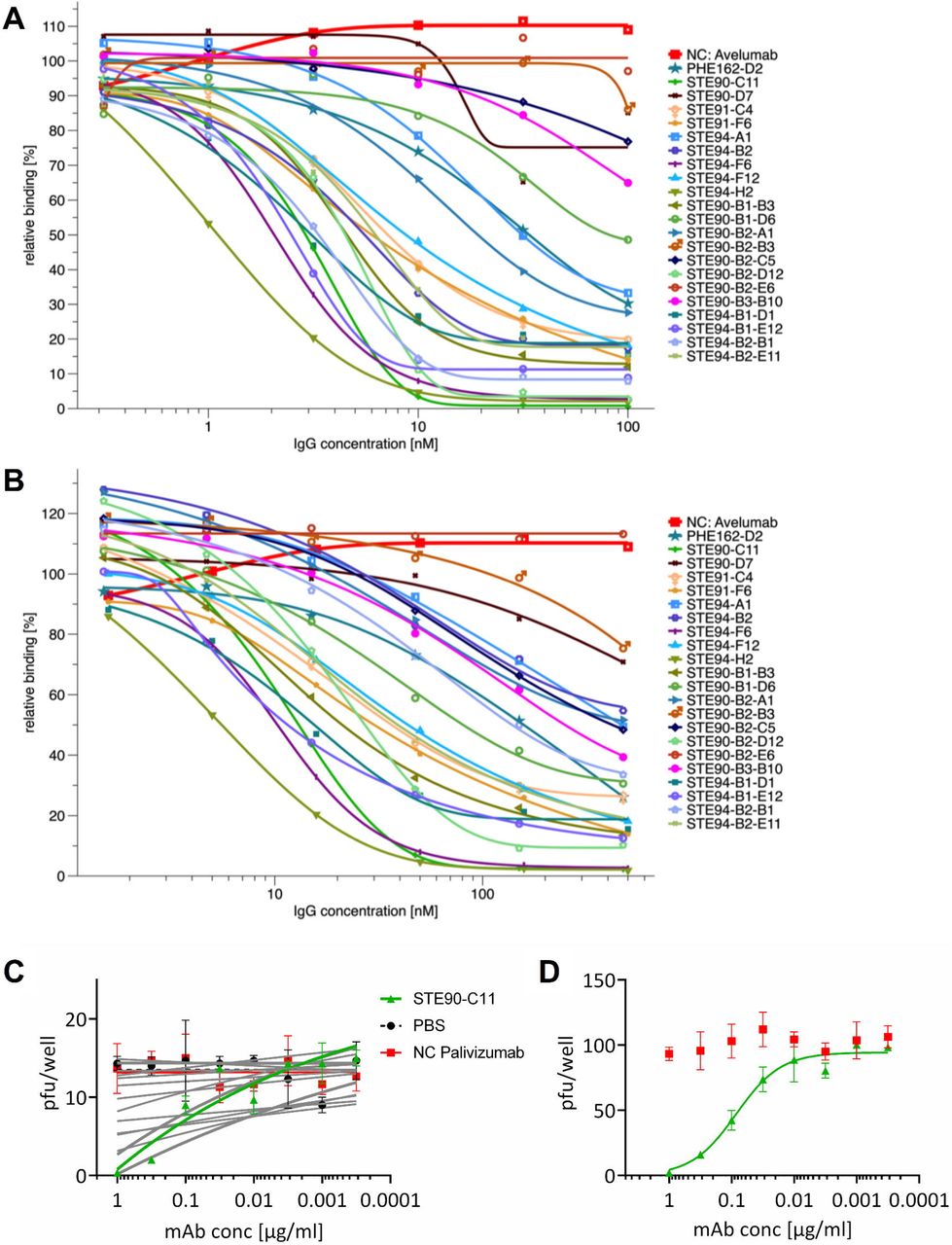
The 30 antibodies were screened in a cytopathic effect (CPE)-based neutralization screening assay (data not shown) and the best neutralizing 19 scFv-Fc were re-cloned and produced as human IgG in 50 mL culture scale, with yields ranging from 12 to 93 mg/L. EC50 values of binding to RBD, S1 or S1-S2 were determined (Figure 2) and are shown in Table 1. Antibodies STE90-C11, STE90-B2-D12, STE94-F6 and STE94-H2 showed EC50 values ranging between 0.2-0.5 nM on all antigens tested. Inhibition of ACE2 binding was assessed at concentrations from 100 nM - 0.3 nM IgG using 10 nM RBD (Figure 3A) or 500 nM - 1.5 nM IgG using 50 nM S1-S2 spike (Figure 3B) in the previously described cell-based inhibition assay. The extent of inhibition by STE90-C11 of both RBD and S1-S2 was further validated in an further cellular assay with ACE2 expressing cells (Supplementary Data 5). The best antibodies showed IC50 values of 2.6-15 nM IgG for 50 nM S1-S2 spike, respectively 1-3 nM IgG for 10 nM RBD (Table 1). For 50% inhibition the best molar ratios were from 0.02-0.3:1 (antibody binding site:antigen).

Binding in titration ELISA of the IgGs to (A) RBD (fusion protein with murine Fc part), (B) S1 (fusion protein with murine Fc part) or (C) S1-S2 (fusion protein with His tag). An unrelated antibody with murine Fc part (TUN219-2C1), human HEK293 cell lysate, BSA and lysozyme were used as controls. Experiments were performed in duplicate and mean ± s.e.m. are given. EC50 were calculated with GraphPad Prism Version 6.1, fitting to a four-parameter logistic curve.

(A) IC50 determination by flow cytometry using 10 nM RBD-mFc and 100 nM - 0.3 nM IgG. (B) IC50 determination by flow cytometry using 50 nM S1-S2-His and 500 - 1.5 nM nM IgG. The binding is given in relation to the binding without the antibody. The inhibition assays were made as single titrations. (C) Neutralization screening of ~15 pfu SARS-CoV-2 by 19 anti-RBD SARS-CoV-2 IgGs. (D) STE90-C11 titration on ~150 pfu SARS-CoV-2 to determine the IC50. Neutralization assays were performed in triplicates, mean ± s.e.m. are given. Palivizumab was used as isotype control in both assays. IC50 were calculated with OriginPro using the Logistic5 Fit.
Antibodies from COVID19 patient libraries neutralize active SARS-CoV-2 virus
To analyze the neutralization capacity of the antibodies, the 19 IgGs were first screened in a plaque titration assay using active ~15 pfu SARS-CoV-2 virus. The antibody Palivizumab was used as control antibody (Figure 3C). The antibody STE90-C11 was chosen to be further analyzed. To confirm its neutralizing activity, a plaque assay using ~150 pfu was performed and the determined IC50 was 0.56 nM in the IgG format (Figure 3D).
