

Abstract
AKT-phosphorylated IWS1 promotes Histone H3K36 trimethylation and alternative RNA splicing of target genes, including the U2AF65 splicing factor-encoding U2AF2. The predominant U2AF2 transcript, upon IWS1 phosphorylation block, lacks the RS-domain-encoding exon 2, and encodes a protein which fails to bind Prp19. Here we show that although both U2AF65 isoforms bind intronless mRNAs containing cytoplasmic accumulation region elements (CAR-E), only the RS domain-containing U2AF65 recruits Prp19 and promotes their nuclear export. The loading of U2AF65 to CAR-Elements was RS domain-independent, but RNA PolII-dependent. Virus- or poly(I:C)-induced type I IFNs are encoded by genes targeted by the pathway. IWS1 phosphorylation-deficient cells therefore, express reduced levels of IFNα1/IFNβ1 proteins, and exhibit enhanced sensitivity to infection by multiple cytolytic viruses. Enhanced sensitivity of IWS1-deficient cells to Vesicular Stomatitis Virus and Reovirus resulted in enhanced apoptotic cell death via caspase activation. Inhibition of this pathway may therefore sensitize cancer cells to oncolytic viruses.
Introduction
AKT regulates alternative RNA splicing (Sanidas et al., 20141, Zhou et al., 20122). Our earlier studies addressing this critical AKT function, identified a pathway that plays a major role in its regulation. The first step in this pathway is the phosphorylation of IWS1 at Ser720/Thr721 by AKT3 and AKT1, but not by AKT2. Following phosphorylation, IWS1 recruits the Histone methyltransferase SETD2 to an SPT6/IWS1/ALY-REF complex, which assembles on the Ser2-phosphorylated C-terminal domain (CTD) of RNA Pol II. During transcription, SETD2 trimethylates Histone H3 on K36 in the body of transcribed target genes, and this is recognized by several H3K36me3 readers, which initiate the process of alternative RNA splicing (Sanidas et al., 20141, Laliotis et al., 20213). One of the genes whose alternative RNA splicing is regulated by this pathway is U2AF2, which encodes the core RNA splicing factor, U2AF65. Phosphorylation of IWS1 by AKT3 and AKT1 promotes the inclusion of this exon 2 in the mature U2AF2 mRNA transcript. This exon encodes part of the RS domain of U2AF65, which is required for U2AF65 binding to Prp19, a member of a seven-member protein complex (PRP19C), with ubiquitin ligase activity, which is also involved in RNA splicing. Importantly, this pathway is cell cycle regulated and some of its target genes are regulators of the cell cycle. As a result, it promotes cell proliferation and tumor growth (Laliotis et al., 20213).
Earlier studies had shown that U2AF65 and Prp19 also regulate the nuclear export of the mRNAs of a set of intronless genes (Lei et al., 20134). The common feature of these mRNAs is that they all possess one or more 10 nucleotide long motifs, which are involved in their nuclear export and are known as Cytoplasmic Accumulation Region Elements (CAR-E) (de Padilla et al., 20145). The functional activity of these elements depends on the binding of the Transcription-Export (TREX) complex, the U2 Associated-Factor 2 (U2AF2)-encoded splicing factor U2AF65, and the pre-mRNA Processing Factor 19 complex (Prp19C) (Lei et al., 20134). Given that this process depends on U2AF65 and Prp19, whose interaction depends on the activity of the AKT/IWS1 axis, we hypothesized that the nuclear export of the mRNAs of these intronless genes will depend on IWS1 and its phosphorylation by AKT.
Prominent among the CAR-E-positive intronless genes are the genes encoding type I Interferons (IFNs) (de Padilla et al., 20145). We therefore reasoned that the expression of type I IFNs may also be regulated by this AKT-dependent pathway. Type I IFNs are members of a large family of cytokines, known for their role in the regulation of innate and adaptive immunity and the antiviral response. They include IFNA, with 13 members and IFNB, with only one member (Frisch et al., 20206) and they engage the heterodimeric receptor IFNAR1/IFNAR2, which is widely expressed (Schreiber et al., 20177). The genes encoding type I IFNs are induced by signals initiated through the activation of Pattern Recognition Receptors (PRRs), the sensors of innate immunity (Acosta et al., 20208). These receptors recognize molecules presented by pathogens (pathogen-associated molecular patterns, PAMPs), such as bacterial lipopolysaccharides, flagellin, bacterial lipoproteins, double-stranded RNA (dsRNA) and cytosolic DNA (Amarante-Mendes et al., 20189). PRR signals regulate multiple signaling pathways, including the IκB Kinase (IKK) pathway, which phosphorylates and activates the transcription factors IRF3 and NF-κB. These factors transactivate the IFNB1 gene (Ablasser et al., 202010) and induce the expression of IFNβ. The latter acts in an autocrine or paracrine manner to activate JAK1 and TYK2, which phosphorylate STAT1 and STAT2 and promote the formation of the trimeric complex STAT1-STAT2-IRF9, known as the Interferon Stimulated Gene Factor 3 (ISGF3). This complex binds palindromic DNA sequences, known as IFN-stimulated Response Elements (ISREs) in the promoters of interferon-stimulated genes (ISGs) and promotes their expression (Aleynick et al., 201911). One of these genes is the gene encoding the transcription factor IRF7, which along with the transcription factors IRF3 and IRF5, is required for the induction of IFNA and the full type I IFN response (Lu et al., 200012, Conzelmann et al., 200513, Lazear et al., 201314).
The regulation of type I IFN signaling has important implications in the regulation of innate and adaptive immunity, along with the control of viral infection and replication. The pathways regulating type I IFN signaling are receiving added attention in recent years, due to the emergence of cytotoxic oncolytic viruses (OVs) as a new class of anti-cancer therapeutics (Park et al., 202015). The first clinically approved OV, Talimogene laherparepvec (TVEC), is a genetically modified type I herpes simplex virus (HSV) that expresses granulocyte-macrophage colony-stimulating factor (GM-CSF) (Rehman et al., 201616) and is now incorporated in the treatment protocols of a select group of patients with melanoma. However, most OVs exhibit only weak antitumor activity, especially when used as monotherapy (Martinez-Quintanilla et al., 201917). This could potentially change by targeting type I IFN signaling and altering the sensitivity of the tumor cells to infection by such viruses.
Earlier studies had shown that the type I IFN response is regulated by multiple signaling pathways. Among them, the AKT pathway regulates the IFN response at multiple levels. By activating the mechanistic target of rapamycin (mTOR), AKT promotes the translation of Interferon-stimulated genes (ISGs) (Kroczynska et al., 201418). Subsequent studies revealed that AKT1 activates β-catenin by phosphorylation at Ser552 and that the activated β-catenin promotes the transcriptional activation of IFNB (Gantneret al., 201219). In addition, we have also shown that AKT1 selectively phosphorylates EMSY at Ser209, relieving the EMSY-mediated repression of IFN-stimulated genes (ISGs) (Ezell et al., 201220).
Data presented in this report fully support the hypothesis that the AKT/IWS1/U2AF2 axis regulates the nuclear export and expression of type I IFN genes, and by doing so, they identify yet another pathway by which AKT regulates IFN gene expression. They also show that this pathway plays a critical role in the regulation of type I IFN gene expression, because inhibiting this pathway essentially blocks the IFN response and dramatically increases the sensitivity of the cells to infection by cytolytic viruses and virus-induced cell death.
Results
IWS1 phosphorylation regulates the nucleocytoplasmic transport of mRNAs transcribed from a set of intronless genes, via a process that depends on the alternative splicing of U2AF2
Our earlier studies had shown that the knockdown of IWS1 and its replacement by the non-phosphorylatable mutant IWS1 S720A/T721A alter the RNA splicing pattern of U2AF2 giving rise to a mature mRNA that lacks exon 2. The U2AF2 splicing variant lacking exon 2 encodes a variant of the U2AF65 core splicing factor with a partial deletion of the RS domain (Laliotis et al., 20213). Importantly, whereas the RS domain-containing U2AF65 binds Prp19, a component of a seven-member complex with ubiquitin ligase activity, which is also involved in the regulation of RNA splicing, the RS domain-deficient U2AF65 does not (R. Hogg et al., 201021, Chanarat S. et al., 201322). More important, U2AF65 and Prp19, along with the TREX complex, bind RNA motifs, designated as cytoplasmic accumulation region elements (CAR-E), which are present in, and promote the nucleocytoplasmic transport of most mRNAs transcribed from naturally intronless genes (Lei et al., 20134).
mRNAs whose nucleocytoplasmic transport is regulated by this mechanism include those transcribed from IFNA1 (encoding IFNα1), IFNB1 (encoding IFNβ1), HSPB3 (encoding Hsp27) and JUN (Lei et al., 20134). We therefore asked whether U2AF2 alternative RNA splicing downstream of IWS1 phosphorylation, regulates the nuclear export of the mRNAs transcribed from these genes. To address this question, we first engineered shControl, shIWS1, shIWS1/wild type IWS1-rescue (shIWS1/WT-R), shIWS1/IWS1-S720A/T721A-rescue (shIWS1/MT-R) and shIWS1/U2AF65α-rescue or shIWS1/U2AF65β-rescue NCI-H522 and NCI-H1299 lung adenocarcinoma cells. Notably, the IWS1 rescue clones were engineered to be shIWS1 resistant (Sanidas et al., 20141, Laliotis et al., 20213). U2AF65α and U2AF65β are encoded by the exon 2-containing, and the exon 2-deficient U2AF2 splice forms, respectively. To validate these cell lines, we analyzed them for the expression of IWS1, phospho-IWS1 and U2AF65 by western blotting, and for the alternative RNA splicing of U2AF2 by RT-PCR (Fig. 1a).

a. Western blots of lysates of NCI-H522 and NCI-H1299 cells, transduced with the indicated constructs and probed with anti-IWS1, anti-phosphor-IWS1, anti-U2AF65 and anti-β-actin antibodies. RT-PCR of U2AF2, using oligonucleotide primers that map in exons 1 and 3 (fifth row).
b. IWS1 phosphorylation regulates the nuclear export of intronless mRNAs. Cells were fractionated into cytoplasmic and nuclear fractions. The abundance of the RNAs of IFNA1, IFNB1, JUN, HSPB3 and GAPDH in each fraction was determined using qRT–PCR and it was normalized relative to the 18S rRNA. Type I IFNs were induced by SeV-GFP infection (MOI 0.5) and cells were harvested at 24 hours from the start of the exposure to the virus. Bars show the mean normalized Cytoplasmic/Nuclear RNA ratio ±SD. To validate the fractionation, we measured the Cytoplasmic/Nuclear ratio of the GAPDH RNA (see supplementary Table 4).
c. Motif analysis of intronless genes identified sets of CAR-E-positive and CAR-E-negative genes. Using q-RT-PCR, we measured the abundance of the RNAs of 25 CAR-E-positive and 24 CAR-E-negative genes in the cytoplasmic and nuclear RNA fractions described in b. The heatmaps were generated from the z-score of Cytosolic/Nuclear RNA ratios.
d. Total RNA was harvested from the NCI-H522 and NCI-H1299 cells in a, and b. The expression of the indicated mRNAs was measured by qRT-PCR, and was normalized to 18S rRNA. Heatmaps were generated from the z-scores of the abundance of the indicated RNAs. Type I IFNs were induced by infection with SeV-GFP.
e. (Left panel) Western blots of lysates of the NCI-H522 and NCI-H1299 cells in a and b were probed with the indicated antibodies. Type I IFNs were induced by infection with SeV-GFP or by treatment with Poly (I:C) {5μg/mL for 6h (NCI-H522) or 12h (NCI-H1299)}. (Right panel) Quantification of the relative abundance of the indicated proteins in the experiment on the left. Bars show relative expression normalized to loading control±SD. All experiments in this figure were done in triplicate, on three biological replicates. n.s : non-significant *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (one-side unpaired t-test).
