

Abstract
Microbial transmission from parent to offspring is hypothesized to be universal in vertebrates. However, evidence for this is limited as many clades remain unexamined. Chondrichthyes, as one of the earliest–branching vertebrate lineages, provide an opportunity to investigate the phylogenetic breadth of this hypothesis. To assess the potential for bacterial transmission in an oviparous chondrichthyan, we used 16S rRNA amplicon sequencing to characterize the microbial communities associated with the skin, gill, and egg capsule of the little skate, Leucoraja erinacea, at six points during ontogeny. We identify site-specific microbiomes dominated by the bacterial phyla Proteobacteria and Bacteroidetes, a composition similar to, but distinct from, that of other chondrichthyans. Our data reveal that the skate egg capsule harbors a highly diverse bacterial community–particularly on the internal surface of the capsule–and facilitates intergenerational microbial transfer to the offspring. Embryonic skin and external gill tissues host similar bacterial communities; the skin and gill communities later diverge as the internal gills and skin denticles develop. Our study is the first exploration of the chondrichthyan microbiome throughout ontogeny and provides the first evidence of vertical transmission in this group, which may be the primary mechanism for the signature of phylosymbiosis previously observed in elasmobranchs.
Introduction
Host-associated microbial communities are often species and tissue-specific due to complex local interactions between hosts and microbes (1,2). In some clades, microbiome composition closely tracts host phylogeny over evolutionary time, resulting in long-term eco-evolutionary relationships (3). This pattern, known as phylosymbiosis, can result from either horizontal microbial transmission facilitated by ecological similarity between related host species or vertical microbial transmission from parents to offspring over many generations (3). Microbes can be horizontally recruited from the surrounding environment through the host’s contact with fluids, substrates, diet, or social interactions, allowing for rapid changes in community composition in response to external conditions over the lifespan of an individual. Alternatively, vertical transmission could offer a significant selective advantage by ensuring the colonization of progeny by symbiotic microbial taxa and may be universal in vertebrates (4), but this hypothesis awaits testing across a wider range of species. The relative contributions of these two transmission modes likely covary with life history traits, balancing the need for intergenerational continuity of genomic information with the capacity for rapid environmental responsiveness.
Next-generation sequencing has facilitated the characterization of a broad range of microbiomes across an increasing diversity of host species; nonetheless, many marine clades remain understudied. Chondrichthyans—the earliest branching of the extant, jawed-vertebrate lineages —constitute one of the major divisions of vertebrates (5). To date, only a limited number of metagenomic studies of chondrichthyan microbiomes have been conducted (6–11). Existing studies of species belonging to subclass Elasmobranchii, which includes sharks, skates, rays, and guitarfish, and are skewed towards the skin or gut microbiota of pelagic sharks (6–10), with the remaining focusing on the skin of select ray species (10,11). These datasets show that elasmobranch skin microbiomes differ from the surrounding environment and are primarily dominated by the phyla Proteobacteria and Bacteroidetes, similar to the skin microbiomes of other marine species (12–14). However, this work is limited to adult elasmobranchs, providing no direct information on juvenile microbiota or intergenerational transmission in chondrichthyans. Collection of data across ontogeny is needed to understand the signature of phylosymbiosis previously reported in elasmobranchs (10).
Oviparity is present in nearly half of chondrichthyan species and may be the plesiomorphic reproductive mode for this clade (5,15). Unlike in viviparous taxa—where microbes can be transferred during birth—oviparity requires indirect microbial transfer through the egg for intergenerational transmission in the absence of parental care. The capacities of different egg types to transmit microorganisms remains an open question (16). Previous work on egg-mediated microbiome establishment in vertebrates provides evidence for mixed models of microbial transmission. In birds and reptiles, some egg-mediated intergenerational transmission occurs, both within the egg (17) and on the eggshell due to maternal contact during incubation (18). In contrast, fish eggs are primarily colonized by microbes from the surrounding water, although vertical transmission cannot be ruled out (19). Rather than relying on egg-mediated transmission, fish may rely on other methods. For example, the gut microbiomes of discus fish are derived from both the surrounding water and the parental mucus fed to offspring (20). These studies suggest that while eggs can facilitate intergenerational microbial transfer, parental care or environmental sources may be more important in many taxa, depending on life history.
