

Abstract
SARS-CoV-2 Spike amino acid replacements in the receptor binding domain (RBD) occur relatively frequently and some have a consequence for immune recognition. Here we report recurrent emergence and significant onward transmission of a six-nucleotide deletion in the S gene, which results in loss of two amino acids: H69 and V70. Of particular note this deletion, ΔH69/V70, often co-occurs with the receptor binding motif amino acid replacements N501Y, N439K and Y453F. One of the ΔH69/V70+ N501Y lineages, B.1.1.7, is comprised of over 1400 SARS-CoV-2 genome sequences from the UK and includes eight S gene mutations: RBD (N501Y and A570D), S1 (ΔH69/V70 and Δ144/145) and S2 (P681H, T716I, S982A and D1118H). Some of these mutations have possibly arisen as a result of the virus evolving from immune selection pressure in infected individuals and possibly only one chronic infection in the case of lineage B.1.1.7. We find the ΔH69/V70 enhances viral infectivity, indicating its effect on virus fitness is independent to the N501Y RBM change. Enhanced surveillance for the ΔH69/V70 deletion with and without RBD mutations should be considered as a priority. Such “permissive” mutations have the potential to enhance the ability of SARS-CoV-2 to generate vaccine escape variants that would have otherwise significantly reduced viral fitness.
Background
SARS-CoV-2’s Spike surface glycoprotein engagement of hACE2 is essential for virus entry and infection1, and the receptor is found in respiratory and gastrointestinal tracts2. Despite this critical interaction and the constraints it imposes, it appears the RBD, and particularly the receptor binding motif (RBM), can tolerate mutations3,4, raising the real possibility of virus escape from vaccine-induced immunity and monoclonal antibody treatments. Spike mutants exhibiting reduced susceptibility to monoclonal antibodies have been identified in in vitro screens5,6, and some of these mutations have been found in clinical isolates7. Due to the susceptibility of the human population to this virus, the acute nature of infections and limited use of vaccines to date there has been limited selection pressure placed SARS-CoV-28; as a consequence few mutations that could alter antigenicity have increased significantly in frequency.
The unprecedented scale of whole genome SARS-CoV-2 sequencing has enabled identification and epidemiological analysis of transmission and surveillance, particularly in the UK9. As of December 18th, there were 270,000 SARS-CoV-2 sequences available in the GISAID Initiative (https:gisaid.org/). However, geographic coverage is very uneven with some countries sequencing at higher rates than others. This could result in novel variants with altered biological or antigenic properties evolving and not being detected until they are already at high frequency.
Studying SARS-CoV-2 chronic infections can give insight into virus evolution that would require many chains of acute transmission to generate. This is because the majority of infections arise as a result of early transmission during pre or asymptomatic phases, and virus adaptation not observed as it is naturally cleared by the immune response. We recently documented de novo emergence of antibody escape mediated by S gene mutations in an individual treated with convalescent plasma (CP)10. Dramatic changes in the prevalence of Spike variants ΔH69/V70 (an out of frame six-nucleotide deletion) and D796H variant followed repeated use of CP, while in vitro the mutated ΔH69/V70 variant displayed reduced susceptibility to the CP and multiple other sera, at the same time retaining infectivity comparable to wild type10. Worryingly, other deletions in the N-Terminal Domain (NTD) have been reported to arise in chronic infections (ref Choi) and provide escape from NTD-specific neutralising antibodies11.
Here we analysed the available GISAID Initiative data for circulating SARS-CoV-2 Spike sequences containing ΔH69/V70. We find, while occurring independently, the Spike ΔH69/V70 often emerges after a significant RBM amino acid replacement that increases binding affinity to hACE2. We present evidence that the Spike ΔH69/V70 is a fitness enhancing change that may be stabilising other S gene mutations. Protein structure modelling indicates this mutation could also contribute to antibody evasion as suggested for other NTD deletions11.
Results
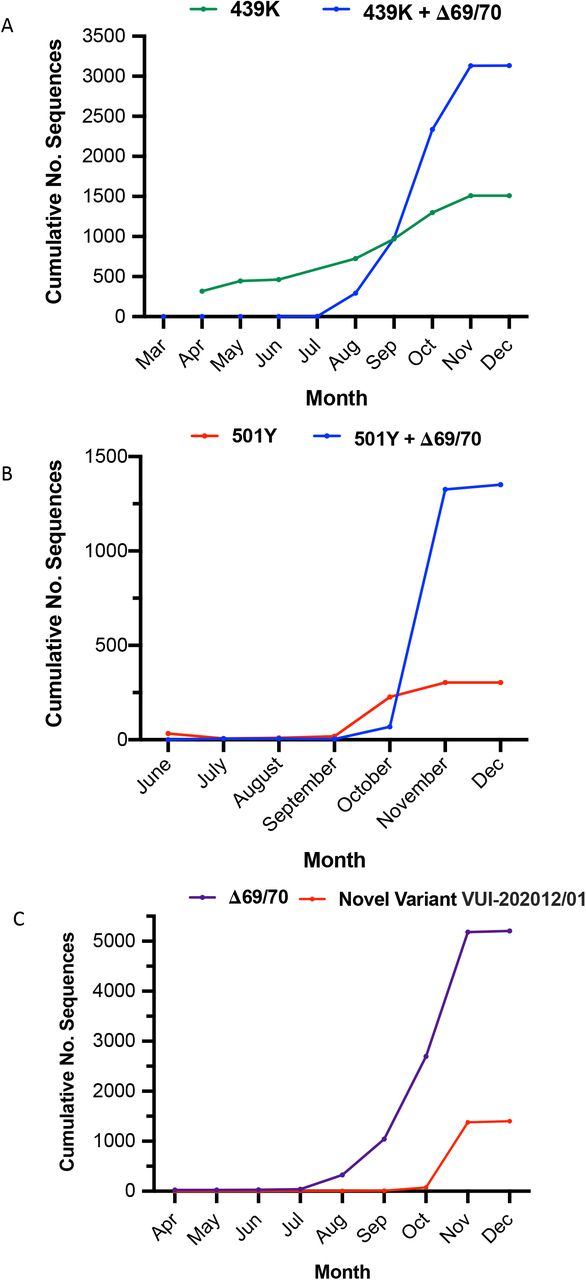
The deletion H69/V70 is present in over 6000 sequences worldwide, 2.5% of the available data (Figure 1), and largely in Europe from where most of the sequences in GISAID are derived (Figure 2A). Many of the sequences are from the UK and Denmark where sequencing rates are high compared to other countries. ΔH69/V70 occurs in variants observed in different global lineages, representing multiple independent acquisitions of this SARS-CoV-2 deletion (Figure 1). The earliest sample that includes the ΔH69/V70 was detected in Sweden in April 2020 and is an independent deletion event relative to others. The prevalence of ΔH69/V70 has since increased in other countries since August 2020 (Figure 2B, C). Further analysis of sequences revealed, firstly, that single deletions of either 69 or 70 were uncommon and secondly, some lineages of ΔH69/V70 alone were present (Figure 1 and Figure 2A), as well as ΔH69/V70 in the context of other mutations in Spike, specifically those in the RBM (Figure 2B and C).