Following validation, type I IFNs were induced in these cells via infection with a GFP-expressing Sendai virus, (SeV-GFP) (Yount et al., 200623, Bedsaul et al., 201624). Twenty-four hours later, and before the emergence of virus-induced cytotoxicity, we fractionated the cells into nuclear and cytoplasmic fractions and we examined the mRNA levels of IFNA1, IFNB1, in the fractions by quantitative RT-PCR. The expression of HSPB3 and JUN was measured, also with qRT-PCR, in the nuclear and cytoplasmic fractions of similarly fractionated uninfected cells. The total mRNA levels of these genes were also measured in unfractionated lysates of the same cells. The results showed that whereas the mRNAs of all four genes are primarily cytoplasmic in the shControl and WT-R cells, they are primarily nuclear in the shIWS1 and MT-R cells. More important, whereas U2AF65α rescued the nuclear retention of these mRNAs in shIWS1 cells, U2AF65β did not (Fig. 1b, left panel).
The preceding data suggested that IWS1 phosphorylation promotes the nuclear export of RNAs transcribed from CAR-Element-positive intronless genes and raised the question whether the RNAs of CAR-Element-negative intronless genes are also targets of this pathway. To address this question, we first used FIMO motif analysis (Grant et al., 201126) to show that among the 1724 intronless genes expressed in epithelial cells (Louhichi et al., 201127) 928 (53.38%) are CAR-E-positive and 796 (46.1%) are CAR-E-negative (p<0.05, q<0.1) (Supplementary Figure 1a, Supplementary Data 1). Following this, we examined the nucleocytoplasmic RNA ratio of 25 CAR-Element-positive and 24 CAR-Element-negative genes in shControl, shIWS1, shIWS1/WT-R and shIWS1/MT-R, as well as in shIWS1/U2AF65α and shIWS1/U2AF65β NCI-H522 and NCI-H1299 cells. The results showed that the export of the RNAs of all these genes was impaired in shIWS1 cells and that the RNA export defect was rescued by wild type IWS1. However, the phosphorylation site mutant of IWS1 rescued the export of only the RNAs of the CAR-Element-negative genes. The shIWS1-induced RNA export defect of only the CAR-Element positive RNAs was also rescued by U2AF65α, but not U2AF65β. These data combined, show that the nuclear RNA export of intronless genes described in the report, is specific for the RNAs of genes that are CAR-Element-positive (Fig. 1c).
Although the cytoplasmic mRNA levels of IFNA1, IFNB1, HSPB3 and JUN were decreased in shIWS1 cells, their total RNA levels were increased. Moreover, their increased expression was rescued by wild type IWS1, but not by the phosphorylation site IWS1 mutant (Fig. 1d). We conclude that IWS1 normally inhibits the expression of these genes at the RNA level via a pathway that depends on its phosphorylation by AKT. Importantly, these results were in agreement with RNA-Seq data derived from 516 lung adenocarcinoma patients in The Cancer Genome Atlas (TCGA) LUAD database, which revealed negative correlations between the expression of IWS1 and JUN, HSBP3 and IFNA1 (Supplementary Figure 1b). Although the rescue of the shIWS1-induced phenotype by wild type IWS1 and the IWS1 phosphorylation site mutant gave the expected results, the outcomes of its rescue by U2AF65α and U2AF65β were unexpected. U2AF65α rescued the upregulation of type I IFNs, as expected, while U2AF65β did not. However, both failed to rescue the upregulation of JUN and HSBP3. We interpret these data to suggest that the upregulation of the mRNAs of IFN genes in shIWS1 cells may be due to different mechanisms than the upregulation of the mRNAs of other intronless genes. We hypothesize that the upregulation of type I IFNs may be due to genomic instability, caused by the downregulation of Sororin, downstream of the exclusion of exon 2 from the U2AF2 mRNA (Laliotis et al., 20213). Genomic instability is known to activate the cGAS/STING pathway. Rescue of the shIWS1 phenotype with U2AF65α prevents the downregulation of Sororin (Laliotis et al., 20213) and as a result, it is expected to prevent genomic instability and the induction of the type I IFN genes. However, it is unlikely to regulate the expression of other intronless genes.
mRNAs sequestered in the nucleus cannot be translated. The preceding data therefore suggest that in the absence of IWS1 phosphorylation, the abundance of the proteins encoded by all four intronless genes in figure 1b would be decreased. This was addressed by probing western blots of total cell lysates with antibodies to the proteins encoded by these genes. The expression of IFNβ1 was examined in cells infected with SeV-GFP, or treated with poly (I:C), both of which induce the expression of type I IFNs, and the expression of c-JUN and HSP27 was examined in uninfected cells growing under normal culture conditions. The results confirmed that shIWS1 downregulates the proteins encoded by these genes and that the downregulation is rescued as by wild type IWS1, but not by the phosphorylation site IWS1 mutant, as expected (Fig. 1e). Importantly, the downregulation was also rescued by U2AF65α, but not U2AF65β (Fig. 1e), suggesting that it is due to the effects of the loss of IWS1 phosphorylation on the alternative RNA splicing of U2AF2. Consistent with these findings were the results of Reverse Phase Protein Assay (RPPA) experiments in human lung adenocarcinomas, which showed that although the expression of intronless genes at the RNA level exhibits a negative correlation with the expression of IWS1, (Fig 1d and Supplementary Figure 1b), their expression at the protein level, and IWS1, exhibit a significant positive correlation (Supplementary Figure 1c).
The phosphorylation of IWS1 by AKT3 is required for the nuclear export of intronless gene mRNAs via U2AF2 RNA splicing
The preceding data show that the AKT-dependent phosphorylation of IWS1 is required for the nuclear export and translation of the mRNAs of naturally intronless genes via a U2AF2/Prp19-dependent pathway. This raised the question whether AKT, which phosphorylates IWS1 at Ser720/Thr721 (Sanidas et al., 20141), is required for the activation of the pathway. To address this question, we first infected NCI-H522 and NCI-H1299 cells with SeV-GFP to induce the expression of type I IFNs. Infected cells (24 hours after the infection) and uninfected cells were then treated with 5 μM of the AKT inhibitor MK2206, a dose that fully inhibits all AKT isoforms (Sanidas et al., 20141, Laliotis et al., 20213). Western blotting of cell lysates harvested 24 hours after the start of exposure to MK2206, confirmed the strong inhibition of AKT and the complete block of IWS1 phosphorylation (Fig. 2a). Moreover, RT-PCR using RNA isolated from the same cell lysates, confirmed that AKT inhibition results in the exclusion of exon 2 from the mature U2AF2 mRNA (Fig. 2a). Lysates of the same cells, before and after treatment with MK2206, were fractionated into nuclear and cytoplasmic fractions. Quantitative RT-PCR, addressing the abundance of the RNAs of IFNA1, IFNB1, JUN and HSBP3 in these fractions, confirmed that the AKT activity is required for the nuclear export of the mRNAs of all four intronless genes (Fig. 2b). We conclude that the phosphorylation of IWS1 at Ser720/Thr721 is indeed required for the nuclear export of these mRNAs and that AKT is the main kinase responsible for the phosphorylation. Probing western blots of total cell lysates of the same cells with antibodies to c-JUN, phosphor-c-JUN (Ser73), HSP27 and IFNβ, revealed that their abundance is dramatically downregulated following AKT inhibition, as expected (Fig. 2c).

a. Inhibiting AKT interferes with the inclusion of exon 2 in mature U2AF2 mRNA transcripts NCI-H522 and NCI-H1299 cells were treated with MK2206 (5μM) or DMSO. Lysates of these cells harvested 4h later, were probed with the indicated antibodies. Total RNA isolated from the same cells was also analyzed by RT-PCR, using oligonucleotide primers mapping in U2AF2 exons 1 and 3.
b. The AKT kinase regulates the nucleocytoplasmic export of intronless mRNAs, through IWS1 phosphorylation. Cells in a were infected with SeV-GFP (MOI 0.25), to induce type I IFN gene expression. Infected and uninfected cell lysates harvested 24h later, were fractionated into cytoplasmic and nuclear fractions, and mRNA levels of the indicated genes in each fraction were measured, using qRT-PCR. RNA levels for each transcript, were normalized to 18S rRNA. Bars show the mean normalized Cytosolic/Nuclear RNA ratio±SD.
c. NCI-H522 and NCI-H1299 lysates of the cells in a, were probed with the indicated antibodies. Type I IFNs were induced again by SeV-GFP infection, as in 2b.
d. The NCI-H522 and NCI-H1299 cells were transduced with lentiviral shAKT3 or shControl constructs. Lysates of these cells were probed with the indicated antibodies. Total RNA was also analyzed by RT-PCR, using primers mapping in U2AF2 exons 1 and 3.
e. Cells in d were infected with SeV-GFP (MOI 0.5), to induce type I IFN expression. Infected and uninfected cell lysates harvested 24h later, were fractionated into cytoplasmic and nuclear fractions, and the abundance of the indicated mRNAs in each fraction was determined, using qRT-PCR. RNA levels, were normalized to 18S rRNA. Bars show the mean normalized Cytosolic/Nuclear RNA ratio ±SD.
f. NCI-H522 and NCI-H1299 lysates of cells in d, were probed with the indicated antibodies. Type I IFNs were induced again by SeV-GFP infection. To validate the cellular fractionation in the experiments in b and e we measured the Cytoplasmic/Nuclear ratio of the GAPDH RNA, as in figure 1b (see supplementary Table 4). Experiment in b and e were done on three biological replicates, in triplicate. n.s: non-significant *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (one-side unpaired t-test).
To determine whether it is the AKT3 isoform, which is responsible for the observed effects of AKT on RNA transport and translation, we transduced NCI-H522 and NCI-H1299 cells with shAKT3 or shControl lentiviral constructs. Following confirmation of the AKT3 knockdown (Fig. 2d), the cells were infected with SeV-GFP. Measuring the abundance of the IFNA1 and IFNB1 mRNAs in the two fractions of the infected cells and the JUN and HSBP3 mRNAs in the two fractions of the non-infected cells, confirmed that the knockdown of AKT3 profoundly inhibits the nuclear export of these mRNAs (Fig. 2e). Western blotting of total lysates of the same cells confirmed that the knockdown of AKT3 is sufficient to significantly lower the abundance of the proteins encoded by these mRNAs (Fig. 2f), as expected. We conclude that AKT3 is the main kinase responsible for the phosphorylation of IWS1 at Ser720/Thr721.