Skates (family: Rajidae) are egg-laying elasmobranchs that protect their embryos inside egg capsules, colloquially known as a mermaid’s purses (21). These capsules are laid on the seafloor and the embryos develop inside for months to years depending upon the temperature (22,23) and species (24). While initially sealed to the environment, slits at the anterior and posterior ends of egg capsule open up part way through development allowing seawater to flow through (25). The potential effects of this environmental shift on the microbiome and development are unknown. Upon hatching, juvenile skates are self-sufficient, with no known parental care (5). These life history traits–long embryonic development and lack of parental care–pose potential obstacles to vertical microbial transmission in members of this clade. Thus, skates can be a powerful system for testing vertical transmission because confounding parental sources are minimized, and any transmitted microbial community must be stable over a substantial period of time.
The little skate, Leucoraja erinacea, is a model system for research in embryology and development (23,26–29) with a sequenced genome (30). Little skates are common in the North Atlantic and easy to obtain though sampling and breeding methods implemented in that region. Little skate embryos have gestational periods inside the egg capsule of 22–54 weeks depending on the season (22). Embryogenesis is divided into thirty-three stages, based on morphological features (31,32). Here, we use 16S rRNA amplicon sequencing to describe and track changes in bacterial diversity throughout little skate ontogeny by sampling the microbiome of the skin and gills, as well as the internal liquid and internal surface of the egg capsule at six developmental stages. These stages are (i) stage 0, when capsules are freshly laid and fertilization cannot be visually confirmed; (ii) stage 16, an early stage when the embryo can be visually identified; (iii) stage 26, by which external gill filaments have formed and the egg capsule is still sealed; (iv) stage 30, when the egg capsule starts to open and the gills remain external; (v) stage 33, by which time the egg capsule is open and the embryo is fully formed with internal gills; and (vi) adult. We present the first evidence of vertical microbial transmission in an oviparous chondrichthyan by tracking community continuity between these stages.
Materials and Methods
Ethics Statement
All procedures were conducted in accordance with Marine Biological Laboratory IACUC protocol 19-42.
Sample Collection
All skates used in this study were housed in 15°C filtered seawater at the Marine Resources Center (MRC) of the Marine Biological Laboratory, Woods Hole, MA. All embryos were laid within the facility tanks. Samples were collected from adult females (n=4) by swabbing the gills and the skin around the cloaca. Egg capsules were sampled at five times points: stages 0, 16, 26, 30, and 33 (n=4 each, n=20 total) as per refs. (31,32). Egg capsules were not associated with any particular adult female. Eggs capsules were opened with a razor blade and the embryos euthanized by cervical transection. At each stage, samples of the internal liquid (n=20) were collected using a 1000 mL pipette and the inside of the egg capsule was swabbed (n=20). All samples at stage 33 were open, as were two samples (A & D) at stage 30. All other samples were closed. Samples where the egg capsule slits were already open to the environment were drained into a collection tube before the egg capsule was opened. At stages 26, 30 and 33, gill filament samples and tail clippings (∼2 cm long) were collected (n=4 each, n=24 total). Control samples included hand swabs of A.S.O and K.M. and bench swabs before sample processing both on the day of sample collection and again on the day of DNA extraction (n= 6 total). A sample of the bench after sterilization (n=1) was taken on the day of sample collection as well. To broadly sample the marine bacteria likely to be encountered by skates in the MRC, 1 mL of water was collected from (i) ambient-temperature water tanks (n=2); (ii) 15°C tanks housing the skates (n=2); (iii) ocean water from the dock neighboring the pump into the MRC (n=2); and (iv) the bucket used to transfer the skate embryos from the MRC to the dissection station was collected before (n=1) and after sampling (n=1). Prior to sample collection, the bench and all dissection tools were sterilized using Clorox bleach, followed by 70% ethanol. These surfaces were resterilized with 70% ethanol between each egg capsule and with bleach and ethanol between every four egg capsules. All skate samples were collected on the same day and egg capsules were opened in a randomly selected order.