STE90-C11 is specific for SARS-CoV-2 and binds RBD independent of known RBD mutations
A detailed characterization of the best neutralizer was performed in respect to cross-reaction to other coronaviruses, binding to known RBD mutations, as well as biochemical and biophysical properties. The binding of STE90-C11 to other coronaviruses SARS-CoV-1, MERS-CoV, HCov-HKU1, HCoV-229E and HCoV-NL63 Spike proteins was tested by titration ELISA (Figure 4A). STE90-C11 bound specifically SARS-CoV-2 and did not show any cross reactions to other human-pathogenic coronaviruses.

(A) Cross-reactivity of STE90-C11 to other spike S1 subunits of SARS-CoV-1, MERS-CoV, HCov-HKU1, HCoV-229E and HCoV-NL63 analzyed by ELISA. S1-HIS SARS-CoV-2 Hi5 was produced inhouse. S1-HIS SARS-CoV-2 HEK and all other coronaviruses S1 domain proteins were obtained commercially. (B) Binding to S1 of different SARS-CoV-2 RBD mutations identified in virus isolates from COVID-19 patients analyzed by ELISA. ELISA experiments were in duplicate and mean ± s.e.m. are given. (C) Flow cytometry using 10 nM S1-His, respectively the mutants, and 100 nM - 0.3 nM IgG to analyze the inhibition of S1 binding to ACE2 presenting cells. The binding is given in relation to the binding without the antibody. The inhibition assays were were performed in triplicates, mean ± s.e.m. are given.
Because first SARS-CoV-2 mutants in patients are being identified (www.gisaid.org and (Shi et al., 2020)) and more mutations within the RBD have been characterized to arise under antibody selection pressure (Baum et al., 2020), S1 subunits harboring seven single point mutations in the RBD were produced and binding of STE90-C11 to those mutants was tested. A “worst-case-scenario” mutant containing all the 7 single point mutations, termed S1-7PM, could also be produced (Figure 4B). All constructs were still able to binding recombinant ACE2 (Supplementary Data 6), indicating correct folding. The antibody STE90-C11 was able to bind to all of the investigated RBD mutations including the “worst-case” S1-7PM mutant. STE90-C11 also inhibited ACE2 binding to all analyzed S1 mutants in the cell-based inhibition assay (Figure 4C).
Biochemical characterization of STE90-C11
Aggregation propensity is an important quality parameter in therapeutic antibody development. STE90-C11 showed no relevant aggregation under normal conditions (pH7.4, 4°C in PBS), heat stress conditions (pH7.4, 45°C, 24h in PBS) and pH stress (pH3, 24h, RT) (Supplementary Data 7). A further test to exclude polyreactivity was performed on different antigens (DNA, LPS, lysozyme and EXPI cell lysate) with the FDA-approved antibody Avelumab as control (Supplementary Data 8). Here, STE90-C11 showed equal or less unspecificity compared to Avelumab. The monovalent affinity of STE90-C11 was determined by Bio-Layer Interferometry (BLI) in three different setups (Supplementary Data 9). In the first setup, RBD-mFc was immobilized using anti-Fc capture tips and the KD of STE90-C11 in monovalent Fab format was determined as 8.1 nM. In the second setup, the IgG was immobilized using a Fab2G capture tip and the KD of monovalent S1-His was determined to be 1.6 nM. In the third assay, the IgG was immobilized using a protein A capture tip, resulting in a measured KD of 6.5 nM to S1-His, conferming the affinity of STE90-C11 is in the low nanomolar range. STE90-C11 does not bind to RBD on immunoblots after treatment at 95°C and SDS-gel separation under reducing conditions, while it was able to detect RBD when separated under non-reducing conditions heated either at 95°C or 56°C (Supplementary Data 10). These data suggest that the antibody recognizes a conformational epitope.
STE90-C11 binds at the ACE2-RBD interface
To get further insight into the neutralizing mechanism of STE90-C11, a complex of STE90-C11 Fab and SARS-CoV-2 RBD (22 kDa fragment) was prepared and subjected to crystallization screening. X-ray diffraction images collected from resulting crystals yielded a dataset to an overall resolution limit of 2.0 Å (Supplementary Data 11). After solving the structure by molecular replacement a model was built into the electron density (Figure 5). Figure 5A shows the binding of STE90-C11 to RBD and Figure 5B the binding of ACE2 to RBD. STE90-C11 binds SARS-CoV2 RBD with an interface area of 1113 Å2 (Figure 5C, Supplementary Data 12). Roughly 60% of this area can be contributed to the VH segment forming up to ten hydrogen bonds at the same time and region dominated by hydrophobic interaction (V99, A100, Y33, Y52) (Figure 5D). The remaining 40% are provided by the VL segment contributing eight additional hydrogen bonds to stabilizing the interaction (Figure 5E). All six CDR loops contribute to the interaction between STE90-C11 Fab and SARS-CoV-2 RBD. The superposition of the RBDs of the STE90-C11:SARS-CoV-2 RBD complex with a ACE2:SARS-CoV-2 RBD complex (Lan et al., 2020) resulted in a low Cα root mean square distance of 0.532 Å (183 atoms) indicating that binding of STE90-C11 did not induce substantial changes in the RBD. The superposition further revealed that the neutralization mechanism of STE90-C11 is based on directly competing for the ACE2 binding side, as the interaction interface on the RBD of both molecules almost completely overlap. Further superposition of the STE90-C11:SARS-CoV-2 RBD complex model onto cryo-EM models of the whole SARS-CoV-2 spike protein (Walls et al., 2020) indicate that due to steric hindrance STE90-C11 like ACE2 is expected to only bind to the open conformation of the spike protein (Supplementary Data 13).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A+B) The structure of the complex between RBD (green) and STE90-C11 (A; heavy chain: Yellow; light chain: pale yellow) or ACE2 (B; red; PDB: 6M0J (Lan et al., 2020)) depicted in cartoon representation after superposition of the RBD. (C) Surface representation of the RBD with the binding surface (cutoff 4Å) of STE90:C11 colored in yellow and of ACE2 colored in red. The competitive binding surface is colored in orange. (D+E) Detailed view of the interactions between RBD and the heavy chain (D) and light chain (E) of STE90-C11. Possible hydrogen bonds are displayed as dashed yellow lines and residues with alternate conformations are marked with an asterisk. (F) Sequence alignment of RBD. Depicted is a part of the sequence alignment of the SARS-CoV-2 and SARS-CoV S-protein (UniProt: P0DTC2; P59594). Identical residues are depicted as bold blue letters. Under the alignment contacts between residus of the S-protein and ACE2 (PDB: 6M0J (Lan et al., 2020)), STE90-C11 (PDB: 7B3O), CB6 (PDB: 7C01 (Shi et al., 2020)), B38 (PDB: 7BZ5 (Wu et al., 2020)) CR3022 (PDB: 6W41 (Yuan et al., 2020)), REGN10933, REGN10987 (PDB: 6XDG (Hansen et al., 2020)) and BD-368-2 (PDB: 7CHF (Cao et al., 2020)) are marked by an A (ACE2), H (heavy chain) or L (light chain). A black letter indicates a distance under 4Å and a red letter under 3.2Å between non-hydrogen atoms of the RBD and the respective protein. Black arrows indicated residues often exchanged in the RBD of SARS-CoV-2, which were tested for their influence on STE90-C11 binding in this work. Distances were calculated with CNS (Brünger et al., 1998) and the representation was prepared utilizing ESPript (Robert and Gouet, 2014).