IWS1 phosphorylation drives the recruitment of Prp19 to mRNA CAR-Elements, by promoting the inclusion of exon 2 in the mature U2AF2 mRNA transcripts
Previous studies had shown that the nuclear export of the mRNA of intronless genes depends on the recruitment of U2AF65 and Prp19 to CAR-Elements (Lei et al., 20134). In addition, they had shown that U2AF65 interacts directly with Prp19 via its exon 2-encoded RS domain (Laliotis et al., 20213, David et al., 201027), suggesting that the recruitment of Prp19 to these complexes may depend on the alternative splicing of U2AF2. Based on these considerations, we reasoned that the IWS1 phosphorylation-dependent inclusion of exon 2 in the U2AF2 mRNA, might promote the nuclear export of these mRNAs by regulating the recruitment of Prp19 to CAR-Elements. This hypothesis was addressed by RNA Immunoprecipitation (RIP) with anti-U2AF65 and anti-Prp19 antibodies in shControl, shIWS1, shIWS1/WT-R, shIWS1/MT-R, shIWS1/U2AF65α and shIWS1/U2AF65β NCI-H522 and NCI-H1299 cells. Immunoprecipitated RNA was detected by qRT-PCR, using sets of primers designed to amplify the CAR-Elements or control regions of the IFNA1, IFNB1, JUN and HSPB3 mRNAs (Supplementary Figure 2a). The results showed that whereas neither of the U2AF65 splice variants binds the control sequences, both bind the CAR-Elements with equal efficiency (Fig. 3a, 3b upper panels). However, Prp19 binding to the same CAR-Elements, was significantly impaired in shIWS1 and shIWS1/MT-R cells, which predominantly express the RS domain-deficient U2AF65β isoform (Fig. 3a lower panels). More important, the impaired Prp19 binding to the CAR-Elements was rescued by U2AF65α, but not U2AF65β (Fig. 3a, 3b lower panels). We conclude that the recruitment of Prp19 to the CAR-Element-associated complexes and the nuclear export of the mRNA of CAR-Element-containing intronless genes, is regulated by IWS1 phosphorylation via the alternative RNA splicing of U2AF2.

a. and b. IWS1 phosphorylation controls the recruitment of Prp19 to CAR-Elements, by regulating U2AF2 alternative RNA splicing. NCI-H522 and NCI-H1299 cells transduced with the indicated constructs, were infected with SeV-GFP (MOI 0.5). 24h later, infected cells and parallel cultures of uninfected cells, were used for RIP assays addressing the binding of U2AF65 (upper panels) and Prp19 (lower panels) to CAR-Elements or to sequences without CAR-Elements in the indicated RNAs. The bars show the mean fold enrichment in U2AF65 and Prp19 binding (anti-U2AF65 or anti-Prp19-IP, vs IgG control-IP)±SD. Data were normalized relative to the input (2%). The map location of the PCR primers used to amplify the binding regions is shown in Supplementary Figure 2a.
c. The IWS1 phosphorylation-dependent nuclear export of the mRNAs of intronless genes is active in human Lung Adenocarcinomas. (Upper panel) Western blots of lysates of 6 human LUAD samples (3 with high and 3 with low IWS1 expression), randomly selected out of LUAD samples previously analyzed (Laliotis et al., 20213), were probed with the indicated antibodies (top three rows). RT-PCR, using primers mapping in U2AF2 exons 1 and 3 (bottom row) (Lower Panel) LUAD tumor samples were fractionated into cytoplasmic and nuclear fractions. mRNA levels of the indicated genes were determined in each fraction, using qRT–PCR. Fractionation was validated as in figures 1b/1c and 2b/2e (Supplementary Table 4). Bars show the mean Cytoplasmic/Nuclear RNA ratio, normalized to the 18S ribosomal RNA ±SD.
d. IWS1 phosphorylation controls the recruitment of Prp19 to CAR-Elements in the mRNAs of CAR-Element-positive intronless genes, in human Lung Adenocarcinomas. RIP assays in the high and low-IWS1 LUADs, shown in c. The bars show the mean fold enrichment in U2AF65 (upper panel) and Prp19 (lower panel) binding to the same CAR-Element-positive and CAR-Element-negative regions as in a and b (anti-U2AF65 or anti-Prp19-IP, vs IgG control-IP)±SD. Data were normalized relative to the input (2%). All experiment in this figure were done in triplicate, on three biological replicates. n.s : non-significant *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (one-side unpaired t-test).
The preceding data raised the question whether the expression and phosphorylation of IWS1 regulates the nuclear export of the mRNAs of CAR-Element-containing intronless genes in human cancer. To address this question, we used qRT-PCR to measure the abundance of the IFNA1, IFNB1, JUN and HSPB3 mRNAs in nuclear and cytoplasmic fractions of tumor cell lysates of three lung adenocarcinomas expressing high and three expressing low levels of IWS1/p-IWS1 (Fig. 3c Upper panel). The results confirmed that the Cytoplasmic/Nuclear ratio of the RNAs transcribed from the IFNA1, IFNB1, JUN and HSPB3 genes in Human Lung Adenocarcinomas, correlates with the expression and phosphorylation of IWS1 and with the inclusion of exon 2 in the mature U2AF2 mRNA (Fig. 3c Lower panel). Next, we carried out RIP assays for U2AF65 and Prp19, using total cell lysates of the same lung adenocarcinomas. The results confirmed that whereas U2AF65 binds equally well the CAR-Elements in the mRNAs of all four genes in both the high and the low p-IWS1 tumors, Prp19 binds efficiently the CAR-Elements of these mRNAs only in the high p-IWS1 tumors. Given that the predominant U2AF65 isoform in high p-IWS1 tumors is the U2AF65α isoform, which binds Prp19, while the predominant isoform in the low p-IWS1 tumors is U2AF65β, which does not interact with Prp19, these data indicate that in lung adenocarcinoma patients, as in cultured tumor cells, the recruitment of Prp19 to the CAR-Elements is mediated by U2AF65. We conclude that the IWS1 phosphorylation-dependent pathway regulating the nuclear export of RNAs transcribed from CAR-Element-containing intronless genes, is active in human lung adenocarcinomas.
The RNA Pol II promoter of type I IFN genes, plays an essential role in the IWS1 phosphorylation-dependent mRNA nuclear export
Earlier studies had shown that U2AF65 binds RNA Pol II and recruits Prp19. During transcriptional elongation, both U2AF65 and Prp19 are loaded to the newly synthesized pre-mRNA, promoting RNA splicing co-transcriptionally (David et. al., 201127). This suggested that U2AF65 and Prp19 might be loaded to the CAR-Elements of the RNAs transcribed from naturally intronless genes, via a similar mechanism. To address this hypothesis, we expressed IFNA1 and IFNB1 from RNA Pol II, or RNA Pol III constructs and we asked whether the mRNAs transcribed form the two different promoters are transported to the cytoplasm with equal efficiency. We reasoned that if U2AF65 and Prp19 are loaded to the CAR-Elements by RNA Pol II during transcriptional elongation, they will probably fail to load to the RNA Pol III transcripts, because U2AF65 and Prp19 do not bind RNA Pol III. As a result, RNA Pol III transcripts will stay in the nucleus, and they will not be translated into protein. To carry out this experiment, we cloned the IFNA1 and IFNB1 cDNAs in the lentiviral vectors pLx304 and pLKO.1, which drive expression through CMV (RNA Pol II-dependent) or U6 (RNA Pol III-dependent) promoters, respectively (Schramm et al., 200228) (Supplementary Figure 3a). We then transduced NCI-H522 and NCI-H1299 cells with shIFNA1 or shIFNB1 lentiviral constructs, and we rescued the IFNA1 and IFNB1 knockdown with the RNA Pol II-driven (pLx304-R) or RNA Pol III-driven (pLKO.1-R) lentiviral constructs of these genes. Following this, the cells were infected with SeV-GFP and the expression of IFNA1 or IFNB1 was measured by qRT-PCR, 24 hours later. The results showed that IFNA1 or IFNB1 were transcribed efficiently from both the RNA Pol II and the RNA Pol III promoters (Fig. 4a Upper panel). However, measuring the abundance of the IFNA1 or IFNB1 mRNAs in the nuclear and cytoplasmic fractions of the same cells by qRT-PCR, revealed that only the mRNAs transcribed from the RNA Pol II promoter were efficiently transported to the cytoplasm (Fig. 4a Lower panel). Western blots of total cell lysates from the IFNB1-transduced NCI-H522 and NCI-H1299 cells, confirmed that only the cells rescued with the RNA Pol II construct (pLx304IFNβ) express IFNβ (Fig. 4b). We interpret these data to suggest that U2AF65 and Prp19, which bind RNA Pol II but not RNA Pol III, are likely loaded to the CAR-Elements co-transcriptionally via RNA Pol II, and that the co-transcriptional RNA loading of these molecules’ controls mRNA nuclear export and translation.

a. Type I IFN mRNAs are transcribed equally well from an RNA Pol II and an RNA Pol III promoter, but only the mRNAs transcribed from the RNA Pol II promoter are exported efficiently to the cytoplasm. (Upper panel) Total RNA was harvested from shControl, shIFNA1 (or shIFNB1), shIFNA1/pLx304-IFNα1 (or shIFNB1/pLx304-IFNβ1) and shIFNA1/pLKO.1-IFNα1 (or shIFNB1/pLKO.1-IFNβ1) NCI-H522 and NCI-H1299 cells. pLx304-IFNA1 and pLx304-IFNB1 drive IFNα1 and IFNβ1 expression respectively, from the CMV (RNA Pol II) promoter, while pLKO.1-IFNA1 and pLKO.1-IFNB1 drive IFNα1 and IFNβ1 expression respectively, from the U6 (RNA Pol III) promoter. The maps of the pLX304 and pLKO.1 constructs are shown in supplementary figure 3. The abundance of the mRNAs of IFNA1 and IFNB1, was determined by qRT-PCR and was normalized to the abundance of 18S rRNA. Heatmaps were generated, based on the z scores of the abundance of the mRNAs of IFNA1 and IFNB1. Type I IFNs were induced by infection with SeV. (Lower panel) The cells in the upper panel were fractionated into cytoplasmic and nuclear fractions and the abundance of IFNA1, IFNB1 and GAPDH mRNAs in each fraction was determined with qRT–PCR Bars show the mean Cytoplasmic/Nuclear ratio of the IFNA1 and IFNB1 mRNAs, normalized to 18S rRNA. ±SD. All assays in were done on three biological replicates, in triplicate for each replicate. ***p<0.001, ****p<0.0001. (one-side unpaired t-test). Cell fractionation was validated as in figures 1b/1c, 2b/2e and 3c (Supplementary Table 4).
b. Western blots of lysates of the shControl, shIFNB1, shIFNB1/pLx304-IFNB1 and shIFNB1/pLKO.1-IFNB1 NCI-H522 and NCI-H1299 cells in A, were probed with anti-IFNβ1 and anti-β-actin (control) antibodies. Cells were harvested 24 hours after infection with SeV-GFP (MOI=0.5).