All sequences containing four major spike mutations (Δ69/70, N439K, Y453F and 501Y) were downloaded from the GISAID database. Duplicate sequences were removed and subsampled. Tree tips are coloured as follows; brown, Δ69/70; cyan Δ69/70 + Y453F; blue, Δ69/70 + N501Y; green Δ69/70 + 439K; remainder in black. Columns showing presence or absence of deletion and amino acid variants N439K, Y453F, and N501Y matching the tip labelling.

All sequences containing the ΔH69/V70 Δetion were extracted from the GISAID database (Accessed 16th Dec 2020) and tabulated according to both reporting country of origin and date in which they were deposited in the database. The frequency of viruses carrying the ΔH69/V70 Δetion rose substantially from August to present. A. Worldwide carriage of ΔH69/V70; most sequences preferentially carry RBD mutations alongside the Δetion. B. Several distinct ΔH69/V70 lineages carrying RBD mutations 439K, 453F and 501Y have begun to emerge, predominantly in Denmark and England. C. Sequences carrying the ΔH69/V70 but in the absence of other major Spike mutations are restricted mostly to Denmark.
To estimate the structural impact of ΔH69/V70, the structure of the NTD possessing the double deletion was modelled. The ΔH69/V70 deletion was predicted to alter the conformation of a protruding loop comprising residues 69 to 76, pulling it in towards the NTD (Figure 3A). In the post-deletion structural model, the positions of the alpha carbons of residues either side of the deleted residues, Ile68 and Ser71, were each predicted to occupy positions 2.9Å from the positions of His69 and Val70 in the pre-deletion structure. Concurrently, the positions of Ser71, Gly72, Thr73, Asn74 and Gly75 are predicted to have changed by 6.5Å, 6.7Å, 6.0Å, 6.2Å and 8Å respectively, with the overall effect of drawing these residues inwards, resulting in a less dramatically protruding loop. The position of this loop in the pre-deletion structure is shown in the context of the wider NTD in Figure 3B. The locations of main RBD mutations observed with ΔH69/V70 are shown in Figure 3C and D. Residues belonging to a similarly exposed, nearby loop that form the epitope of a neutralising, NTD-binding epitope are also highlighted.

A) Prediction of conformational change in the spike N-terminal domain due to Δetion of residues His69 and Val70. The pre-Δetion structure is shown in cyan, except for residues 69 and 70, which are shown in red. The predicted post-Δetion structure is shown in green. Residues 66-77 of the pre-Δetion structure are shown in stick representation and coloured by atom (carbon in cyan, nitrogen in blue, oxygen in coral). Yellow lines connect aligned residues 66-77 of the pre- and post-Δetion structures and the distance of 6 Å between aligned alpha carbons of Thr73 in the pre- and post-Δetion conformation is labelled. B) Surface representation of spike homotrimer in open conformation (PDB: 7C2L) with each monomer shown in different shades of grey. On the monomer shown positioned to the right, the exposed loop consisting of residues 69-77 is shown in cyan and the neutralising antibody (4A8) binding NTD epitope described by Chi et al. is shown in magenta C) Surface representation of spike homotrimer in closed conformation (PDB: 6ZGE., 2020) homotrimer viewed in a ‘top-down’ view along the trimer axis. The locations of RBD mutations at residues 439, 453 and 501 and the Δetion at Tyr144 are highlighted in red and labelled on a single monomer. D) Spike in open conformation with a single erect RBD (PDB: 6ZGG) in trimer axis vertical view with the locations of Δeted residues His69 and Val70 in the N-terminal domain and RBD mutations highlighted as red spheres and labelled on the monomer with erect RBD. Residues 71-75, which form the exposed loop undergoing conformational change in A, are omitted from this structure.
We next examined the lineages where S gene mutations in the RBD were identified at high frequency, in particular co-occurring with N439K, an amino acid replacement reported to be defining variants increasing in numbers in Europe and detected across the world3 (Figure 4A, Supplementary figure 1). N439K appears to have reduced susceptibility to a small subset of monoclonals targeting the RBD, whilst retaining affinity for ACE2 in vitro3. The proportion of viruses with ΔH69/V70 only increased from August 2020 when it appeared with the second N439K lineage, B.1.1413 (Figure 4A). As of November 26th, remarkably there were twice as many cumulative sequences with the deletion as compared to the single N439K indicating it may be contributing to the success of this lineage (Figure 4A). Due to their high sampling rates the country with the highest proportion of N439K+ΔH69/V70 versus N439K alone is England. The low levels of sequencing in most countries indicate N439K’s prevalence could be relatively high3. In Scotland, where early growth of N439K was high (forming N439K lineage B.1.258 that subsequently went extinct with other lineages after the lockdown3), there is now an inverse relationship with 546 versus 177 sequences for N439K and N439K+ΔH69/ΔV70 respectively (as of November 26th). These differences therefore likely reflect differing epidemic growth characteristics and timings of the introductions the N439K variants with or without the deletion.