Discussion
For the treatment of COVID-19 but also to protect risk groups, human anti-SARS-CoV-2 antibodies are a promising therapeutic option. Human recombinant antibodies were successfully used for the treatment of other viral diseases. Antibody mAb114 (Corti et al., 2016) and the currently FDA approved three-antibody cocktail REGN-EB3 (Pascal et al., 2018) showed a good efficiency in clinical trials against Ebola virus especially in comparison to Remdesivir (Mulangu et al., 2019). For the treatment of a severe respiratory infection of infants caused by the respiratory syncytial virus (RSV), the antibody Palivizumab is approved by EMA and FDA (van Mechelen et al., 2016; Subramanian et al., 1998). These anti-viral antibodies can be used as blueprint to develop therapeutic antibodies against SARS-CoV-2. Currently, monoclonal antibodies against SARS-CoV-2 were selected by rescreening memory B-cells from a SARS patient (Pinto et al., 2020), selected from COVID-19 patients by single B-cell PCR (Cao et al., 2020; Shi et al., 2020) or FACS sorting (Kreer et al., 2020), selected from transgenic mice followed and from patients by single B-cells FACS sorting (Hansen et al., 2020) or using phage display using universal or patient libraries including different antibody formats (Bertoglio et al., 2020; Li et al., 2020; Liu et al., 2020b; Zeng et al., 2020).
In this work, antibody phage display immune libraries were constructed using lymphocytes from six local convalescent COVID-19 patients. 197 unique antibodies were selected against RBD. We intently focused on RBD aiming to find antibodies which directly interfer with the interaction between spike and ACE2, to avoid potential antibody dependent enhancement (ADE). ADE during coronavirus entry has been described for both MERS (Wan et al., 2020) or SARS (Wang et al., 2014). It is defined as “enhancement of disease severity in an infected person or animal when an antibody against a pathogen…worsens its virulence by a mechanism that is shown to be antibody-dependent” (Arvin et al., 2020). Furthermore, immune dysregulation and lung inflammation was also caused by anti-spike antibodies in an acute SARS-CoV infection (Liu et al., 2019). On the other hand, Quinlan et al. (Quinlan et al., 2020) reported that animals immunized with RBD SARS-CoV-2 did not mediate ADE. A therapeutic antibody directed against RBD and the usage of a Fc-part which is not binding to Fc-gamma receptors (“silent Fc”) would reduce the risk of ADE and increase safety for human patients, in particular because ADE cannot be fully predicted from in vitro studies or from animal models (Arvin et al., 2020).
The selected anti-RBD antibodies were analyzed for RBD::ACE2 inhibition in the scFv-Fc format. The advantage of the IgG like bivalent scFv-Fc format is that it speeds up analysis by requiring only one cloning step and provides high expression yields in small scale format (Bertoglio et al., 2020; Jäger et al., 2013; Wenzel et al., 2020a). 30 scFv-Fc antibodies inhibited the binding of the spike protein to ACE2-expressing living cells. Interestingly, the majority of the VH V-genes of the selected anti-RBD antibodies and 28 of these 30 inhibiting antibodies is the VH3-66 V-gene showing strong enrichment of this V-gene in the immune response of COVID-19 patients against RBD. This is in accordance with Cao et al. (Cao et al., 2020) who found that enriched use of VH3-53 or VH3-66. VH3-53 and VH3-66 V-genes are closely related. Robbiani et al. (Robbiani et al., 2020) showed an enrichment of VH3-30, VH3-53 and VH3-66 and Hansen et al. (Hansen et al., 2020) reported a bias towards VH3-53. The VH3-53 and VH3-66 V-genes are closely related. In contrast, Ju et al. (Ju et al., 2020) identified mainly VH1-2, VH3-48 and VH3-9 V-genes for their selected anti-RBD antibodies from one patient and Brouwer et al. (Brouwer et al., 2020) identified a strong enrichment of VH1-69, VH3-30 and VH1-24 in three patients, but also an enrichment of VH3-53 and VH3-66. VH3-66 antibodies against RBD were also selected from the naive HAL9/10 antibody gene libraries made long before the SARS-CoV-2 outbreak (Kügler et al., 2015) but not significantly enriched (Bertoglio et al., 2020). The selected inhibiting antibodies in our study (Table 1) all have a high germinality index and are therefore very similar to the human germline, predicting low immunogenicity when used as a therapeutic agent. Only some light chains have shown a major difference to their closest related V-gene. Kreye et al. (Kreye et al., 2020) also described that their antibodies selected from COVID-19 patients are very near to the germline genes. Interestingly, the VH V-genes of these immune libraries are closer to their germline compared to the anti-RBD antibodies selected from the naive HAL9/10 antibody gene libraries (Bertoglio et al., 2020).
In the cell based inhibition assay, some of the antibodies show a molar antibody:spike monomer ratio lower then 1:1. This was also shown by Bertoglio et al. (Bertoglio et al., 2020) and can be explained by the three RBDs on the same spike trimer that can be in different conformations (“up” and “down”) and only the RBDs in “up” positions are able to bind ACE2 (Walls et al., 2020) and therefore, the “down” RBDs are not fully accessible. The monovalent affinity of STE90-C11 was measured with 1.6 nM-8.1 nM depending on the BLI setup. This is in the same range as BD-368-2 with 0.82 nM affinity to RBD (Cao et al., 2020), 3.3 nM for REGN10933 in the setup with the monovalent RBD (Hansen et al., 2020). 70 nM for B38 (Wu et al., 2020) or 0.8-4.7 nM for CB6 (Shi et al., 2020).