The IWS1 phosphorylation-dependent U2AF2 alternative RNA spicing is required for the RNA nuclear export function of the Cytoplasmic Accumulation Region Elements (CAR-E)
The preceding data suggest that the nuclear export of the mRNAs of CAR-E-positive intronless genes, depends on IWS1 phosphorylation by AKT, which regulates the loading of U2AF65/Prp19 complexes to CAR-Elements in these mRNAs. To confirm that IWS1 phosphorylation and U2AF2 alternative RNA splicing regulate CAR-E function, we employed previously described pCMV promoter β-globin constructs, in which 16 tandem copies of the most conserved CAR-Element (CCAGTTCCTG element of JUN) or its mutated inactive version (CAR-Emut), were inserted in the 5’ UTR of the β-globin cDNA (Fig. 3a) (Lei et al., 20134). As controls, we used a pCMV promoter / β-globin cDNA construct and a pCMV promoter / β-globin gene construct. All constructs were transiently transfected in shControl, shIWS1, shIWS1/WT-R, shIWS1/MT-R, shIWS1/U2AF65α-R, and shIWS1/U2AF65β-R NCI-H522 and NCI-H1299 cells and the expression of globin in the transfected cells was monitored by Western blotting. Earlier studies had shown that whereas the β-globin mRNA transcribed from the wild type β-globin gene, efficiently accumulates in the cytoplasm, the β-globin mRNA transcribed from the cDNA, is degraded in the nucleus (Dias et al., 201029, Lei et al., 201130, Valencia et al., 200831). In agreement with the results of these studies, our data showed that whereas shControl cells transduced with the cDNA construct do not express β-globin, shControl cells transduced with the wild type β-globin gene construct, do (Fig. 5b, Supplementary Figure 4). More important, the failure of the cDNA construct to direct the expression of β-globin was rescued with the insertion of the array of wild type, but not the mutant CAR-Elements in its 5’ UTR (Fig. 5b, Supplementary Figure 4 upper panels). Since expression of the β-globin gene depends on the ability of its mRNA to exit the nucleus, the data in shControl cells, confirm the earlier observations on the function of CAR-Elements. The experiment in shIWS1, shIWS1/WT-R and shIWS1/MT-R cells showed the CAR-Element array is not functional in shIWS1 cells and that whereas rescue with wild type IWS1 restores its function, rescue with the phosphorylation site mutant of IWS1 does not (Fig. 5b, Supplementary Figure 4 upper and middle panes). Moreover, the shIWS1 induced defect in the CAR-Element function was rescued by the RS domain-containing U2AF65α, but not by the RS domain-deficient U2AF65β (Fig. 5b, Supplementary Figure 4 lower panels). We conclude that the function of the CAR-Elements depends on IWS1 phosphorylation, which controls the function of the CAR-Elements, by regulating the alternative splicing of U2AF2.

a. Schematic of the pCMV-HA-β-globin cDNA construct. The sequences of the CAR-E and CAR-Emut are shown and the map position of their insertion is indicated with a solid arrow (Lei et al., 20134). The transcription initiation site, the translation initiation codon (ATG), the HA epitope tag, the bovine growth hormone (BGH) polyA signal (pA) and the sizes of the exons in base pairs are also shown. The schematic for β-globin CAR-E reporter was created with Biorender.com under the third-party publication license permission OO22MW9YPA.
b. IWS1 phosphorylation is required for CAR-E function. shControl, shIWS1, shIWS1/WT-R, shIWS1/MT-R, shIWS1/U2AF65α-R and shIWS1/U2AF65β-R NCI-H522 cells, were transfected transiently with pCMV-based constructs of HA-β-globin cDNA, HA-β-globin Gene, HA-β-globin cDNA-CAR-E and HA-β-globin cDNA-CAR-Emut. Transfected cells were harvested 48h later, and their lysates were probed with the indicated antibodies.
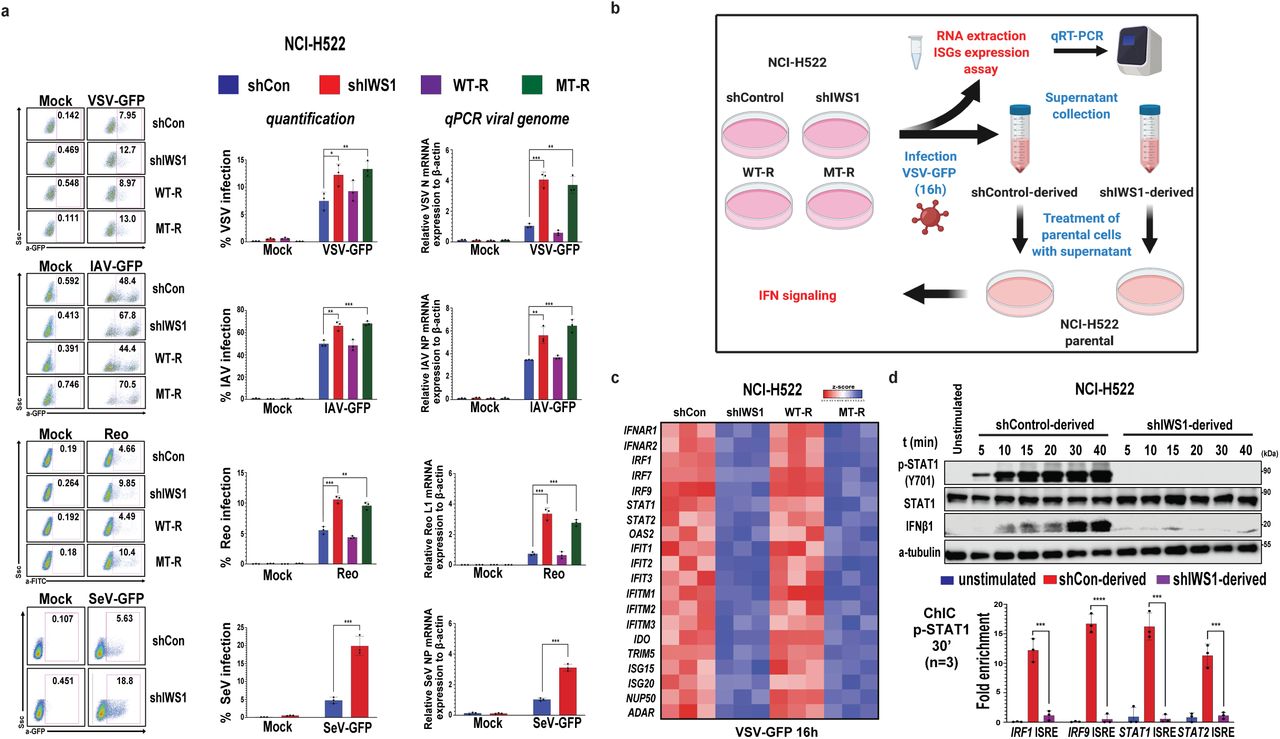
The low expression of type I IFNs in shIWS1 and shIWS1/MT-R cells enhances their sensitivity to viral infection
Type I IFNs regulate innate and adaptive immunity and orchestrate the cellular antiviral response (Lazear et al., 201932). Cells failing to induce type I IFNs in response to viral infection and cells, which fail to respond to type I IFNs are more sensitive to infection. Given that the IWS1 phosphorylation-dependent alternative RNA splicing of U2AF2 regulates the nuclear export and translation of type I IFN genes, we hypothesized that cells in which IWS1 was knocked down and cells in which the IWS1 knockdown was rescued with the phosphorylation site mutant of IWS1 will be more sensitive to viral infection. To address this hypothesis, we infected shControl, shIWS1, shIWS1/WT-R and shIWS1/MT-R NCI-H522 and NCI-H1299 cells with Vesicular Stomatitis Virus (MOI=0.5) or Influenza A virus (MOI=0.5), engineered to express GFP (VSV-GFP and IAV-GFP). NCI-H522 cells, but not NCI-H1299 cells, were also infected with the Reovirus (MOI=1). Finally, shControl and shIWS1 NCI-H522 and NCI-H1299 cells were infected with GFP-expressing Sendai virus (SeV-GFP) (MOI=0.5), Twenty four hours later, cells were harvested and the percentage of infected cells was determined by flow cytometry (Chesarino et al., 201533, Kenney et al., 201934, Sermersheim et al., 202035). The results showed that the percentage of infected cells with all four viruses was higher in shIWS1 than in shControl cells and that the shIWS1 phenotype was rescued with WT IWS1 (shIWS1/WT-R cells), but not with the phosphorylation site mutant of IWS1 (shIWS1/MT-R cells), as expected (Fig. 6a, Supplementary Figure 5a). To assess viral replication changes in the shIWS1 and shIWS1/MT-R NCI-H522 and NCI-H1299 cells, we employed qRT-PCR, using the viral oligonucleotide primers listed in the methods section. The results showed that the knockdown of IWS1, and its rescue with the phosphorylation site IWS1 mutant, resulted in a significant increase of the abundance of replicating viral genomes in virus-infected cells (Fig. 6a Right panels, Supplementary Figure 5a, Supplementary Data 2). These results confirmed that the loss of phosphorylated IWS1 increases the sensitivity of the cells to virus infection and they were fully consistent with the results of the flow-cytometry experiments. The IWS1 knockdown in the experiments in this section was carried out with a short hairpin RNA constructs in a pGIPZ vector, we modified by deleting the GFP cassette, as described in the methods section.

a. Loss of IWS1 expression and phosphorylation, enhance the sensitivity of cells to viral infection. shControl, shIWS1, shIWS1/WT-R and shIWS1/MT-R NCI-H522 cells were infected with VSV-GFP, Influenza A-GFP (IAV-GFP) or Reovirus. In addition, shControl and shIWS1 NCI-H522 cells were infected with SeV-GFP. All infections and analyses were carried out as described in the methods. (Left panels) Flow-cytometric analyses, representative of at three independent experiments. (Middle panels) Quantification of the data from all three experiments. Bars show the percentage of infected cells ±SD. (Right panels) The expression of viral genes in cells infected with the same viruses was quantified by qRT-PCR, as described in the methods Bars show the relative expression of viral genes ±SD.
b. Design of the experiment in c and d. The indicated cells were infected with VSV-GFP (MOI=0.5). 16h later, total RNA was harvested and analyzed by qRT-PCR for the expression of the mRNAs of 20 ISGs. In parallel, the supernatants of shControl and shIWS1 cultures were harvested and used in a bioassay for the abundance of biologically active IFN-type-I. Naïve NCI-H522 cells were treated with the supernatants and examined for STAT1-phosphorylation and the binding of phosphor-STAT1 to the ISREs of four ISGs. The schematic was created with Biorender.com under the third-party publication license permission UR22MWA47E.
c. Heatmaps showing the relative expression of 20 ISGs in the indicated cells, as determined by qRT-PCR. ISG expression was normalized to 18S rRNA. Heatmaps were based on z scores, derived from experiments on three biological replicates, done in triplicate.
d. (Upper panel). Naïve NCI-H522 cells were treated with culture supernatants as described in 6b. Cell lysates were harvested at the indicated time-points and probed with the indicated antibodies. (Lower panel) The cells in the upper panel were harvested at 30’ from the start of the exposure to the supernatants, and the lysates, were used to carry out ChIC assays for p-STAT1 binding. The bars show the mean fold enrichment in p-STAT1 binding to the ISREs of four ISGs±SD, (anti-p-STAT1 IP vs IgG-IP). All assays were done on three biological replicates. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (one-side unpaired t-test).
The data presented in this report collectively suggest that the increased sensitivity of shIWS1 and shIWS1/MT-R cells to viral infection, is due to the impaired induction of type I IFNs. To address this question, we infected shControl shIWS1, shIWS1/WT-R and shIWS1/MT-R NCI-H522 cells with VSV-GFP. Sixteen hours later, the cells and their culture supernatants were harvested (Fig. 6b). Quantitative RT-PCR addressing the abundance of a set of IFN-stimulated genes (ISGs) in RNA isolated from lysates of these cells, revealed robust induction in shControl, but not in shIWS1 cells. Moreover, the defect in ISG induction in shIWS1 cells was rescued by wild type IWS1, but not by the phosphorylation site IWS1 mutant (Fig. 6c). These findings and the results of the preceding experiments are in full agreement, and they collectively suggest that IWS1 phosphorylation is required for the induction of type I IFNs in virus-infected cells.