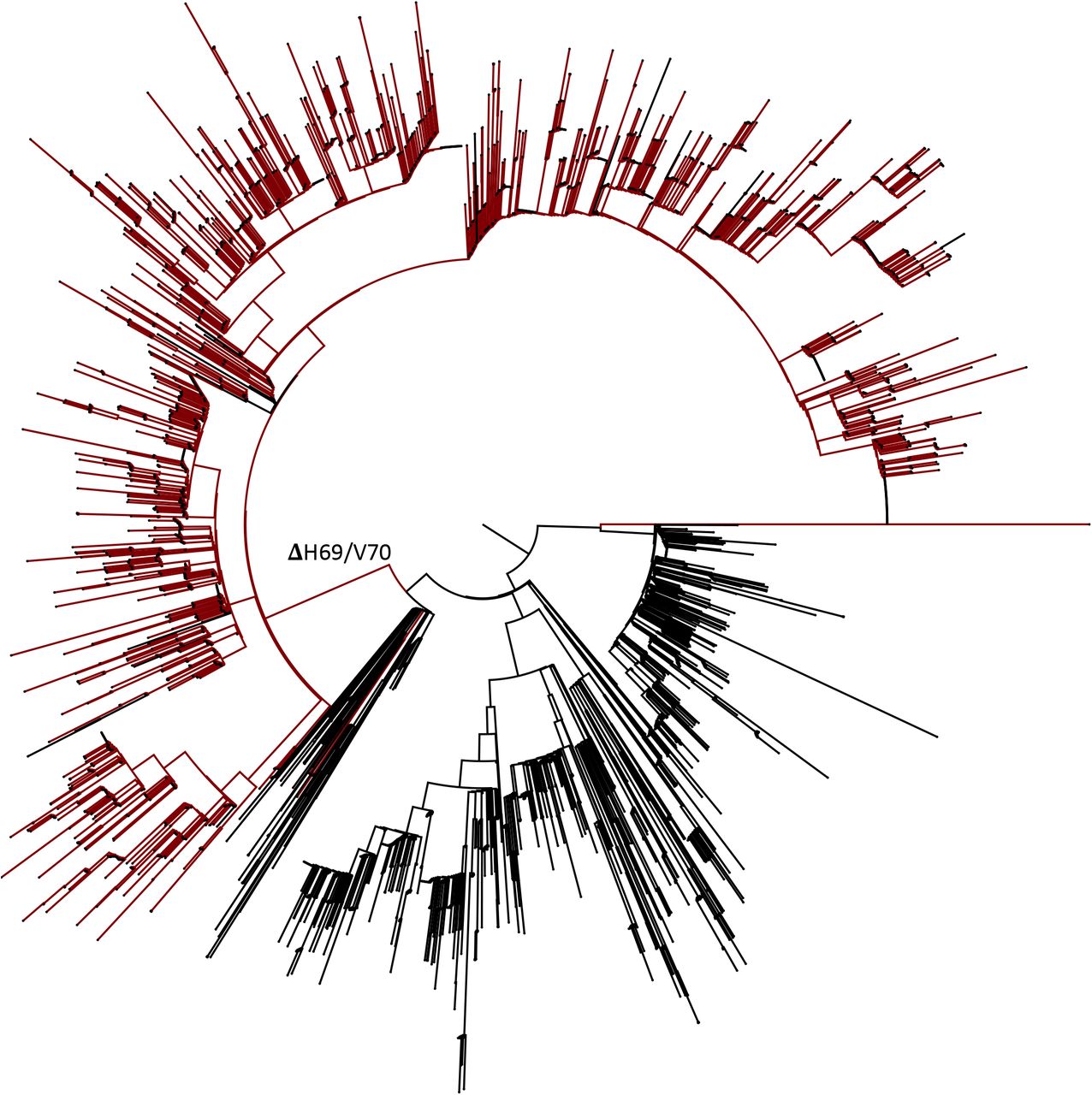
All sequences in the GISAID database containing S:439K (3820 sequences, 26th November 2020) were realigned to Wuhan-Hu-1 using MAFFT. Viruses carrying the Spike double Δetion ΔH69/V70 (red) emerged and expanded from viruses with S:439K (black).
In lineages carrying 439K and 501Y, viruses co-carrying the Δ69/70 Δetion became dominant between September-October 2020 in terms of cumulative cases. The novel variant, lineage B.1.1.7 carrying eight linked Spike mutations is increasing in frequency.
The second significant cluster with ΔH69/V70 and RBD mutants involves Y453F, another RBD mutation that increases binding affinity to ACE24 and has been found to be associated with mink-human infections12. In one SARS-CoV-2 mink-human sub-lineage, termed ‘Cluster 5’, Y453F and ΔH69/V70 occurred with F486L, N501T and M1229I and was shown to have reduced susceptibility to sera from recovered COVID-19 patients (https://files.ssi.dk/Mink-cluster-5-short-report_AFO2). The ΔH69/V70 was first detected in the Y453F background on August 24th and thus far appears limited to Danish sequences (supplementary figure 3).