The analysis of the binding STE90-C11 to other human coronaviruses showed specificity for SARS-CoV-2. Comparison between the epitope of STE90-C11 and other published antibodies (Figure 5F) revealed that it binds in the same region as CB6 (Shi et al., 2020) and B38 (Wu et al., 2020). The selectivity of STE90-C11 and CB6 for SARS-CoV-2 over SARS-CoV RBD can mainly be attributed to the interaction with residues 473-476, as they are not involved in ACE2 binding and SARS-CoV RBD folds into a different conformation of this loop causing steric hindrance for antibody binding (Lan et al., 2020).
The binding of STE90-C11 to S1 containing RBD mutations from COVID-19 patients was analyzed, showing a binding to all mutants including a “worst case” mutant which combined all seven analyzed mutations. As the cell based inhibition analysis demonstrate, STE90-C11 is able to inhibiting all analyzed mutants, validating a tolerance for RBD mutations. REGN10933 (Hansen et al., 2020), which is also binding at the RBD-ACE2 interface, showed a loss of neutralization in an assay using pseudoviral particles for the F486V and a reduced neutralization for both G485D and E484K mutations (Baum et al., 2020). Loss of binding to F486A is also described for VH-Fc ab8 (Li et al., 2020). In the epitope analysis, REGN10933 has more molecular interactions within the region aa483-aa486 compared to STE90-C11. Even though these RBD mutations are currently not spread widely (www.GISAID.org), the imminent use of therapeutic antibodies will inevitably result in escape mutants as already demonstrated by Baum et al. (Baum et al., 2020) in vitro. Therefore, an antibody combination like REGN10933 + REGN10987 where both are sensitive to some but different mutations, or an antibody like STE90-C11 which is tolerant to mutations is needed.
We hypothesize that STE90-C11 is still binding to the analyzed RDB mutants because of both the wide interface area of 1133 Å2 and the extensive light chain contacts which may compensate the exchange of several amino acids. The calculated interface area of STE90-C11 VL is more than twice the area of REGN10933 and REGN10987 and slightly larger compared to CB6 or CR3022 (Supplementary Data 12).
In summary, the patient-derived SARS-CoV-2 neutralizing anti-RBD antibody STE90-C11 binding to the RBD-ACE2 interface maintains high similarity to the human germline V-genes VH3-66, the same family of many isolated anti-SARS-CoV-2 neutralizing antibodies. Unlike other monoclonals, STE90-C11 is tolerant to known RBD mutants and therefore an excellent candidate for therapeutic development.
STAR Methods
Key resources table
Resource Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Hust (m.hust{at}tu-bs.de)
Materials Availability
All requests for resources and reagents should be directed to the Lead Contact author. This includes antibodies, plasmids and proteins. All reagents will be made available on request after completion of a Material Transfer Agreement.
Data and Code Availability
The structure data and the antibody sequence is available at PDB 7B3O.
Experimental models and subject detail
SARS-CoV-2-infected individuals and sample collection
For the generation of human immune antibody libraries against SARS-CoV-2, blood from COVID-19 convalescent patients were collected from local hospitals. This was performed in accordance with the Declaration of Helsinki. All the voluntary donors were informed about the project and gave their consent. The use of blood samples for the development of antibody phage display libraries was approved by the ethical committee of the Technische Universität Braunschweig (Ethik-Kommission der Fakultät 2 der TU Braunschweig, approval number FV-2020-02). The donors have given their consent for study publication. A control serum was obtained from LADR Braunschweig.
Method Details
Production of antigens in insect cells
The antigens were produced and purified as described before (Bertoglio et al., 2020; Bleckmann et al., 2019). In brief, different domains or subunits of the Spike protein (GenBank: MN908947) were produced Baculovirus-free in High Five cells (Thermo Fisher Scientific, Schwerte, Germany) by transient transfection. High Five cells were cultivated at 27°C, 110 rpm in ExCell405 media (Sigma Aldrich, Munich, Germany) and kept at a cell density between 0.3 – 5.5 ×106 cells/mL. For transfection cells were centrifuged and resuspended in fresh media to a density of 4×106 cells/mL and transfected with 4 μg plasmid/mL and 16 μg/mL of PEI 40 kDa (Polysciences). After 4 h to 24 h after transfection cells were diluted to a final density of 1× 106 cells/mL. At 48 h after transfection, the culture volume was doubled. Cell supernatant was harvested five days after transfection in a two step centrifugation (4 min at 180xg and 20 min at above 3500xg) and 0.2 μm filtered for purification.
Protein purification
Protein purification was performed as described before (Bertoglio et al., 2020) depending on the production scale in either 24 well filter plate with 0.5 mL resin (10 mL scale) or 1 mL column on Äkta go (Cytiva), Äkta Pure (Cytiva) or Profina System (BIO-RAD). MabSelect SuRe or HiTrap Fibro PrismA (Cytiva) was used as resin for Protein A purification. For His-tag purification of Expi293F supernatant HisTrap FF Crude column (Cytiva) and for His-tag purification of insect cell supernatant HisTrap excel column (Cytiva) was used. All purifications were performed according to the manufacturer’s manual. Indicated antigens were further purified by size exclusion chromatography by a 16/600 Superdex 200 kDa pg (Cytiva). All antigens, antibodies and scFv-Fc were run on Superdex 200 Increase 10/300GL (Cytiva) on Äkta or HPLC (Techlab) on an AdvanceBio SEC 300Å 2.7 μm, 7.8×300 mm (Agilent) for quality control.
Serum ELISA
For COVID-19 convalescent serum analysis, sera were titrated from 1:314 in 11 steps (dilution ratio 1:3.14) on 100 ng/well of RBD-His. As unspecificity controls, sera were tested on 100 ng/well BSA. All immobilization steps were performed in 0.05 M carbonate buffer (pH 9.6). Serum IgGs were detected using goat-anti-hIgG(Fc)-HRP (1:70,000, A0170, Sigma). Titration assays were performed in 96 well microtiter plates (Costar).