To confirm the failure of VSV-GFP to induce biologically active type I IFNs in IWS1 knockdown cells, we used the culture supernatants harvested from shControl and shIIWS1 NCI-H522 cells to stimulate naïve NCI-H522 parental cells (Fig. 6b). Immunoblotting of protein lysates harvested at multiple time points from the start of the stimulation, revealed rapid robust phosphorylation of STAT1 (Y701) and rapid increase in the expression of IFNβ1 in the lysates of cells stimulated with the shControl, but not the shIWS1 culture supernatants (Fig. 6d Upper panel). The phosphorylation of STAT1, a known target of type I IFNs, combined with the data in figure 5c, further support the selective induction of type I IFNs in virus-infected, IWS1 phosphorylation proficient cells. The increase in the abundance of IFNβ1, within 10 minutes from the start of the stimulation, was surprising because it was too rapid to be due by the induction of the IFNB1 gene. Previous studies had shown that IFNβ1 undergoes endocytosis and that it can be siloed in endosomes, where it can be detected for days following IFN treatment (Altman et al., 202034). Based on this information, we hypothesized that IFNβ1 detected in this experiment was endocytosed from the culture supernatants of shControl cells. To address this hypothesis, we treated the parental NCI-H522 cells with recombinant human IFNβ1, and we probed the cell lysates harvested at sequential time points from the start of the treatment with antibodies to IFNβ1. The results confirmed the rapid accumulation of the recombinant IFNβ1 in the harvested cell lysates (Supplementary Figure 5c).
Phosphorylated STAT1 and STAT2, in combination with IRF9, form a trimeric complex known as Interferon Stimulated Gene Factor 3 (ISGF3), which is required for the induction of ISGs. This suggests that the phosphorylation of STAT1 in cells treated with the culture supernatants of VSV-GFP-infected shControl NCI-H522 cells is critical for the induction of type I IFNs. To determine whether the phosphorylated STAT1 indeed contributes to ISG induction in these cells (Wang et al., 201735), we performed Chromatin ImmunoCleavage (ChIC) assays in NCI-H522 cells in the upper panel, harvested at 30 minutes from the start of the stimulation. Unstimulated cells were used as controls. Consistent with the STAT1 phosphorylation pattern, the results showed increased binding of p-STAT1 (Y701) to the ISREs of the ISGs IRF1, IRF9, STAT1, and STAT2, only in cells stimulated with the culture supernatants of the VSV-GFP-infected shControl cells (Fig. 6d, lower panel).
If the phosphorylation of IWS1 by AKT3 is required for the induction of type I IFNs, as the data presented in this report indicate, inhibition of AKT prior to viral infection should block the induction of ISGs. To address this hypothesis, we treated NCI-H522 cells with 5 μM MK2206, or with the vehicle (DMSO). 4 hours later, cells were infected with VSV-GFP (MOI=1), and they were harvested 16 hours later. Using RNA isolated from the harvested cell lysates and qRT-PCR, we examined the expression of the same set of 20 ISGs as in the experiment in figure 5C. The results confirmed that AKT inhibition, similarly to the knockdown of IWS1 blocks the induction of ISGs in virus-infected cells (Supplementary Figure 5c), as expected. These data collectively show that by regulating the expression of type I IFNs, IWS1 phosphorylation by AKT enhances the resistance to viral infection
Inhibition of the AKT/p-IWS1 axis sensitizes lung adenocarcinoma cells to cytolytic virus-induced apoptotic cell death
Data presented in this report show that the knockdown of IWS1 or the inhibition of its phosphorylation, interferes with the expression of type I IFNs by virus-infected cells and enhances the sensitivity of the cells to viral infection. Given that infection by cytolytic viruses induces cell death, infection of human tumors with engineered strains of “oncolytic viruses” emerged in recent years as a new therapeutic tool in Oncology. Such viruses not only kill tumor cells, but they also alter the relative abundance and function of different types of immune cells in the tumor microenvironment. As a result, they can be effective either as a monotherapy, or in combination with other anti-cancer therapeutics.
Variants of two of the viruses we used in the preceding experiments, VSV-GFP and Reovirus, are evaluated as potential virotherapy agents in a variety of solid tumors, including lung cancer (Schreiber et al., 201938, Villalona-Calero et al., 201639). We therefore tested whether inhibition of the AKT/p-IWS1 axis enhances their cytolytic activity against tumor cells. In addition, we examined the mechanism by which they induce cell death. To address these questions, we first infected shControl, shIWS1, shIWS1/WT-R, and shIWS1/MT-R NCI-H522 and NCI-H1299 cells, with VSV-GFP or Reovirus, at logarithmically increasing multiplicities of infection (MOI), and we monitored the percentage of surviving cells at 16 hours (VSV-GFP) or at 48 hours (Reovirus) after infection. The results revealed that both the knockdown of IWS1 and its rescue with the phosphorylation site IWS1 mutant, dramatically enhance cell death by both VSV-GFP and Reovirus (Fig. 7a, Supplementary Figure 7a). A repeat of the experiment in naive parental NCI-H522 and NCI-H1299 cells, pretreated with the AKT inhibitor MK2206, gave similar results (Fig. 7b, Supplementary Figure 7b), as expected. We conclude that blocking the IWS1 phosphorylation pathway by inhibiting AKT, can be used as a tool to increase the killing efficiency of oncolytic viruses.

a. The knockdown of IWS1 and its rescue with the phosphorylation site IWS1 mutant sensitizes lung adenocarcinoma cell lines to virus-induced cell death. ShControl, shIWS1, shIWS1/WT-R and shIWS1/MT-R NCI-H522 cells were infected with VSV-GFP or Reovirus at the indicated MOIs. The percentage of surviving cells was measured 16 hours later (for VSV-GFP) and 48 hours later (for Reovirus), using the resazurin reduction protocol described in the methods section under “virus-induced cell death”. The cell survival curves show the mean percent survival values at each MOI±SD (n=3) and they are representative of two independent experiments.
b. The inhibition of AKT, which is required for the phosphorylation of IWS1, sensitizes lung adenocarcinoma cells to virus-induced cell death. Parental NCI-H522 cells were treated with DMSO or with the AKT inhibitor MK2206 (5μΜ) and they were infected with VSV-GFP or Reovirus at the indicated MOIs. Infected and uninfected cells were harvested 16 hours later (VSV-GFP), or 48 hours later (Reovirus) and their survival was measured as in a. The cell survival curves show the mean percent survival values at each MOI ±SD (n=3) and they are representative of two independent experiments.
c. The knockdown of IWS1 accelerates the caspase-dependent cleavage of PARP1 in virus-infected lung adenocarcinoma cell lines. ShControl and shIWS1-transduced NCI-H522 cells were infected VSV-GFP (MOI 1) and they were harvested at the indicated time points. Western blots of the harvested cell lysates were probed with antibodies to IWS1, cleaved PARP, or α-tubulin.
To address the mechanism of cell death following infection with cytolytic viruses, we infected shControl and shIWS1 NCI-H522 and NCI-H1299 cells with VSV-GFP (MOI=1). Cells were harvested before and at different time points from the start of the infection and the cleavage of PARP along with the abundance of IWS1 was monitored in the cell lysates by Western blotting. The results showed a dramatic upregulation of cleaved PARP in the lysates of the shIWS1 cells, starting at 2 hours (NCI-H522 cells) or 6 hours (NCI-H1299 cells) from the start of the infection (Fig. 7c, Supplementary Figure 7c). Given that the cleavage of PARP, is a hallmark of Caspase-mediated cell death (Chaitanya et al., 201040), we conclude that cell death induced by these cytolytic viruses is due to caspase activation-induced apoptosis.
Model of the pathway by which the AKT3/IWS1/U2AF2 axis promotes nuclear export of CAR-Element-containing intronless gene mRNAs, and resistance to viral infection
The phosphorylation of IWS1 at S720/T721 by AKT controls the epigenetic regulation of the alternative RNA splicing of U2AF2, promoting the inclusion of exon 2 in the mature U2AF2 mRNA. The RS domain-containing U2AF65α isoform encoded by the exon 2 containing U2AF2 mRNA, binds CAR-Elements in the mRNA of type I IFN, and other intronless genes, and recruits Prp19. The U2AF65/Prp19 complex assembled on the CAR-Elements is required for the nuclear export and translation of these mRNAs. Overall, IWS1 expression and phosphorylation by AKT, promotes the expression of intronless genes, including type I IFNs and increases the resistance of the cells to infection by cytolytic viruses (Fig. 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The phosphorylation of IWS1 at S720/T721 by AKT controls the epigenetic regulation of the alternative RNA splicing of U2AF2, promoting the inclusion of exon 2 in the mature U2AF2 mRNA. The RS domain-containing U2AF65α encoded by the exon 2-containing U2AF2 mRNA, is loaded to CAR-Elements in the mRNA of type I IFNs, and other CAR-Element-positive intronless genes via RNA Pol II, and recruits Prp19. The U2AF65/Prp19 complex assembled on the CAR-Elements is required for the nuclear export of these mRNAs. Overall, IWS1 expression and phosphorylation by AKT, enhances the abundance of the proteins encoded by CAR-Element-positive intronless genes, including type I IFN genes and increases the resistance of the cells to infection by cytolytic viruses. The schematic was created with Biorender.com under the third-party publication license permission SC22PV8Z05.
Discussion
Data presented in this report show that a pathway initiated by the AKT3-mediated phosphorylation of IWS1 promotes the nucleocytoplasmic export of the mRNAs of a set of intronless genes and controls the expression of the proteins encoded by these mRNAs. Genes regulated by this pathway include the type I IFN-encoding genes IFNA1 and IFNB. Inhibition of the pathway by knocking down IWS1, or by rescuing the IWS1 knockdown with the IWS1 mutant IWS1-S720A/T721A, resulted in low expression of type I IFNs and in cellular sensitization to viral infection and replication and virus-induced, caspase-mediated cell death. As expected, inhibition of the pathway sensitized the cells to a broad array of viruses, including Vesicular Stomatitis Virus (VSV), Influenza virus, Sendai virus and Reovirus. In addition to the type I IFN genes, other genes also regulated by this mechanism, include JUN and the HSP27-encoding HSBP3.
The rationale of the experiments linking the IWS1 phosphorylation pathway to the nucleocytoplasmic export of the mRNAs of intronless genes and the sensitization to viral infection, was based on the integration of two earlier observations. First, it had been shown that the mRNAs of the majority of naturally intronless genes, including type I IFNs, JUN and HSPB3, contain 10 nucleotide consensus CAR-Elements, which provide the docking site for the assembly of nucleocytoplasmic export complexes, containing members of the TREX complex, U2AF65 and Prp19. Second, we had shown previously that IWS1 phosphorylation by AKT3 promotes transcription-coupled chromatin modifications, which regulate the alternative RNA splicing of U2AF2. The predominant U2AF2 transcript in the absence of IWS1 phosphorylation, is exon 2-deficient. Given that U2AF2 exon 2 encodes the RS domain of U2AF65, which is the domain of interaction between U2AF65 and Prp19, we hypothesized that in the absence of phosphorylated IWS1, the interaction between U2AF65 and Prp19 would be impaired, and this would affect the binding of Prp19 to CAR-Elements in naturally intronless mRNAs. In addition, if this interaction plays a critical role in the nucleocytoplasmic export of the mRNAs of intronless genes harboring CAR-Elements, the partial loss of phosphorylated IWS1 would also impair the nuclear export of these mRNAs. The data in this report fully support this hypothesis.
In the experiments presented in this report, we show that the RS domain-deficient U2AF65β, which is encoded by exon 2-deficient U2AF2, continues to bind the CAR-Elements in the mRNAs of type I IFNs. However, Prp19, whose recruitment is required for the nuclear export of these mRNAs (Lei et al., 20134), failed to bind RNA CAR-Elements in cells expressing U2AF65β, indicating that its recruitment to these elements depends on its interaction with U2AF65α. These findings strongly suggest that the IWS1 phosphorylation-dependent alternative RNA splicing of U2AF2 is a direct regulator of the nuclear export phenotype of the mRNAs of CAR-E-positive intronless genes. These findings make it unlikely that the RNA nuclear export phenotype is due to secondary effects of IWS1 on RNA splicing.