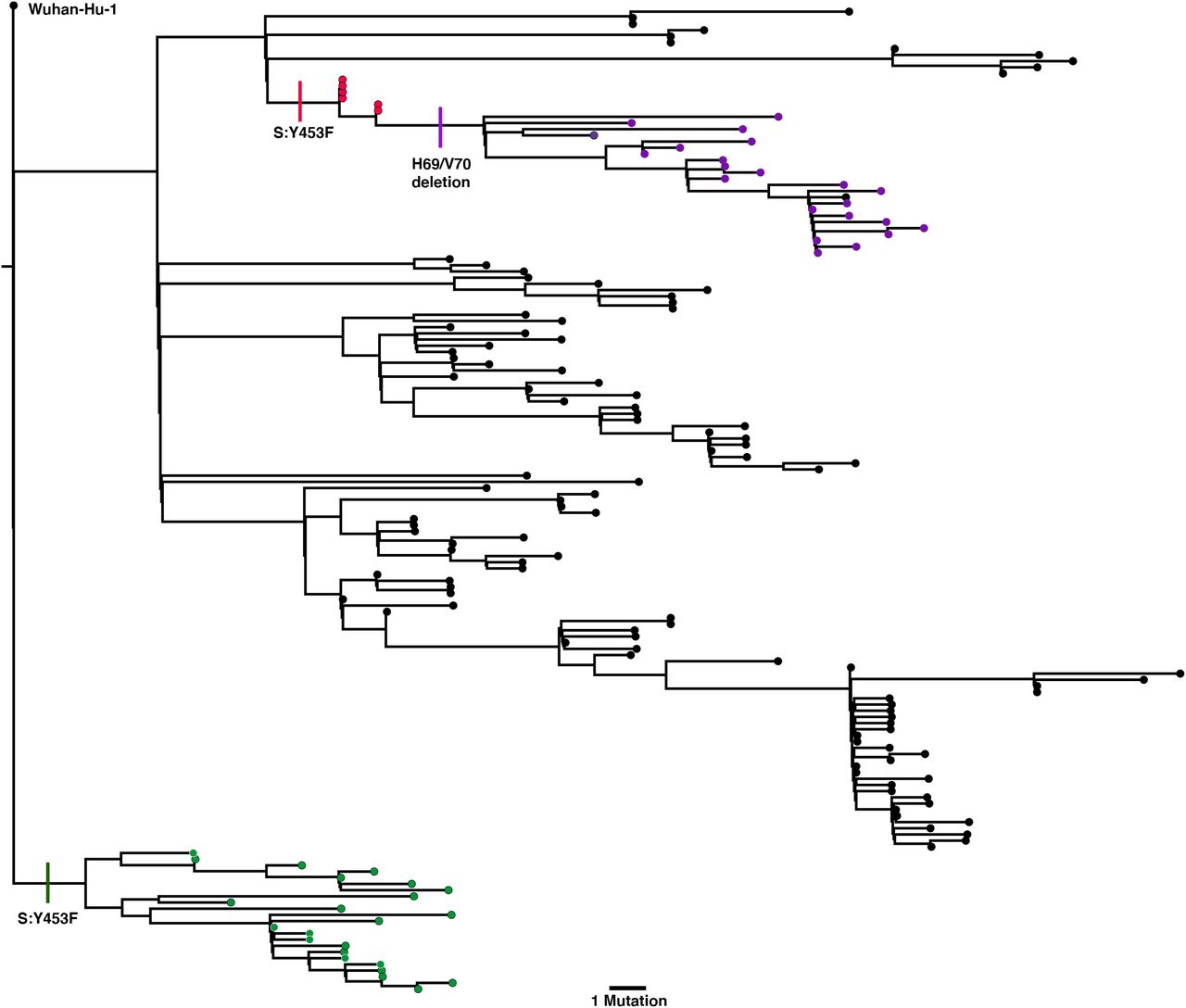
A third lineage containing the same out of frame deletion ΔH69/V70 has arisen with another RBD mutation N501Y (Figure 4B, Figure 5, supplementary figure 2). Based on its location it might be expected to escape antibodies similar to COV2-24995. In addition, when SARS-CoV-2 was passaged in mice for adaptation purposes for testing vaccine efficacy, N501Y emerged and increased pathogenicity13. Early sequences with N501Y alone were isolated both in Brazil and USA in April 2020. N501Y + ΔH69/V70 sequences appear to have been detected first in the UK in September 2020, with the crude cumulative number of N501Y + ΔH69/V70 mutated sequences now exceeding the single mutant N501Y lineage (Figure 4B). Of particular concern is a lineage (B.1.1.7) associated with relatively high numbers of infections and currently around 1400 sequences (Figure 4C, Figure 5) with six S mutations across the RBD (N501Y, A570D) and S2 (P681H, T716I, S982A and D1118H) as well as the ΔH69/V70 and Δ144 in England14 (Figure 6). This lineage has a very long branch (Figure 5 and supplementary figure 3), suggestive of possible within host evolution.

All sequences in the GISAID database containing S:501Y were downloaded and realigned to Wuhan-Hu-1 using MAFFT. Sequences were broadly split into four major clades; sequences carrying the Spike double Δetion ΔH69/V70 (red) formed an entirely separate clade from non-carriers. Sequences carrying 501Y but an absence of ΔH69/V70 formed a second lineage and appeared to expand only in Wales (green). Another major clade (blue) was limited entirely to Australia and finally a fourth clade (black) was limited to several African countries and Brazil.

All 753 sequences in the GISAID database (accessed 14th December 2020) were downloaded and realigned to Wuhan-Hu-1 using MAFFT. Two distinct lineages carrying the mink-associated Spike Y453F mutations can be seen in Danish (red) sequences, with a separate lineage isolated only in Netherlands (green). After acquiring the Y453F mutation, Danish mink also appeared to acquire the Spike Δetion ΔH69/V70 (purple).