COVID-19 convalescent patient library construction
For the generation of human immune antibody libraries against SARS-CoV-2, blood from 6 donors showing a good antibody titer against RBD was used. The library construction was performed as described previously with minor modifications (Kügler et al., 2018). In brief, the peripheral blood mononuclear cells (PBMC) were extracted from the blood via Ficoll (GE Healthcare, Freiburg, Germany) and RNA isolated with TRIzol LS reagent (Life Technologies, Carlsbad, USA) and Direct-zol RNA Miniprep Plus kit (Zymo Research, Freiburg, Germany). For the plasma B-cell sorted library, plasma B-cells were double-stained with mouse α-CD19 APC-conjugated (MHCD1905, Thermo Fisher Scientific, Schwerte, Germany) and mouse α-CD138 (FITC conjugated) antibody (DL-101, BioLegend, San Diego, USA), sorted via FACSMelody (BD, Franklin Lakes, USA). Sorted cells were directly collected in TRIzol LS reagent and the RNA was extracted as indicated above. RNA was converted into cDNA using Superscript IV (Thermo Fisher Scientific, Waltham, USA) according to the manufacturer’s instructions. Library construction was achieved as described previously (Wenzel et al., 2020a) with slight modifications. In brief, specific primers for the VH chain, kappa light chain and lambda light chain were used to amplify antibody genes from the cDNA. These resulting PCR products were purified and again amplified with primers adding specific restriction sites for further cloning into the E. coli expression vector pHAL52. The vector pHAL52 is derived from the vector pHAL30 (Kügler et al., 2015) with an AscI restriction site in the VL stuffer and a SalI restriction site in the VH stuffer for removal of uncut vector backbone. First, the light chain was cloned between the restriction sites MluI-HF and NotI-HF. In a second step the VH chain was cloned with the restriction sites HindIII-HF and NcoI-HF into previously cloned pHAL52-VL, additionally digested with AscI. The efficiency of library cloning was tested by colony PCR and the rate of complete scFv insertion was determined. The libraries were packaged with Hyperphage (Rondot et al., 2001; Soltes et al., 2007). Antibody phage were precipitated with PEG-NaCl and resuspended in phage dilution buffer (10 mM TrisHCl pH7,5, 20 mM NaCl, 2 mM EDTA). Resulting phage titer was determined by infection of E. coli XL1 blue MRF’.
Antibody selection using phage display
Antibody selection was performed as described previously with modifications (Russo et al., 2018). In brief, 5 μg of of S1-S2-His or RBD-His (produced in High Five cells) was diluted in carbonate buffer (50 mM NaHCO3/Na2CO3, pH 9.6) and coated onto the wells of a High binding 96 well microtiter plate (High Binding, Costar) at 4 °C overnight. Next, the wells were blocked with 350 μL 2% MBPST (2% (w/v) milk powder in PBS; 0.05% Tween20) for 1 h at RT and then washed 3 times with PBST (PBS; 0.05% Tween20). Before adding the libraries to the coated wells, the libraries (5×1010 phage particles) were preincubated with 5 μg of an unrelated scFv-Fc and 2% MPBST on blocked wells for 1 h at RT, to deprive libraries of human Fc fragment binders. The libraries were transferred to the antigen coated wells, incubated for 2 h at RT. After 10 washes, bound phages were eluted with 150 μL trypsin (10 μg/mL) at 37°C, 30 minutes and used for the next panning round. The eluted phage solution was transferred to a 96 deep well plate (Greiner Bio-One, Frickenhausen, Germany) and incubated with 150 μL E. coli TG1 (OD600 = 0.5) firstly for 30 min at 37°C, then 30 min at 37°C and 650 rpm to infect the phage particles. 1 mL 2xYT-GA (1.6% (w/v) Tryptone; 1% (w/v) Yeast extract; 0.5% (w/v) NaCl (pH 7.0), 100 mM D-Glucose, 100 μg/mL ampicillin) was added and incubated for 1 h at 37°C and 650 rpm, followed by addition of 1×1010 cfu M13KO7 helper phage. Subsequently, the infected bacteria were incubated 30 min at 37°C followed by 30 min at 37°C and 650 rpm before centrifugation for 10 min at 3220xg. The supernatant was discarded and the pellet resuspended in fresh 2xYT-AK (1.6% (w/v) Tryptone; 1% (w/v) Yeast extract; 0.5% (w/v) NaCl (pH 7.0), 100 μg/mL ampicillin, 50 μg/mL kanamycin). The antibody phage were amplified overnight at 30°C and 650 rpm and used for the next panning round. In total four panning rounds were performed. In each round, the stringency of the washing procedure was increased (20x in panning round 2, 30x in panning round 3) and the amount of antigen was reduced (2.5 μg in panning round 2, 1.5 μg in panning round 3). After third panning round single clones were analyzed for production of RBD specific scFv by screening ELISA.
Screening of monoclonal recombinant binders using E. coli scFv supernatant
Soluble antibody fragments (scFv) were produced in 96-well polypropylene MTPs (U96 PP, Greiner Bio-One) as described before (Russo et al., 2018; Wenzel et al., 2020a). Briefly, 150 μL 2xYT-GA was inoculated with the bacteria bearing scFv expressing phagemids. MTPs were incubated overnight at 37°C and 800 rpm in a MTP shaker (Thermoshaker PST-60HL-4, Lab4You, Berlin, Germany). A volume of 180 μL 2xYT-GA in a PP-MTP well was inoculated with 20 μL of the overnight culture and grown at 37°C and 800 rpm for 90 minutes (approx. OD600 of 0.5). Bacteria were harvested by centrifugation for 10 min at 3220xg and the supernatant was discarded. To induce expression of the antibody genes, the pellets were resuspended in 200 μL 2xYT supplemented with 100 μg/mL ampicillin and 50 μM isopropyl-beta-D-thiogalacto-pyranoside (IPTG) and incubated at 30°C and 800 rpm overnight. Bacteria were pelleted by centrifugation for 20 min at 3220xg and 4°C.
For the ELISA, 100 ng of antigen was coated on 96 well microtiter plates (High Binding, Costar) in PBS (pH 7.4) overnight at 4°C. After coating, the wells were blocked with 2% MPBST for 1 h at RT, followed by three washing steps with H2O and 0.05% Tween20. Supernatants containing secreted monoclonal scFv were mixed with 2% MPBST (1:2) and incubated onto the antigen coated plates for 1 h at 37°C followed by three H2O and 0.05% Tween20 washing cycles. Bound scFv were detected using murine mAb 9E10 which recognizes the C-terminal c-myc tag (1:50 diluted in 2% MPBST) and a goat anti-mouse serum conjugated with horseradish peroxidase (HRP) (A0168, Sigma) (1:42000 dilution in 2% MPBST). Bound antibodies were visualized with tetramethylbenzidine (TMB) substrate (20 parts TMB solution A (30 mM Potassium citrate; 1% (w/v) Citric acid (pH 4.1)) and 1 part TMB solution B (10 mM TMB; 10% (v/v) Acetone; 90% (v/v) Ethanol; 80 mM H2O2 (30%)) were mixed). After stopping the reaction by addition of 1 N H2SO4, absorbance at 450 nm with a 620 nm reference was measured in an ELISA plate reader (Epoch, BioTek). Monoclonal binders were sequenced and analyzed using VBASE2 (www.vbase2.org) (Mollova et al., 2010) and possible glycosylation positions in the CDRS were analyzed according to Lu et al (Lu et al., 2019).