Given that the recruitment of the TREX complex to CAR-Elements is U2AF65 independent (Lei et al., 20134), the changing pattern of U2AF2 mRNA splicing in cells deficient in phosphorylated IWS1, should only affect the binding of Prp19, which should be responsible for the defective nuclear export of intronless CAR-Element-containing mRNAs in these cells. Prp19 may ultimately be affecting the composition and/or the functionality of the complex. The exact mechanism by which it is regulating the nuclear export of this class of mRNAs will be addressed in future studies.
The loading of the TREX complex to RNA is co-transcriptional (Sträßer et al., 200241), RNA Pol II-dependent and RNA splicing independent and as a result, it contributes to the nuclear export of the mRNA of naturally intronless genes. To address the mechanism of U2AF65 and Prp19 loading to the RNAs of naturally intronless genes we hypothesized that U2AF65 is also loaded co-transcriptionally and, given that U2AF65 binds RNA Pol II, its loading may also be RNA Pol II-dependent. We therefore placed IFNB1 under the control of an RNA Pol III promoter, and we examined its expression at the RNA and protein levels. The results showed that although RNA Pol III efficiently transcribed IFNB1, mRNA transcribed from the RNA Pol III promoter was not exported efficiently form the nucleus, and failed to be translated, as evidenced by the fact that the expression of IFNB1 at the protein level did not parallel its expression at the RNA level. Given that RNA Pol III is not known to bind U2AF65, we interpret these data to suggest that the loading of the U2AF65/Prp19 complex to CAR-Elements in the mRNA of naturally intronless genes is also co-transcriptional and RNA Pol II-dependent.
In the preceding paragraphs we pointed out that the downregulation of IWS1 and the block of IWS1 phosphorylation inhibit the expression of intronless genes that harbor CAR-Elements, because IWS1 phosphorylation is required for the nuclear export and translation of their mRNAs. However, the abundance of RNA transcripts of these genes is increased, perhaps because of a feedback mechanism activated by sensing the downregulation of their protein products. This finding is also in agreement with the RNA-Seq data of lung adenocarcinoma patients in the TCGA database, which show that the expression of IWS1 exhibits a negative correlation with the expression of type I IFN genes. A potential mechanism by which AKT inhibition may promote the transcriptional activation of type I IFNs was suggested by recent studies showing that AKT phosphorylates the human cyclic GMP-AMP synthase (cGAS) on Ser305, inhibiting its activity and the induction of type I IFNs (Seo et al., 201542). If inhibition of AKT induces partial G2/M arrest and genomic instability, as suggested by our earlier studies, it would also activate the cGAS/STING pathway and the transcriptional activation of the type I IFN genes and this may be facilitated by the absence of cGAS phosphorylation. An apparent paradox in these data is the observation that despite the downregulation of expression of type I IFNs at the protein level in cells with low abundance of phosphorylated IWS1, the expression of a set of IFN-stimulated genes (ISGs) is increased. We propose here that the paradox is due to the PRR-mediated activation of IRF3 and NF-κB, which promote the expression of IFN-independent ISGs.
AKT kinase may regulate the expression of and the response to type I IFNs by multiple mechanisms. Specifically, it may stimulate the expression of ISGs by promoting the translation of their mRNAs downstream of mTOR activation. In addition, AKT1 may stimulate the expression of IFNB downstream of β-catenin phosphorylation and the expression of ISGs via phosphorylation of EMSY, which relieves the EMSY-mediated ISG repression (Ezell et al., 201220). Moreover, a recent report provided evidence that AKT may be activated in Reovirus-infected cells via Clathrin-mediated endocytosis and that this activates the PI3K/AKT1/EMSY pathway and inhibits viral replication (Tian et al., 201543). The data in this report identify yet another pathway by which AKT1 and AKT3 regulates the IFN response and the sensitivity to viral infection and replication. The fact that the selective inhibition of some of these pathways, such as the EMSY or the IWS1 pathway, had profound effects on the sensitivity of the cells to viral infection, suggests that these pathways may not function independently of each other and that their roles may not be additive, but synergistic. The potential crosstalk between these AKT-regulated pathways will be addressed in future studies.
The data in this report may have significant implications in cancer treatment. First and foremost, they show that inhibition of the IWS1 phosphorylation pathway enhances the sensitivity to viral infection and replication and promotes virus-induced, caspase-mediated apoptosis. We interpret these data to suggest that inhibition of the pathway will enhance the therapeutic potential of oncolytic viruses. Oncolytic viruses may have direct anti-tumor activity, because of their ability to kill tumor cells, which tend to be more sensitive to viral infection than normal cells (Xia et al., 201644, Bommareddy et al., 201845). In addition, they may modulate innate immunity in the tumor microenvironment, enhancing the anti-tumor immune response, or the anti-tumor effects of cancer immunotherapies. For example, intratumoral reovirus administration enhanced the effects of PD-1 blockade in mice inoculated subcutaneously with B16 melanoma cells, by promoting tumor infiltration with CD8+T cells and by increasing the ability of NK cells to kill reovirus-infected tumor cells (Rajani et al., 201646). Immunomodulation by oncolytic viruses may be enhanced by using viruses engineered to deliver immunomodulatory molecules to the tumor microenvironment. For example, the first approved oncolytic virus TVEC, is an HSV1, which was genetically modified to express GM-CSF (Rehman et al., 201616). Another HSV-based oncolytic virus οHSV G47Δ, engineered to deliver IL-12, induced long-term durable cures in two syngeneic mouse models of GBM, when combined with anti-CTLA-4 and anti-PD-1 treatment. Anti-tumor effects were mediated by a profound increase in the ratio of T effector to Tregs in the tumor microenvironment (Saha et al., 201747). Multiple clinical trials addressing the effectiveness of oncolytic viruses or combinations of oncolytic viruses with immunomodulatory treatments are currently in progress with promising results, in patients with lung cancer (NCT03029871, NCT00861627) (NCT02263508, NCT02307149, NCT03153085) (Bishnoi et al., 201848). Based on the data presented in this report, we propose that all oncolytic virus-based anticancer treatments could potentially benefit by the inhibition of the IWS1 phosphorylation pathway.
The data in this report may also be relevant for the design of strategies to prevent or overcome the resistance of EGFR mutant lung adenocarcinomas to EGFR inhibitors. Our earlier studies had shown that the IWS1 phosphorylation pathway is active in human lung adenocarcinomas (Sanidas et al., 20141, Laliotis et al., 20213). Moreover, IWS1 phosphorylation and exon 2 inclusion in the U2AF2 mRNA were shown to correlate positively with tumor stage, histologic grade, and metastasis, and to predict poor survival in patients with EGFR mutant, but not KRAS mutant tumors. More important, a recent publication provided evidence, linking resistance to EGFR inhibitors to the upregulation of type I IFN signaling (Gong et al., 202049). This suggests that by promoting IFN signaling, the IWS1 phosphorylation pathway may promote resistance to EGFR inhibitors, contributing to the poor prognosis of these tumors. Inhibition of the IWS1 phosphorylation pathway for all the preceding anti-cancer applications can be achieved by inhibiting selectively AKT1 and AKT3, by blocking the interaction between phosphorylated IWS1 and SETD2 and by using antisense oligonucleotides or pharmacologic modulators of the splicing machinery (Obeng et al., 201950), to modulate the alternative RNA splicing of U2AF2.
In conclusion, data presented in this report describe a novel pathway by which AKT regulates the nucleocytoplasmic transport of the mRNAs of intronless genes harboring CAR-Elements and the effects of this process on the translation of these mRNAs. Type I IFNs are encoded by genes that belong to this gene set. By regulating their expression via this pathway, AKT regulates the sensitivity of the cells to viral infection and replication. The data presented in this report, may have significant implications in cancer therapeutics.
Methods
Cells, culture conditions, stimulation and inhibitors
NCI-H522, NCI-H1299 cells were grown in RPMI-1640 medium (Sigma-Millipore, Cat No. D8758). HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium (Sigma-Millipore, Cat No. D5796). Both types of media were supplemented with penicillin and streptomycin (Corning, Cat No. 30-002-CI), nonessential amino acids (Corning, Cat No. 25-025-CI), glutamine (Corning, Cat No. 25-005-CI), plasmocin 2.5ng/uL (Invivogen, Cat No. ant-mpp) and 10% fetal bovine serum. Cells were used for up to 5 passages. The human NCI-H522 non–small cell lung cancer adenocarcinoma cell line originated from an NCI-60 cell line panel obtained from Daniel Haber at Massachusetts General Hospital. The NCI-H1299 non–small cell lung cancer adenocarcinoma cell line originated from Dr Carbone’s Lab at the Ohio State University. The HEK-293T human embryonic kidney cell line originated from Richard Van Etten’s laboratory at Tufts Molecular Oncology Research Institute. The MDCK, HeLa and Vero cells were purchased from American Type Culture Collection (ATCC) (Cat No. CCL-34, CCL2 and CCL81 respectively). Cell lines were also periodically checked for mycoplasma, using the PCR mycoplasma detection kit (ABM, Cat No. G238). All experiments were carried out in mycoplasma-free cultures. To inhibit AKT, cells growing in complete media were treated with the AKT inhibitor MK2206 (MERCK) (5 μM) for 4 hours. At this concentration, MK2206 inhibits all three AKT isoforms. For stimulation of NCI-H522 and NCI-H1299 cells, 1000μg/mL hIFNβ1 (Bio-Rad Cat No. OBT1547) was used. For adequate expression of type I IFNs, NCI-H522 and NCI-H1299 cells were treated with 5 μg/mL Poly I:C sodium salt (Cell Signaling Technologies Cat No. 61401) for 6 and 12 hours respectively.
shRNA and Expression constructs
shRNAs and expression constructs are listed in Supplementary Table 3. The pLx304 IFNα1-V5 and pLx304 IFNβ1-V5 constructs were obtained by the DNAsu Plasmid Repository (DNAsu Plasmid Repository Clone : HsCD00436920 and HsCD00436917). To transfer IFNβ1-V5 and IFNα1-V5 from the pLX304 to the pLKO.1 lentiviral vector, we amplified the pLX304 inserts, using oligonucleotide primers flanked by AgeI and EcoR1 restriction endonuclease sites. Amplified DNA fragments were separated in 1% agarose gels, and they were gel-purified using the NucleoSpin Gel and PCR Clean-Up kit (M&N, Cat. No. 740609.50). Following purification, they were recombined into the AgeI-EcoRI-digested pLKO.1-TRC cloning vector (Addgene #10878), using T4 DNA ligase (Thermofisher, Cat No EL0011). To remove the GFP casette from the pGIPZ shIWS1 construct, we initially amplified the CMV Promoter/Enhancer region (Vector map position 2707-3385) by PCR. Following gel purification, we inserted NotI sites to the ends of the amplified DNA fragment using a PCR-based strategy, and following a second purification, we digested it with XbaI and NotI. In parallel, we treated the full pGIPZ shIWS1 construct also with XbaI-NotI, to remove the DNA fragment from map position 2707 to map position 4100 and following gel purification we recombined the two DNA fragments together, using T4 DNA ligase. The pGIZP GFP vector map used to design the vector modification strategy described above, can be downloaded from : https://www.snapgene.com/resources/plasmid-files/?set=viral_expression_and_packaging_vectors&plasmid=pGIPZ. The primers used for the cloning strategies described above, are listed in Supplementary Table 2. All the constructs were sequenced in the Genomics Shared Resource (GSR) of OSUCCC. https://cancer.osu.edu/for-cancer-researchers/resources-for-cancer-researchers/shared-resources/genomics, prior to use.