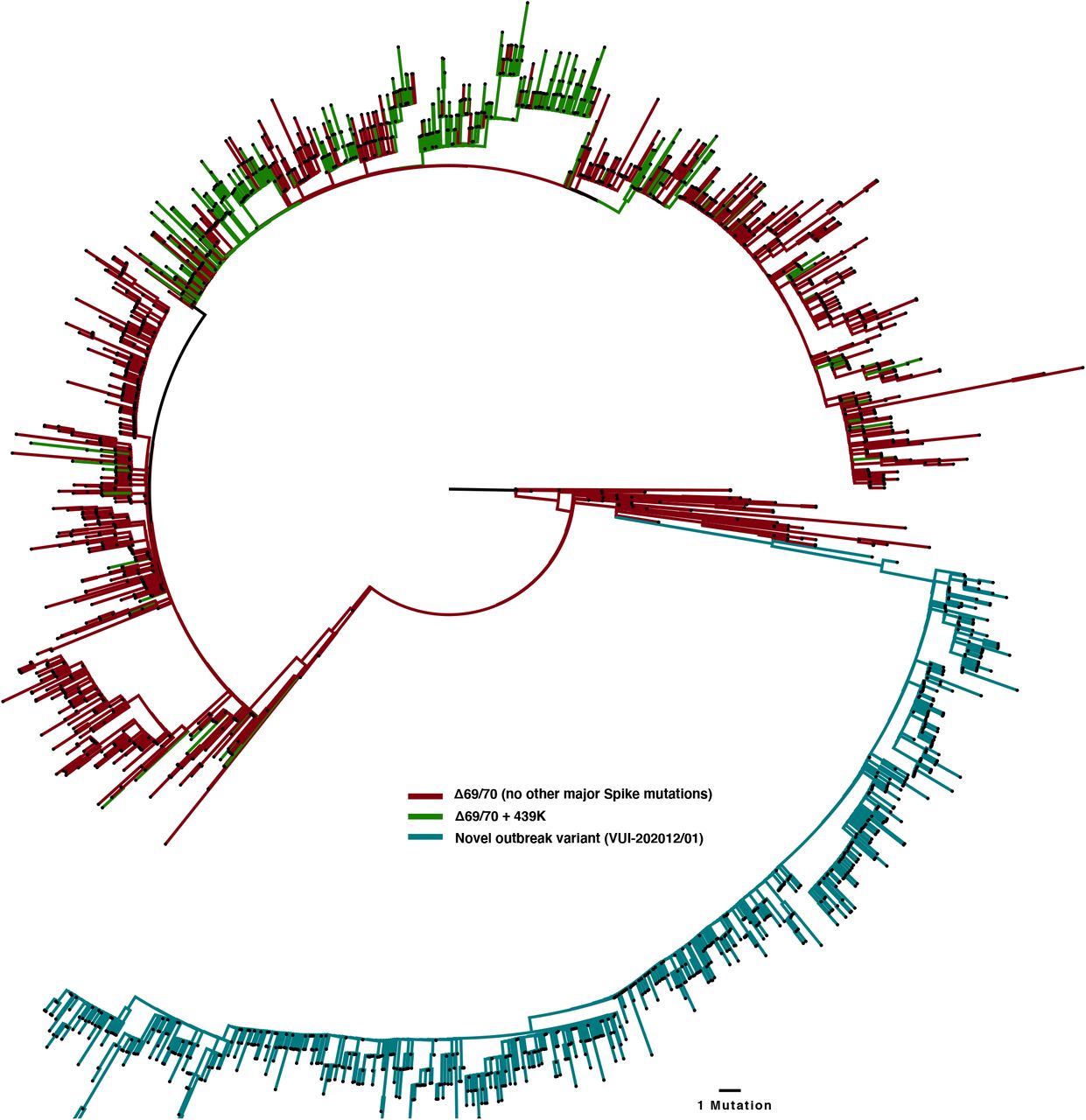
All sequences carrying the double-Δetion were downloaded from the GISAID database (5000 sequences, 16th Dec) and aligned to the Wuhan-Hu-1 reference strain using MAFFT. All duplicate sequences were removed to create a representative sub-sample. The inferred phylogeny clearly shows several distinct lineages carrying ΔH69/V70 in the absence of other major Spike mutations (red) and alongside RDB mutation 439K (green). N501Y lineages cluster within the outbreak variant lineage. Y453F sequnces are lost after de-depulication. The novel outbreak lineage currently circulating in England carries eight co-occurring Spike mutations (cyan) in the RBD (N501Y, A570D), S1 (ΔH69/V70 and Δ144/145) and S2 (P681H, T716I, S982A and D1118H).
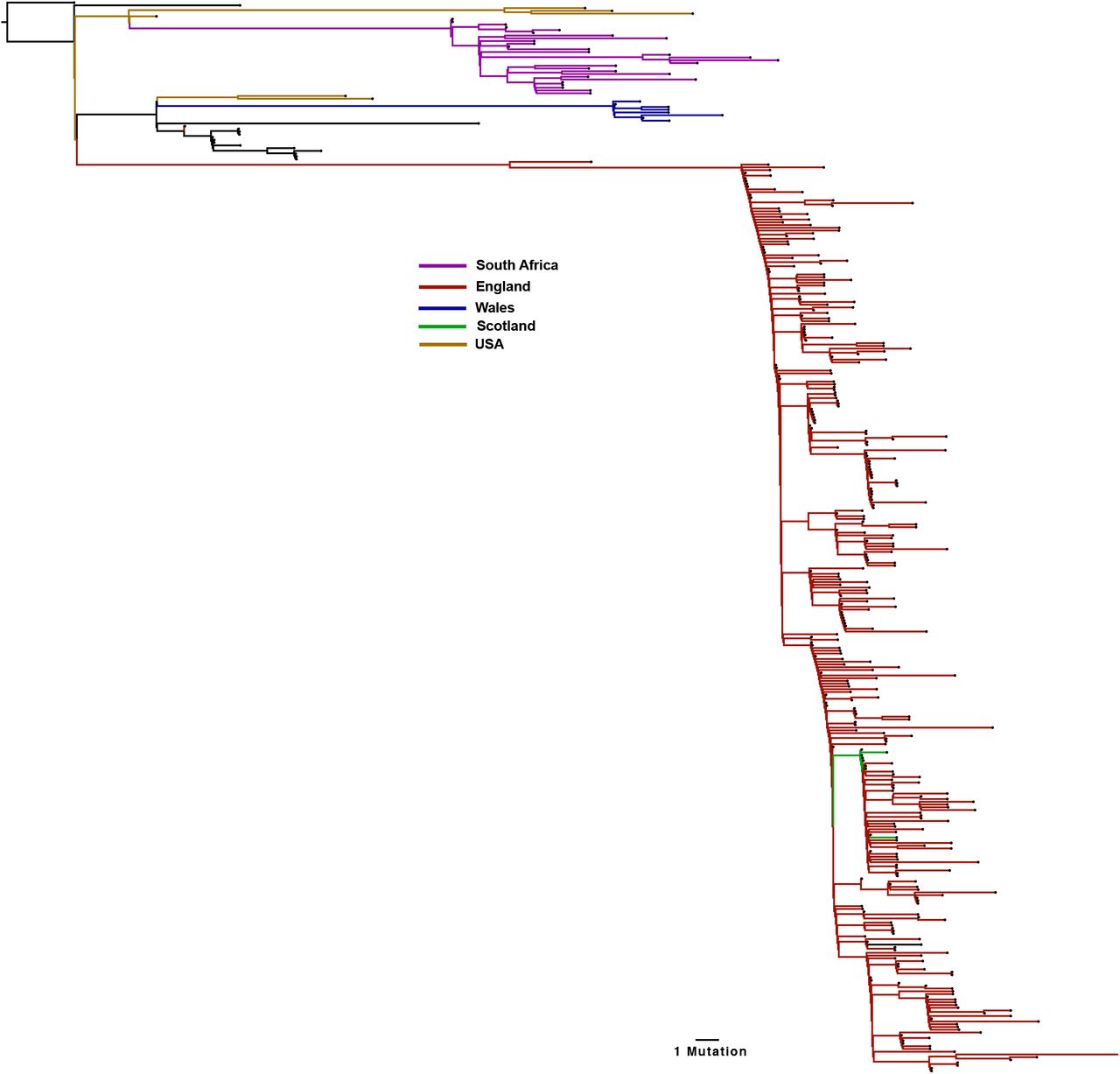
A global maximum likelihood phylogeny of all SARS-CoV-2 sequences bearing the 501Y mutations were downloaded from the GISAID database (2000 sequences, accessed 16th December 2020). All duplicate sequences were removed to create a