ScFv-Fc and IgG production
Unique scFv sequences isolated by antibody-phage display were subcloned into pCSE2.7-hIgG1-Fc-XP using NcoI/NotI (New England Biolabs, Frankfurt, Germany) for mammalian production in Expi293F cells as scFv-Fc (Wenzel et al., 2020a). For IgG production, the variable domains were recloned into the IgG vectors human IgG1 format by subcloning of VH in the vector pCSEH1c (heavy chain) and VL in the vector pCSL3l/pCSL3k (light chain lambda/kappa) (Steinwand et al., 2014) adapted for Golden Gate Assembly procedure with Esp3I restriction enzyme (New England Biolabs, Frankfurt, Germany). For the antibodies CB6 (pdb 7C01) and REGN10933 and REGN10987 (pdb 6XDG) the public available amino acid sequences were used and the V-Genes were ordered as GeneArt Strings DNA fragments (Thermo Fisher Scientific, Schwerte, Germany) and recloned in the above indicated vectors. A “silenced” Fc part with following point mutations described Armour et al (Armour et al., 1999) and Shields et al (Shields et al., 2001) were used: E233P, L234V, L235A, deletion of G236, D265G, A327Q and A330S. Expi293F cells were cultured at 37°C, 110 rpm and 5% CO2 in Gibco FreeStyle F17 expression media (Thermo Fisher Scientific) supplemented with 8 mM Glutamine and 0.1% Pluronic F68 (PAN Biotech). At the day of transfection cell density was between 1.5 - 2×106 cells/mL and viability at least above 90%. For formation of DNA:PEI complexes 1 μg DNA/mL transfection volume and 5 μg of 40 kDa PEI (Polysciences) were first diluted separately in 5% transfection volume in supplemented F17 media. DNA (1:1 ratio of the vectors for IgG production) and PEI was then mixed and incubated ~25 min at RT before addition to the cells. 48 h later the culture volume was doubled by feeding HyClone SFM4Transfx-293 media (GE Healthcare) supplemented with 8 mM Glutamine. Additionally, HyClone Boost 6 supplement (GE Healthcare) was added with 10% of the end volume. One week after transfection supernatant was harvested by 15 min centrifugation at 1500xg.
Inhibition of S1-S2 and RBD binding to ACE2 expressing cells using flow cytometry
The inhibition tests in flow cytometry on EXPI293F cells were performed based on a previously published protocol (Bertoglio et al., 2020). Briefly, Expi293F cells were transfected according to the protocol above using pCSE2.5-ACE2fl-His and 5% eGFP plasmid. Two days after transfection, purified S1-S2-His was labelled using Monolith NTTM His-Tag Labeling Kit RED-tris-NTA (Nanotemper) according to the manufacturer’s protocol. In this setup 50 nM antigen was incubated with min. 1 μM of different scFv-Fc and the ACE2 expressing cells. The resulting median antigen fluorescence of GFP positive living single cells was measured. For comparison of the different scFv-Fc first the median fluorescence background of cells without antigen was subtracted, second it was normalized to the antigen signal where no antibody was applied. ScFv-Fc showing an inhibition in this first setup were further titrated as IgGs (max. 500 nM-0.5 nM) on S1-S2-His, S1-His or on RBD-mFc (max. 100 nM-0.1 nM). S1-His and the corresponding mutants were detected with mousen anti-penta His (Qiagen) and goat anti-mFc APC-conjugated antibody (Dianova). RBD-mFc was detected directed with the goat anti-mFc APC-conjugated antibody (Dianova). Measurments were performed with MACSQuant Analyzer (Milteny Biotech). The IC50 was calculated using the equation f(x)=Amin+(Amax-Amin)/(1+(x0/x)^h)^s and parameters from OriginPro (2019).
Dose dependent binding of IgG in titration ELISA
For titration ELISA, purified IgGs were titrated from 3.18 μg/mL- 0.001 ng/mL on 30 ng/well of the following antigens: S1-S2-His (High Five cell produced), RBD-mFc (High Five cell produced), S1-mFc (High Five cell produced) and TUN219-2C1-mFc (as control for unspecific Fc binding). In addition, all scFv-hFc were also tested only at the highest concentration (3.18 μg/mL) for unspecific cross-reactivity on Expi293F cell lysate (104 cells/well), BSA (1% w/v) and lysozyme. IgGs were detected using goat-anti-hIgG(Fc)-HRP (1:70000, A0170, Sigma). Titration assays were performed using 384 well microtiter plates (Greiner Bio-One) using Precision XS microplate sample processor (BioTek), EL406 washer dispenser (BioTek) and BioStack Microplate stacker (BioTek). EC50 were calculated with by GraphPad Prism Version 6.1, fitting to a four-parameter logistic curve. Titration ELISAs on other coronaviruses and S1-HIS mutants were performed as described above.
Screening and titrating monoclonal antibodies for SARS-CoV-2 neutralization in cell culture
VeroE6 cells (ATCC CRL-1586) were seeded at a density of 6*104/well onto cell culture 96-well plates (Nunc, Cat.#167008). Two days later, cells reached 100% confluence. For titration, antibodies were diluted in 1/√10 steps and mixed with a fixed inoculum of SARS-CoV-2/Münster/FI110320/1/2020 (kind gift of Stephan Ludwig, University of Münster, Germany) (10-20, respectively 100-150 pfu) in a total volume of 500 μl of Vero E6 medium (DMEM, 10% FCS, 2 mM glutamine, penicillin, streptomycin). After one hour incubation at 37°C, cells were infected with the antibody/virus mix, incubated for one hour and then overlaid with Vero E6 medium containing 1.5% methyl-cellulose. Three days postinfection, wells were imaged using a Sartorius IncuCyte S3 (4x objective, whole-well scan) and plaques were counted from these images. Image data was quantified with the IncuCyte S3 GUI tools measuring the decrease of confluence concomitant with the cytopathic effect of the virus in relation to uninfected controls and controls without antibody and analyzed with Origing using the Logistic5 fit.