Transfections and Retroviral/Lentiviral infections
Lentiviral constructs were also packaged in HEK-293T cells by transient co-transfection with the packaging constructs psPax2 (Addgene #12260) and pMΔ2.G (Addgene #12259). Transfections were carried out using 2x HEPES Buffered Saline (Sigma, Cat. No 51558) and CaCl2 precipitation. Forty-eight hours later, culture supernatant was collected and filtered. Infections were carried out in the presence of 8 μg/ml polybrene (Sigma, Cat. No. 107689). At 48 hours, cells were selected for resistance to puromycin (Gibco, Cat. No. A11138) (10 μg/mL, or blasticidin (Gibco, Cat. No A1113903) (5 μg/mL), depending on the selection marker in the vector. Cells infected with multiple constructs, were selected for infection with the first construct, prior to the next infection. Transfection of lung adenocarcinoma cell lines with the pCMV HA-β-globin constructs were carried out, using the Lipofectamine 3000 Transfection Reagent (Invitrogen, Cat. No. 13778) and Opti-MEM Reduced Serum Medium (Gibco, Cat. no. 11058021), according to the manufacturer’s protocol.
Viruses, Virus propagation and titration
Vesicular stomatitis virus (Kenney et al., 201934), expressing GFP (VSV-GFP) was propagated and tittered in HeLa cells. Sendai virus (Kenney et al., 201934, Sermersheim et al., 202035), expressing GFP (SeV-GFP) was propagated in 10-day-old embryonated chicken eggs at 37°C for 40 hours and tittered on Vero cells. Influenza virus A/PR/8/1934 (H1N1) expressing GFP from promoter x (PR8-GFP) was propagated in 10-day-old embryonated chicken eggs (Charles River Laboratories) for 48 hours at 37°C and tittered in MDCK cells. Reovirus (Kenney et al., 201934, Sermersheim et al., 202035), was propagated in Vero cells for 16 hours and tittered also in Vero cells.
Virus infection and detection of infected cells by flow cytometry
NCI-H522 cells were infected with VSV-GFP or SeV-GFP (MOI 0.5) and they were harvested 16 hour later. Alternatively, they were infected with the Influenza A, PR8-GFP strain or the Reovirus (MOI 1.0) and they were harvested 24 hours later. NCI-H1299 cells were infected with VSV-GFP or SeV-GFP at an (MOI 0.25), or with the Influenza A, PR8-GFP strain (MOI 1.0) and they were harvested 24 hours later. Harvested cells infected with VSV-GFP, SeV-GFP and Influenza PR8-GFP, were fixed in 4% paraformaldehyde (Thermo Scientific), permeabilized with 0.1% Triton X-100 in PBS, and resuspended in 2% fetal bovine serum in PBS. Following permeabilization, reovirus-infected cells were stained with the T3D sigma 3 anti-reovirus antibody (DSHB Cat No. 10G10), followed by staining with an anti-mouse Alexa488-conjugated secondary antibody (Thermo Scientific, A-11029). VSV, SeV and Influenza A-PR8, infection rates were measured by counting cells expressing virus-encoded GFP. Flow cytometry was performed, using a FACSCanto II cell analyzer v2.3 (BD Biosciences) (https://www.bdbiosciences.com/eu/instruments/clinical/cell-analyzers/bd-facscanto-ii/m/744689/overview). Data were analyzed using the FlowJo software v9.3.3 (DB, Ashland, OR). The gating strategy is provided in Supplementary Figure 6.
Analysis of viral genome
Virus-infected shControl, shIWS1, shIWS1/WT-R and shIWS/MT-R NCI-H522 and NCI-H1299 cells were lysed and total RNA was extracted. Using these RNAs and virus genome specific oligonucleotide primers, we carried out qRT-PCR assays, as described in the corresponding sections. The virus-specific primers are listed in Supplementary Table 2. The sequences and genomic coordinates of the viral genes whose abundance was monitored by these assays are provided as Supplementary Data 2.
Subcellular Fractionation
5×106 cells were trypsinized, following 2 washes with ice-cold 1x PBS. The cell pellet obtained by a 5 min centrifugation at 12,000 x g, was resuspended in 1mL 1x PBS. Following this, the cells were washed twice with TD buffer (135mM NaCl, 5mM KCl, 0.7mM Na2HPO4, 25mM Tris-HCl) and then lysed using TD/1% NP-40/RVC (Ribonucleoside-Vanadyl Complex, NEB, Cat. No. S1402) in the presence of RNaseOUT™ Recombinant Ribonuclease Inhibitor (Thermo Fisher, Cat. No. 10777019). Following 10 minutes incubation on ice and centrifugation at 21,000 x g for 1 minute, the supernatant, which contains the cytosolic fraction, was aspirated, and kept on ice. The nuclear fraction was washed with TD/0.5% NP-40/RVC twice. Then, we used Trizol and a mixture of phenol-chloroform-isoamyl alcohol to isolate the RNA from both fractions. Isolated RNA was ethanol precipitated overnight in -80°C. cDNA was synthesized from 1.0 μg of total RNA, using oligo-dT priming and the QuantiTect Rev. Transcription Kit. Cytosolic and nuclear RNAs were measured by quantitative RT-PCR performed in triplicate, using the iTaq™ Universal SYBR® Green Super mix and a StepOne Plus qRT-PCR instrument. To validate the fractionation, we calculated the cytoplasmic/nuclear ratio of the GAPDH RNA. The exact values can be found in Supplementary Table 4. Data were normalized to 18S ribosomal RNA, which was used as an internal control. The primer sets used for all the all the quantitative RT-PCR assays are listed on the Supplementary Table 2.
RT-PCR and qRT-PCR
Total cell RNA was extracted using the PureLink RNA Kit (Invitrogen, Cat. No 12183018A). cDNA was synthesized from 1.0 μg of total RNA, using oligo-dT priming and the QuantiTect Rev. Transcription Kit (QIAGEN, Cat No. 205310). To monitor the RNA splicing of U2AF2, cDNA was used for RT-PCR. The abundance of RNA transcribed from a given gene, or from a given exon that may be alternatively spliced, was measured by quantitative RT-PCR. PCR reactions were performed in triplicate, using the iTaq™ Universal SYBR® Green Super mix (Biorad, Cat No. 1725121) and a StepOne Plus qRT-PCR instrument (Thermofisher), as described above. Data were normalized to hGAPDH or human 18S rRNA, which were used as internal controls. The primer sets used are listed in the Supplemental Table 2.
Immunoblotting
Cells were lysed in RIPA lysis buffer {50 mM Tris (pH 7.5), 0.1% SDS, 150 mM NaCl, 5 mM EDTA, 0.5% Sodium deoxycholate, 1% NP-40 and fresh 1x Halt™ Protease and Phosphatase Inhibitor Cocktails (Thermofisher, Cat. No 78444)}. Lysates were sonicated twice for 30 seconds and clarified by centrifugation at 18,000×g for 15 min at 4°C. The clarified lysates were electrophoresed (20μg protein per lane) in SDS-PAGE. Electrophoresed lysates were transferred to polyvinylidene difluoride (PVDF) membranes (EMD Millipore Cat No. IPVH00010) in 25 mM Tris and 192 mM glycine. Following blocking with 5% nonfat dry milk in TBS and 0.1% Tween-20, the membranes were probed with antibodies (at the recommended dilution), followed by horseradish peroxidase-labeled secondary antibodies (1:2500), and they were developed with Pierce ECL Western Blotting Substrate (Thermo Scientific, cat. no 32106). Antibodies we used for western blotting and chromatin immunocleavage (see below) are listed in Supplemental Table 1.
RNA Immunoprecipitation
Cell monolayers in 10cm plates were treated 1% formaldehyde (Sigma, Cat. No F8775) at 37oC for 15 minutes, to cross-link RNA to associated proteins. The cross-linking reaction was stopped by treatment with 0.125M Glycine for 5 minutes at room temperature. Cells were then scraped in 1 ml of a buffer consisting of 1x Phosphate Buffered Saline (PBS) / Nuclear Isolation Buffer (1.28M sucrose, 40mM Tris-HCl, 20mM MgCl2 , 4% Triton-X 100) / H2O (1:1:3 ratio). Following two additional washes, cell were lysed with RIP buffer {(150mM KCl, 25mM Tris-HCl, 5mM EDTA, 0.5mM DTT, 0.5% NP-40, supplemented with fresh 1x Halt™ Protease and Phosphatase Inhibitor Cocktails (Thermofisher, Cat. No 78444) and RNaseOUT™ Recombinant Ribonuclease Inhibitor (Thermo Fisher, Cat. No. 10777019) and the lysates were incubated on ice for 10 minutes. The lysates were clarified by centrifugation at 14,000 rpm, for 30 minutes at 4oC. A fraction of each sample was then precleared by incubation with protein A and salmon sperm DNA-bound agarose beads (Cell Signaling, Cat. No 9863), for 1 hour in 4oC. The precleared lysates were then incubated at 40C overnight with the immunoprecipitating antibody (Supplementary Table S1) or with the IgG isotype control {Rabbit Isotype Control (Thermofisher, Cat. No 10500C or Mouse Isotype Control (Thermofisher, Cat. No 10400C) and for 4 additional hours with Pierce™ Protein A/G Magnetic Beads (Thermofisher, Cat. No 88803). Beads were then washed four times with the RIP buffer and resuspended in 100uL RIP buffer. Next, the protein-RNA cross-linking was reversed by incubating the samples for 60 minutes at 70°C and following this, the RNA-protein complexes were eluted by treatment with 0.1% SDS 100 μL RIP buffer and treatment with proteinase K for 60 minutes at 55°C. The RNA was then extracted using a phenol-chloroform-isoamyl alcohol mixture and it was ethanol precipitated at -80°C overnight, in the presence of yeast tRNA (10mg/mL). The recovered RNA was reverse-transcribed with random hexamers and the abundance of cDNA corresponding to the RNA of specific target genes (IFNA1, IFNB, c-JUN and HSPB3) was measured by quantitative PCR using different sets of primers (Supplementary Table 2). PCR reactions in the RNA recovered from the immunoprecipitates and in 2% input RNA were carried out in triplicate, using the iTaq™ Universal SYBR® Green Super mix (Bio-Rad, Cat No. 1725121) and a StepOne Plus qRT-PCR instrument (Thermofisher). The data were analyzed the analysis file provided online by Sigma-Aldrich. (https://www.sigmaaldrich.com/technical-documents/articles/biology/chip-qpcr-data-analysis.html). SNRNP-70 binding in the human U1 snRNP gene, using the primers F: 5’-GGG AGA TAC CAT GAT CAC GAA GGT-3’, R : 5’-CCA CAA ATT ATG CAG TCG AGT TTC CC-3’, was used as the control for RNA IPs. The detailed protocol and buffer preparation can be found in the online protocols’ depository (Laliotis et al., 202151).