representative sub-sample of sequences. Distinct sub-lineages of the Spike 501Y variants are currently circulating in the UK (predominantly England, red), with significant expansion also occurring in South Africa (pink).

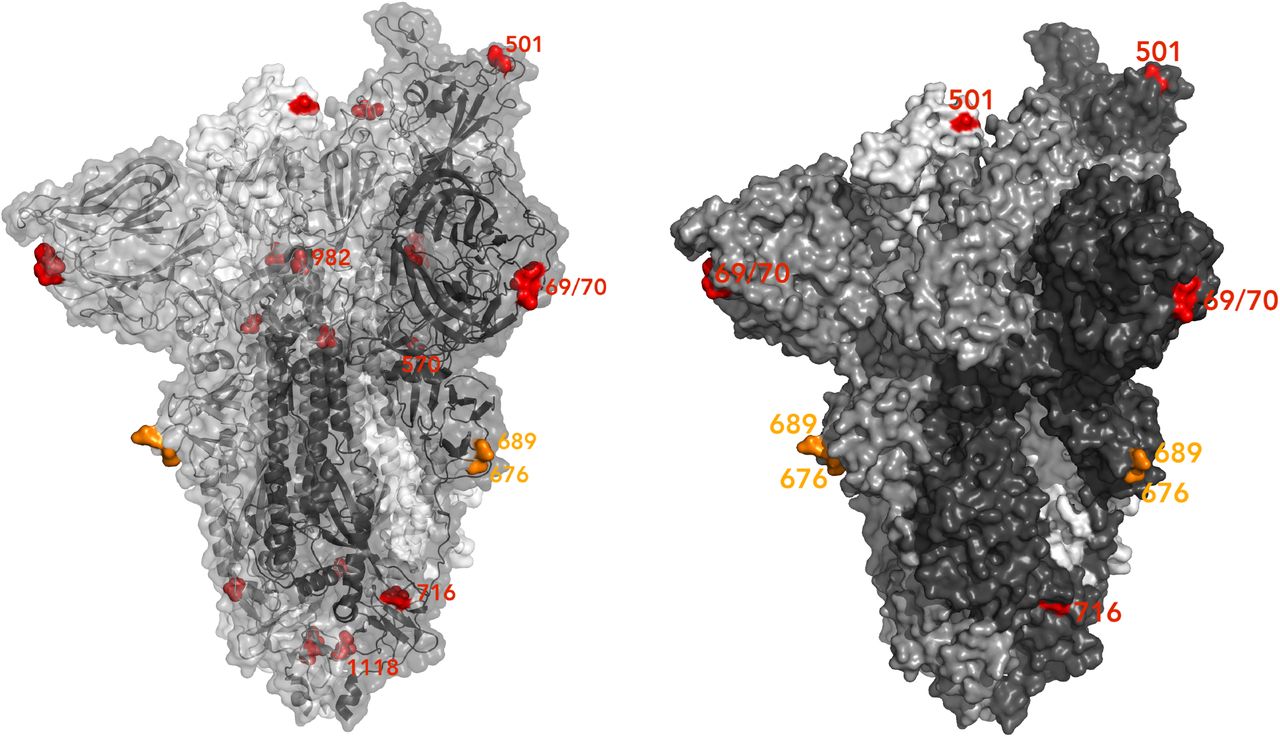
Spike homotrimer in open conformation with one upright RBD (PDB: 6ZGE, Wrobel et al., 2020) with different monomers shown in shades of grey. To the left, surface representation overlaid with ribbon representation and to the right, opaque surface representation accentuating the locations of surface-exposed residues. The Δeted residues 69 and 70 and the residues involved in amino acid substitutions (501, 570, 716, 982 and 1118) are coloured red. The location of an exposed loop including residue 681 is absent from the structure, though the residues either side of the unmoΔled residues, 676 and 689, are coloured orange. On the left structure, highlighted residues are labelled on the monomer with an upright RBD; on the right structure, all visible highlighted residues are labelled.