Specificity assay
To test specificity of the antibody candidates an ELISA on DNA, LPS, lysozyme and cell lysate was performed under standard conditions (see above). In brief, 10 μg/mL of the respective antigen was immobilized on 96 well microtiter plates (High binding, Costar) in PBS (pH 7.4) overnight at 4°C. After blocking 10 μg/mL of STE90-C11 IgG, Avelumab, Palivizumab and IVIG respectively were incubated and later detected using goat-anti-hIgG(Fc)-HRP (1:70000, A0170, Sigma). TMB reaction took place for 30 min and absorbance at 450 nm with a 620 nm reference was measured in an ELISA plate reader (Epoch, BioTek). All signals were normalized to the absorbance of Avelumab.
Analytical size exclusion chromatography (SEC)
All purified antigens and indicated antibodies were run on Superdex 200 Increase 10/300GL column (Cytiva) on Äkta pure system (Cytiva) according to the manufactures protocol.
Affinity measurement by Bio-Layer Interferometry
The affinity was measured by Bio-Layer Interferometry in three different assays using the Octet qKe (Fortebio/Sartorius GmbH, Göttingen, Germany).
In the first assay, anti-Mouse Fc-Capture (AMC) sensors were activated for 10 min in PBS. After that, the sensors were equilibrated in assay buffer (PBS containing 1% BSA and 0.05% Tween 20) for 60 s before RBD-mFc (Sino Biologicals) was loaded onto the sensors at 10 μg/ml for 180 s. After a stable baseline measurement was established (60 s), antigen-loaded sensors were transferred to an 8-point antibody dilution series (500, 158, 50, 15, 5, 1.5, 0.5 and 0 nM). Association of the Fab antibody to the antigen was measured for 300 s. After that, the sensors were transferred into assay buffer were the dissociation was measured for 900 s. Significant binding of the antibody to an unloaded sensor was not detected. For data analysis, the reference measurement (0 nM) was subtracted from the other measurements and data traces ranging from 158 to 1.5 nM were used for modeling of the kinetic data using a 1:1 binding model.
In the second assay, anti-human Fab (FAB2G) sensors were activated for 10 min in PBS. After that, the sensors were equilibrated in assay buffer (PBS containing 1% BSA and 0.05% Tween 20) for 60 s before the IgG antibody was loaded onto the sensors at 2.5 μg/ml for 180 s. After a stable baseline measurement was established (60 s), antibody-loaded sensors were transferred to an 8-point S1-HIS antigen dilution series (500, 158, 50, 15, 5, 1.5, 0.5 and 0 nM). Association of the S1 antigen to the antibody was measured for 300 s. After that, the sensors were transferred into assay buffer were the dissociation was measured for 900 s. Significant binding of the antigen to an unloaded sensor was not detected. For data analysis, the reference measurement (0 nM) was subtracted from the other measurements and data traces ranging from 50 to 5 nM were used for modeling of the kinetic data using a 1:1 binding model in the Data Analysis HT 11.0 software tool.
And in the third assay, protein A sensors were activated for 10 min in PBS. Before use, the sensors were regenerated for 5 cycles in 10 mM Glycine buffer (pH2.0) followed by neutralization in PBS. Each step was performed for 5 s. After that, the regenerated sensors were equilibrated in assay buffer (PBS containing 1% BSA and 0.05% Tween 20) for 60 s before the IgG antibody was loaded onto the sensors at 2.5 μg/ml for 180 s. After a stable baseline measurement was established (60 s), antibody-loaded sensors were transferred to an 8-point S1-HIS antigen dilution series (500, 158, 50, 15, 5, 1.5, 0.5 and 0 nM). Association of the S1 antigen to the antibody was measured for 300 s. After that, the sensors were transferred into assay buffer were the dissociation was measured for 900 s. Significant binding of the antigen to an unloaded sensor was not detected. For data analysis, the reference measurement (0 nM) was subtracted from the other measurements and data traces ranging from 50 to 0.5 nM were used for modeling of the kinetic data using a 1:1 binding model.
Immunoblot analysis
For the immunoblot analysis RBD-His was disrupted either by incubation for 10 minutes at 56°C without β-Mercaptoethanol, at 95°C without β-Mercaptoethanol or at 95°C in Laemmli sample buffer (Laemmli, 1970) with β-Mercaptoethanol. RBD was separated by 12% SDS-PAGE and blotted onto a nitrocellulose membrane (Amersham Protan 0.2 μm NC, GE HealthCare). The membrane was blocked with 2% M-PBST for 1 h at RT. For the detection of RBD-His, 10 μg/mL STE90-C11 IgG was used. For control, RBD-His (95°C + β-Mercaptoethanol) was detected with 2μg/mL mouse anti-His (Dia-900-200, Dianova, Hamburg, Germany) for 1.5 h at RT. After 3x washing with PBST the secondary antibody goat anti-human Fc AP conjugated (1:20,000, Jackson ImmunoResearch, Cambridge House, UK) was used for the detection of STE90-C11 and goat anti-mouse IgG AP conjugated (1:30,000, 115-055-071, Dianova) was used for the anti-His antibody and incubated for 1 h. Finally, the membrane was washed 2x with PBST and 4x with PBS. Staining and visualization of specific proteins was performed with 5-Brom-4-Chlor-3-Indolyl-Phosphat/Nitro Tetrazolium Blue Chloride (NBT-BCIP, Thermo Fisher Scientific GmbH, Dreieich, Germany) according to standard protocols.
Fab and RBD22 production for co-crystallization
The production of STE90-C11 Fab fragment was done by transient co-transfection of plasmids encoding the heavy and the light chain in Expi293F cells cultivated at 37°C, 5% CO2 and 100rpm in Expi Expression Medium. The transfection was performed at a density of 3*10^6 cells/ml by adding 1μg/ml culture mixed plasmids and 4 μg/ml culture PEI 40 kDa (Polyscience). After 24h of incubation CHO CD efficient feed A (Thermo) to a final concentration of 0.5% was added following incubation for an additional 48h. The supernatant was harvested by centrifugation (30min, 3000g) and sterile filtration (0.2μm). The Fab-fragment was purified by affinity chromatography using 1ml HisTrap Excel column (GE Healthcare) followed by a size exclusion chromatography on a 10/300 Superdex200 Increase column (GE Healthcare) according to manufactures manual.