Chromatin Immuno-Cleavage (ChIC)
The binding of p-STAT1 on the ISREs in the promoters of the IFN-stimulated genes IRF1, IRF9, STAT1 and STAT2, was addressed by chromatin Immuno-cleavage (Skene et al., 201852, Laliotis et al., 20213). 2.5×105 cells were washed several times with wash buffer (20mM HEPES (pH 7.5), 150mM NaCl, 0.5mM Spermidine) in the presence of fresh 1x Halt™ Protease and Phosphatase Inhibitor Cocktails. Magnetic Biomag Plus Concanavalin A Beads (Bangs Laboratories, Cat. No. BP531) were activated with multiple washes using a binding buffer (20mM HEPES-KOH (pH 7.9), 10mM KCl, 1mM CaCl2 , 1mM MnCl2 ). Prior to use, the immunoprecipitation antibodies (Supplementary Table 1) or the Rabbit Isotype Control (Thermofisher, Cat. No 10500C), were diluted in 1:50 dilution in 50 μL antibody buffer (2mM EDTA (pH 8.0), 0.1% (wt/vol) digitonin diluted in wash buffer). Then, the activated beads were resuspended with the antibody buffer, containing the immunoprecipitated antibody, and mixed with the cell fraction. Following overnight incubation at 4°C, the immunoprecipitated were subjected to multiple washes with the wash buffer. Similarly, to the primary immunoprecipitating antibody, the Guinea Pig anti-Rabbit IgG (Heavy & Light Chain) secondary antibody (Antibodies-Online, Cat. No. ABIN101961) was diluted in 1:50 dilution in 50μL antibody buffer, was mixed with the immunoprecipitates and incubated for 4 hours at 4°C. Subsequently, the immunoprecipitates were subjected to multiple washes with the wash buffer and mixed with the CUTANA™ pAG-MNase (EpiCypher, Cat No. SKU: 15-1116) at 700 ng/mL. The targeted digestion was activated with 100mM CaCl2 and occurred by incubation on ice for 30 minutes. The reaction was terminated with addition of 2x stop buffer (340mM NaCl, 20mM EDTA (pH 8.0), 4mM EGTA, 0.1% (wt/vol) digitonin, 0.2 mg RNAse A, 0.02 mg Glycogen) and the chromatin fragments were released by incubation at 37°C for 10 minutes. Subsequently, the chromatin fragments were extracted with DNA Purification Buffers and Spin Columns (Cell Signaling Technologies, Cat. No 14209). Real-time PCR using different sets of primers (Supplementary Table 2) to amplify the ISRE genomic loci of several ISGs was carried out in the immunoprecipitated DNA, as well as in the IgG-immunoprecipitated DNA, by using the iTaq™ Universal SYBR® Green Super mix (Bio-Rad, Cat No. 1725121) and a StepOne Plus qRT-PCR machine (Thermofisher). The data were analyzed using the analysis substrate file provided online by Sigma-Aldrich, calculating the fold enrichment. (https://www.sigmaaldrich.com/technical-documents/articles/biology/chip-qpcr-data-analysis.html). This is based on the previously published protocol of ChIC assays.
Virus-induced cell death
To quantitatively measure virus-induced cell death, NCI-H522 and NC-H1299 cells were plated in 24-well plates and they were infected with VSV-GFP, or Reovirus at increasing MOI. Sixteen hours later (VSV-GFP), or 48 hours later (Reovirus), infected cells, and control uninfected cells were treated with resazurin, which is reduced to resorufin in viable cells, in a reaction that can be easily monitored because resorufin is fluorescent (λabs/λem = 571/585 nm). Control virus-infected and uninfected cells were treated with DMSO and their cell viability was measured with alamarBlue™ HS Cell Viability Reagent (Thermo Fisher Cat. No A50100). Cell death measured, based on the fluorescence emitted following the reduction of resazurin, in virus-infected and uninfected cells was normalized, relative to the cell death detected in DMSO-treated control cells. The acquisition was performed at 570 nm using a SpectraMax iD5 v1.6 (Molecular Devices San Jose, CA). The percentage of surviving cells, following virus infection with different MOI was determined based on the comparison of the normalized values in virus infected and uninfected cells. The results were plotted using non-linear regression in GraphPad Prism 8.4
Intronless genes and motif analysis
The sequences of all human intronless genes were retrieved from Ensembl (Howe et al., 202153) using biomaRt R (Durinck et al., 200954). Precisely, by using biomaRt, we selected all human genes which had a single exon reported on Ensembl. Then, we annotated all these genes by using org.Hs.eg.db R package (Carlson et al., 201955). All the non-annotated genes and low count genes in lung epithelial cells derived from ENCODE were filtered. Finally, we made a custom FASTA file containing the exon sequence of all annotated intron-less genes by using an ad hoc R script. For the motif analysis, the FASTA file was analyzed with the FIMO algorithm of the MEME suite (https://meme-suite.org/meme/tools/fimo) (Grant et al., 201126) for the identification of CAR-E, using the known motif : [B][CA][AT]GH[AT][CG][CG][AT][CG], following standard parameters. The results of the motif analysis are available as Supplementary Data 1.
Image acquisition and figure preparation
Western blot images were acquired, using the Li-Cor Fc Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE). Using a linear acquisition detection method, images were acquired in the 700 nm channel (protein ladder), in the 800 nm channel reduced background and increased sensitivity) and chemiluminescent channel (protein bands). For the DNA agarose gels, images were acquired using again the Li-Cor Fc Odyssey Imaging System and a linear image acquisition method in the 600 nm channel. All the images in this report were similarly acquired to ensure that the analysis is unbiased. The images were exported in high-quality image files (600 dpi png files), which were imported in in Adobe Illustrator 2021 (Adobe, San Jose, CA) for the preparation of the figures.
Human Tumor Samples
The 6 Lung Adenocarcinoma samples used in this report have been previously described in our earlier studies for the role of IWS1 in lung cancer (Sanidas et al., 20141, Laliotis et al., 20213). The tissues were obtained from the Tissue Bank of The Ohio State University, under the universal consenting and biobanking protocol, Total Cancer Care (TCC) and from the tissue bank of Tufts Medical Center, after written consent and review of the bioethics board committee. TCC is the single protocol used by the Oncology Research Information Exchange Network (ORIEN), which was formed through a partnership between the OSUCCC – James and Moffitt Cancer Center in Tampa, FL. For more information, please advise the Biospecimen Core Services facility of The Ohio State university Comprehensive Cancer Center (https://cancer.osu.edu/for-cancer-researchers/resources-for-cancer-researchers/shared-resources/biospecimen-services) and ORIEN project (https://cancer.osu.edu/for-cancer-researchers/resources-for-cancer-researchers/orien). The tumor samples in this study were provided as unidentified samples.
TCGA/RPPA analysis
TCGA data were downloaded from https://portal.gdc.cancer.gov/. Overall 658 TCGA-LUAD patients (all stages) were available. In addition to the RNASeq data, 516 of the 658 patients also had RPPA data on p-c-JUN S73 and IWS1. Heatmaps showing the distribution of the relative abundance of IWS1, IFNA1, IFNB1, JUN and HSPB3 and correlation co-efficience graphs were generated using the visualization tools of the Xena browser (http://xena.ucsc.edu) and GraphPad Prism 8.4.
Statistical analyses
All the statistical analysis were performed, using GraphPad Prism. Details on the statistical tests used, can be found in the corresponding figure legend. All the statistical analyses reports can be found in the Mendeley dataset where the source data of this report were deposited. (Laliotis et al., 202156, doi: 10.17632/853gfbbx7m.3)
Code availability
All the code used for the analysis in this report is derived from previously published reports. It is also explained and cited in the appropriate methods section.
Data availability
All the raw data underlying figures 1-8 (uncropped gel images, chart and bar data, qPCR, Flow Cytometry and plate reader data), derived from this report have been deposited in a publicly available Mendeley dataset (Laliotis et al., 202156, doi: 10.17632/853gfbbx7m.3). Specific P values are also included in this dataset. The list of human intronless transcripts, along with the results of the FIMO motif analysis for the position of CAR-E in each gene, are provided as Supplementary Data 1. The FASTA file of all the selected human intronless genes for the motif analysis are provided in the Mendeley dataset (Laliotis et al., 202156, doi: 10.17632/853gfbbx7m.3). The latest version of Ensembl database can be downloaded through the Ensembl project (http://useast.ensembl.org/info/data/ftp/index.html) or github (https://github.com/Ensembl/ensembl-hive). All the uncropped WB and PCR gels are provided as Supplementary Data 3. All the chart and bar data are provided as Supplementary Data 4.
Third Party Images
The schematic for β-globin CAR-E reporter (Figure 5), the cartoon outlining the experiment of ISG induction by infection of NCI-H522 cells with VSV-GFP and the effect of the treatment of naïve NCI-H522 cells with culture supernatants of the VSV-GFP-infected NCI-H522 cells (Fig. 6b), as well as the cartoon summarizing the data in this report (Fig. 8), were created with BioRender.com (https://biorender.com). Georgios I. Laliotis created the images. Philip N. Tsichlis and Georgios I. Laliotis edited them in their final version. The authors used a paid student plan promo (legacy) with the Agreement Numbers OO22MW9YPA, UR22MWA47E, SC22PV8Z05 for Figure 5, 6 and 8, respectively, for granted publication access.
Statistics and reproducibility
The experiments in Fig. 1a-d, 2a-f, 3a-b, 4a-b, 6a-d, Supplementary Figure 5a-c, 7a-b, Supplementary Figure 7a-c, were performed at least in 3 independent biological experiments. The data in figure 4b (patients’ data) were performed once, using 3 patients/group. The human β-globin CAR-E reporter transient transfection in Figure 5 and Supplementary Figure 4 and the Poly (I:C) and IFNβ1 stimulation in Figure 1d and Supplementary Figure 5, respectively, were performed twice. All the attempts at replications were successful. All the statistical analysis was performed in GraphPad Prism, as described in the corresponding section. All the statistical analysis reports can be found in the Mendeley dataset (Laliotis et al., 202156, doi: 10.17632/853gfbbx7m.3).
Author Contributions
G.I.L. Conceptualization, overall experimental design. Performed experiments, analyzed the data, prepared figures and wrote the manuscript. A.D.K. Designed and performed all the infections with viral strains, optimized, performed and analyzed the flow cytometry experiments and edited the manuscript E.C. Designed and performed with G.I.L the time point interval experiment for p-STAT1 activation and Caspase-mediated death in Figure 6 and Figure 7, edited the manuscript A.O. Assisted to the time point interval experiment for Caspase-mediated death and edited the manuscript A.K.K Performed the cloning of the type I IFN vectors for RNA Pol II and III promoter expression, performed RT-PCR experiments A.L.F. Bioinformatics analyses of TCGA data and intronless genes mining, edited the manuscript. V.A. Assisted to the time point interval experiment for p-STAT1 activation and edited the manuscript J.D.B Advised on the design of experiments and edited the manuscript C.T. Advised on the design of experiments and edited the manuscript. L.S. Advised on the design of experiments and edited the manuscript V.C. Contributed to overall experimental design, edited the manuscript. J.S.Y Designed the viral infection experiments and contributed to overall experimental design, edited the manuscript P.N.T. Conceptualization, overall experimental design, project supervision, manuscript writing and editing.
Competing Interests
The authors declare no competing interests.
Acknowledgments
The authors wish to thank all the members of the Tsichlis Lab for helpful discussions. We also thank Dr Samir Achaya for reviewing the manuscript before the submission. This work was supported by the NIH grant R01 CA186729 to P.N.T., the NIH grant R01 CA198117 to P.N.T and V.C, by start-up funds from the OSUCCC to P.N.T, from the National Center for Advancing Translational Sciences grant KL2TR002734 to L.S. G.I.L was supported by the Pelotonia Post-Doctoral fellowship from OSUCCC.
Footnotes
↵10 Lead Contact
References