The B.1.1.7 lineage (termed VUI 202012/01 by Public Health England) has some notable features. Firstly the Δ144 mutation that could lead to loss of binding of the S1 binding antibody 4A811. Secondly the P681H mutation lies within the furin cleavage site. Furin cleavage is a property of some more distantly related coronaviruses, and in particular not found in SARS-CoV-115. When SARS-CoV-2 is passaged in vitro it results in mutations in the furin cleavage site, suggesting the cleavage is dispensable for in vitro infection16. The significance of furin site mutations may be related to potential escape from the innate immune antiviral IFITM proteins by allowing infection independent endosomes17. The significance of the multiple S2 mutations is unclear at present, though D614G, also in S2 was found to lead to a more open RBD orientation to explain its higher infectivity18. T716I and D1118H occur at residues located close to the base of the ectodomain (Figure 6) that are partially exposed and buried, respectively. The residue 982 is buried and located centrally, in between the NTDs, at the top of a short helix (approximately residues 976-982) that is completely shielded by the RBD when spike is in the closed form, though becomes slightly more exposed in the open conformation. Residue 681 is part of a spike region that is unmodelled in multiple published structures [Chi et al. 2020, Wrobel et al. 2020). The surface-exposed locations of modelled residues 676 and 689 (orange in Figure 6) suggest the unmodelled residues 677-688 form a prominently-exposed loop that may be assumed to show significant structural flexibility given the difficulties experienced in attaining an accurate structural model of this region.
Given the association between ΔH69/V70 and other S gene mutations, we hypothesised similar to our chronic infection10, that this deletion is enhancing virus infectivity. In the absence of virus isolates we used a lentiviral pseudotyping approach to test the impact of ΔH69/V70 on virus Spike protein mediated infection. A D614G bearing Spike protein expressing DNA plasmid was co-transfected in HEK 293T producer cell lines along with plasmids encoding lentiviral capsid and genome for luciferase. A mutant Spike bearing ΔH69/V70 was also expressed in and infectious titres measured in supernatants from producer cells (Figure 7). There was a significant difference in infectivity observed in the ΔH69/V70 virus compared to wild type across multiple virus dilutions. When we adjusted infectious titres to account for the amount of virus produced by wild type versus mutant in the supernatants, a robust two fold increase of ΔH69/V70 over wild type was observed (Figure 7).
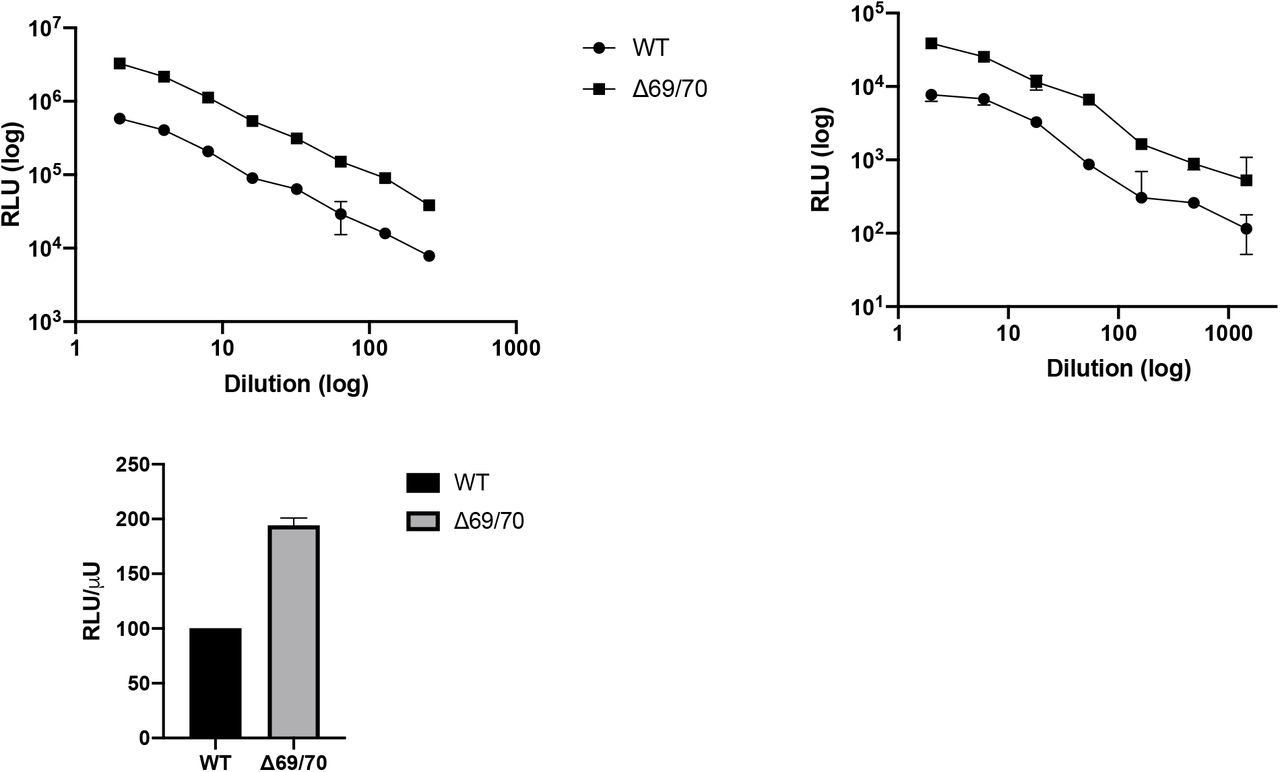
A and B. Single round Infection of target cells A and B by luciferase expressing lentivirus pseudotyped with SARS-CoV-2 Spike protein (WT versus mutant) on A. 293T cells co-transfected with ACE2 and TMPRSS2 plasmids and B. Hela cells stably expressing ACE2. C. Data showing Infectivity normalized for virus input using reverse transcriptase activity in virus supernatants. RLU – relative light units; U – unit of reverse transcriptase activity. Data are representative of 2 independent experiments.