For the production of RBD22 High Five cells grown in Ex-Cell 405 serum-free medium (Sigma) were transiently transfected with 5μg/mL plasmid and 20μg/mL PEI 40 kDa (Polyscience) at a cell density of 5*10^6 cells/ml. After 4h incubation at 27°C and 100rpm the cells were diluted to a density of 1*10^6 cells/ml and further incubated for three more days. Following centrifugation (5000xg 30min) and sterile filtration (0.2μm) the supernatant was loaded on a 5ml HisTrap Excel column (GE Healthcare). After washing and elution with imidazole the RBD22 containing fractions were further purified by size exclusion chromatography on a 26/6000 Superdex 200 pg using 20mM Tris-HCl pH8 and 150mM NaCl as a buffer. Aliquots were snap-frozen in liquid nitrogen and stored at −80°C till further use.
Crystallization, data collection and structure determination
The Fab fragment of STE90-C11 was incubated overnight with a 1.2 molar excess of purified RBD22 at 4°C. The Complex was isolated by size exclusion chromatography on a Superdex 200 Increase 10/300GL (GE Healthcare) with 20mM Tris-HCl pH8 and 150mM NaCl as a running buffer. Fractions containing the complex were concentrated utilizing a Vivaspin2 ultrafiltration unit (10,000 MWCO; Sartorius). Crystallizations trails were set up in 96-well sitting-drop vapor diffusion plates (Intelli 96-3 plates, Art Robbins Instruments) with a pipetting robot (Crystal Gryphon, Art Robbins Instruments) mixing 200nl reservoir solution with 200nl of protein solution (14mg/mL, 7mg/mL) and equilibrated against 60 μl of reservoir solution. As initial screens the Cryos Suite and the JCSGplus Suite (Qiagen) were chosen and crystal growth was monitored in a crystal hotel (RockImager, Formulatrix). Initial hits were further optimized by a random screen assembled with a Formulator pipetting robot (Formulatrix). Best diffracting crystals grew in 13.3% (w/v) polyethylene glycol 6,000, 0.1M MES pH 5.6 and 0.24M tri sodium citrate. Crystals were harvested with nylon loops and soaked in reservoir solution mixed with 2,3-(R,R)-butandiol (Alfa Aeser) to a final concentration of 10% (v/v) prior to flash cooling in liquid nitrogen. 3600 diffraction images with an oscillation angle of 0.1° per image were collected at the beamline P11 at PETRA III (DESY, Hamburg, Germany) (Burkhardt et al., 2016) on a Pilatus 6M fast detector (Dectris) and processed with XDS (Kabsch, 2010) and Aimless (Evans and Murshudov, 2013) yielding a dataset with a resolution cut off of 2.0 Å based on a CC1/2 value greater than 0.5. Initial phases were determined by molecular replacement with Phaser (McCoy et al., 2007). As a search model the coordinates of a Fab fragment and the RBD was used (PDB: 7bwy) (Ju et al., 2020). The model was further improved by manual rebuilding in Coot (Emsley et al., 2010) and computational refinement with phenix. refine (Afonine et al., 2012) including placement of water, TLS refinement and riding hydrogens in the final steps of the procedure. Depictions of the model were generated with PyMol molecular graphics system (Schrödinger LLC; version 2.3.2). Data processing and model refinement statistics can be found in Supplementary Data 11. The final model can be accessed under the PDB code 7B3O.
Quantification and statistical analysis
Flow cytometry experiments with MACSQuant® VYB (MQ3140) were analyzed using the internal MACSQuantify software. BLI experiments were analzyed using the Octet qKe Data Analysis HT 11.0 software. EC50 were calculated with GraphPad Prism Version 6.1, fitting to a four-parameter logistic curve (Figure xy). IC50 were calculated with OriginPro using the Logistic5 Fit (Figure xy). The structure model was improved by manual rebuilding in Coot (Emsley et al., 2010) and computational refinement with phenix.refine (Afonine et al., 2012) including placement of water, TLS refinement and riding hydrogens in the final steps of the procedure. Depictions of the model were generated with PyMol molecular graphics system (Schrödinger LLC; version 2.3.2). Antibody-RDB, respectively ACE2-RBD, interface areas according analyzed using “Protein interfaces, surfaces and assemblies” (PISA) at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) (Krissinel and Henrick, 2007).
Author contributions
F.B., A.F., T.S., S.D., J.v.H. L.Ĉ-Ŝ., M.S., M.H. conceptualized the study. F.B., V.F., M.R., P.A.H, U.R., T.K., D.M., N.L, S.S., R.B., K-T.S., K.D.R.R., P.R., D.S., A.K., S.Z.-E., M.B., Marg.S., G.M.S.G.M., E.V.W., G.R., M.S. performed and designed experiments. F.B., D.M. N.L., S.S., U.R., L.S., P.K., J.v.H., A.F., L.Ĉ-Ŝ., M.S., M.H. analyzed data. H.S.P.G., S.C., A.G. provided patient samples and support. S.D., L.Ĉ-Ŝ., M.H. conceived the funding. P.K., E.V.W., Gi.R., A.H., V.F., S.D., Gü.R., A.F., J.v.H., M.S., M.H. advised on experimental design and data analysis. F.B., T.K., S.D., L.Ĉ-Ŝ., M.S., M.H. wrote the manuscript.
Declaration of interests
The authors declare a conflict of interest. F.B., D.M., N.L., S.S., P.A.H., R.B., M.R., K.T.S., K.D.R.R., S.Z.-E., M.B., V.F., S.T., M.S. and M.H. are inventors on a patent application on blocking antibodies against SARS-CoV-2. A.H., A.F. and T.S. are officers of CORAT Therapeutics GmbH, a company founded by YUMAB GmbH for the clinical and regulatory development of STE90-C11 (COR-101). A.F., T.S., S.D. and M.H. are shareholders of YUMAB GmbH.
Acknowledgements
We kindly acknowledge the financial support of MWK Niedersachsen (14-76103-184 CORONA-2/20) for antibody generation. We also acknowledge support of the European Union for the ATAC (“antibody therapy against corona”, Horizon2020 number 101003650) consortium. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRAIII and we would like to thank Johanna Hakanpää for assistance in using the P11 beamline. We are deeply grateful to Adelheid Langner, Andrea Walzog, Bettina Sandner, Cornelia Oltmann and Wolfgang Grassl for constant help and support during the pandemic.
Footnotes
↵* shared first authors
↵# shared last authors
In Brief Human antibodies were selected from convalescent COVID-19 patients using antibody phage display. The antibody STE90-C11 is neutralizing authentic SARS-CoV-2 virus and the crystal structure of STE90-C11 in complex with SARS-CoV-2-RBD revealed that this antibody is binding in the RBD-ACE2 interface. S1 binding of STE90-C11 and inhibition of ACE2 binding is not blocked by known RBD mutations.
References