Discussion
We have presented data demonstrating multiple, independent, and circulating lineages of SARS-CoV-2 variants bearing a Spike ΔH69/V70. This deletion spanning six nucleotides, is mostly due to an out of frame deletion of six nucleotides, has frequently followed receptor binding amino acid replacements (N501Y, N439K and Y453F that have been shown to increase binding affinity to hACE2 and reduce binding with monoclonal antibodies) and its prevalence is rising in parts of Europe.
A recent analysis highlighted the potential for enhanced transmissibility of viruses with deletions in the NTD, including ΔH69/V7011. Here we show that the ΔH69/V70 deletion increases Spike mediated infectivity by two-fold over a single round of infection. Over the millions of replication rounds per day in a SARS-CoV-2 infection this is likely to be significant. The potential for SARS-CoV-2 mutations to rapidly emerge and fix is exemplified by D614G, an amino acid replacement in S2 that alters linkages between S1 and S2 subunits on adjacent protomers as well as RBD orientation, infectivity, and transmission18–20. The example of D614G also demonstrates that mechanisms directly impacting important biological processes can be indirect. Similarly, a number of possible mechanistic explanations may underlie ΔH69/V70. For example, the fact that it sits on an exposed surface and is estimated to alter the conformation of a particularly exposed loop might be suggestive of immune interactions and escape, although allosteric interactions could alternatively lead to higher infectivity.
The finding of a sub-lineage of over 1400 sequences bearing seven S gene mutations across the RBD (N501Y, A570D), S1 (ΔH69/V70 and Δ144) and S2 (P681H, T716I, S982A and D1118H) in UK requires careful monitoring. The detection of a high number of novel mutations suggests this lineage has either been introduced from a geographic region with very poor sampling or viral evolution may have occurred in a single individual in the context of a chronic infection10. This variant bears some concerning features; firstly the ΔH69/V70 deletion which we show to increase infectivity by two fold. Secondly the Δ144 which may affect binding by antibodies related to 4A811. Thirdly, the N501Y mutation that may have higher binding affinity for ACE2 and a second RBD mutation A570D that could alter Spike RBD structure. Finally, a mutation at the furin cleavage site could represent further adaptative change.
Given the emergence of multiple clusters of variants carrying RBD mutations and the ΔH69/V70 deletion, limitation of transmission takes on a renewed urgency. Continued emphasis on testing/tracing, social distancing and mask wearing are essential, with investment in other novel methods to limit transmission21. In concert, comprehensive vaccination efforts in the UK and globally should be accelerated in order to further limit transmission and acquisition of further mutations. If geographically limited then focussed vaccination may be warranted. Research is vitally needed into whether lateral flow devices for antigen and antibody detection can detect emerging strains and the immune responses to them, particularly given reports that S signal in PCR based tests are frequently negative in the new variant. Finally, detection of the deletion and other key mutations by rapid diagnostics should be a research priority as such tests could be used as a proxy for antibody escape mutations to inform surveillance at global scale.
Conflicts of interest
RKG has received consulting fees from UMOVIS lab, Gilead Sciences and ViiV Healthcare, and a research grant from InvisiSmart Technologies.
Methods
Phylogenetic Analysis
All available full-genome SARS-CoV-2 sequences were downloaded from the GISAID database (http://gisaid.org/)22 on 26th November. Duplicate and low-quality sequences (>5% N regions) were removed, leaving a dataset of 194,265 sequences with a length of >29,000bp. All sequences were realigned to the SARS-CoV-2 reference strain MN908947.3, using MAFFT v7.473 with automatic flavour selection and the --keeplength --addfragments options23. Major SARS-CoV-2 clade memberships were assigned to all sequences using the Nextclade server v0.9 (https://clades.nextstrain.org/).
Maximum likelihood phylogenetic trees were produced using the above curated dataset using IQ-TREE v2.1.224. Evolutionary model selection for trees were inferred using ModelFinder25 and trees were estimated using the GTR+F+I model with 1000 ultrafast bootstrap replicates26. All trees were visualised with Figtree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/), rooted on the SARS-CoV-2 reference sequence and nodes arranged in descending order. Nodes with bootstraps values of <50 were collapsed using an in-house script.
Pseudotype virus preparation
Viral vectors were prepared by transfection of 293T cells by using Fugene HD transfection reagent (Promega). 293T cells were transfected with a mixture of 11ul of Fugene HD, 1μg of pCDNAΔ19Spike-HA, 1ug of p8.91 HIV-1 gag-pol expression vector27,28, and 1.5μg of pCSFLW (expressing the firefly luciferase reporter gene with the HIV-1 packaging signal). Viral supernatant was collected at 48 and 72h after transfection, filtered through 0.45um filter and stored at −80°C as previously described29. Infectivity was measured by luciferase detection in target TZMBL transduced to express TMPRSS2 and ACE2.
Normalisation of virus titre by SG-PERT to measure RT activity in lentivirus preparation
Supernatant was subjected to SG-PERT as previously described30.
Structural modelling
The structure of the post-deletion NTD (residues 14-306) was modelled using I-TASSER31, a method involving detection of templates from the protein data bank, fragment structure assembly using replica-exchange Monte Carlo simulation and atomic-level refinement of structure using a fragment-guided molecular dynamics simulation. The structural model generated was aligned with the spike structure possessing the pre-deletion conformation of the 69-77 loop(PDB 7C2L32) using PyMOL (Schrödinger). Figures prepared with PyMOL using PDBs 7C2L, 6ZGE28 and 6ZGG33.
Acknowledgements
RKG is supported by a Wellcome Trust Senior Fellowship in Clinical Science (WT108082AIA). DLR is funded by the MRC (MC UU 1201412). WH is funded by the MRC (MR/R024758/1). We thank Dr James Voss for the kind gift of HeLa cells stably expressing ACE2.
Footnotes
Author affiliation has been corrected





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}