The DNeasy PowerSoil Kit (Qiagen, Hilden Germany) was used for isolation of the microbial DNA from each sample. FLOQSwabs (COPAN, Murrieta CA) were used to collect all swab samples. Swabs trimmed to fit or tissue samples from the gills and tail were placed directly in the PowerSoil Kit PowerBead tubes after collection. For all liquid samples, 200 µL of the sample was added to the corresponding tube. After collection, samples were left at −20°C overnight prior to completion of the extraction protocol. Extraction continued according to the manufacturer’s instructions. The negative control and post-cleaning bench swab failed to amplify, suggesting our sterilization technique was effective. Pre-cleaning bench samples were thus unlikely to have contaminated other samples and were excluded from further analysis.
Sequencing and Library Preparation
The V4-V5 region of the 16S gene was amplified and sequenced at the Keck Environmental Genomics Facility at the Marine Biological Laboratory as described (33) and uploaded to the Visualization and Analysis of Microbial Population Structures (VAMPS) website (https://vamps2.mbl.edu/) (34). Denoising, classifying, and alpha and beta diversity methods were all implemented using QIIME2 version 2019.10 (35). Demultiplexed paired-end sequencing data from VAMPS was denoised without any trimming using the DADA2 (36) plugin to QIIME2 and the resulting amplicon sequence variants (ASVs) were classified by training Naive Bayes classifier using the SILVA (132 release) 16S only, 99% identity clustered sequences (37–39). ASVs were collapsed to a maximum specificity of seven taxonomic levels, which corresponds to the species level.
Microbial Community Analysis
Data were normalized using transform_sample_counts in Phyloseq (40,41) by dividing the count per ASV within a sample by the total count for that sample, followed by multiplying this ratio by 10 000. QIIME2 was used to calculate Shannon and Chao1 alpha diversity indices (42) and the Bray-Curtis dissimilarity index for beta diversity (43). Between-group significance levels for alpha and beta diversity were assessed using Kruskal-Wallis (44) and PERMANOVA (45) tests, respectively. Taxa comprising the common core microbiome (46) of the egg capsule, combined external gill and embryonic skin, internal gill, and adult skin were identified using the feature-table core-features function in QIIME2 at a threshold of 75% presence. This threshold was chosen to prioritize taxa with high levels of occupancy in tissues of interest. Given our small sample size for each tissue and stage, thresholds higher than 75% result in very few core taxa, while lowering this threshold results in rapid increases in the number of core taxa. Significantly abundant taxa were identified using LEfSe (Linear Discriminant Analysis (LDA) Effect Size) implemented in the Galaxy web application (http://huttenhower.org/galaxy/) (47) using a P-value cut-off of 0.05, an LDA score cut-off of 2, and a one-against-all strategy. Microbial source tracking for each stage was performed in R v3.6.2 (48) using FEAST with pooled source samples from the preceding stage (unless otherwise noted), water, and the investigators’ hands (49).
Metagenome Prediction and Differential Abundance Testing
PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2) was used to predict Kyoto Encyclopedia of Genes and Genomes (KEGG) ortholog (KO) groups for each sample (50). A pairwise PERMANOVA test was implemented using the RVAideMemoire (ver. 0.9-77) R package with 100 000 permutations to test for significant differences in the predicted metagenome between sample types (51). Significantly over-represented pathways were identified using LEfSe with samples normalized to 1 million, a P-value cut-off of 0.05, and LDA score cut-off of 2. Pathways were identified using the KEGG Mapper –Reconstruct Pathway online tool (https://www.genome.jp/kegg/tool/map_pathway.html). R Studio (Version 1.2.5033) with the ggplot2 package (52) was used to produce all figures.
Data Availability
Raw sequencing data is available at the NCBI Short Read Archive (PRJNA688288). All scripts are available online at https://github.com/kmmika/Intergenerational-microbial-transmission-in-the-little-skate-Leucoraja-erinacea.
Results
Eighty-four out of eighty-eight samples successfully amplified and were sequenced to produce a total of 3 516 842 reads, which mapped to 41 486 ASVs (Supplementary Table 1). These ASVs collapsed into 2 255 unique classifications. Each sample contained between 170 and 147 651 reads, with a median value of 36 480. ASV assignments ranged from 1 to 33 211 reads, with a median value of 29.
Taxonomic characterization of the L. erinacea microbiome
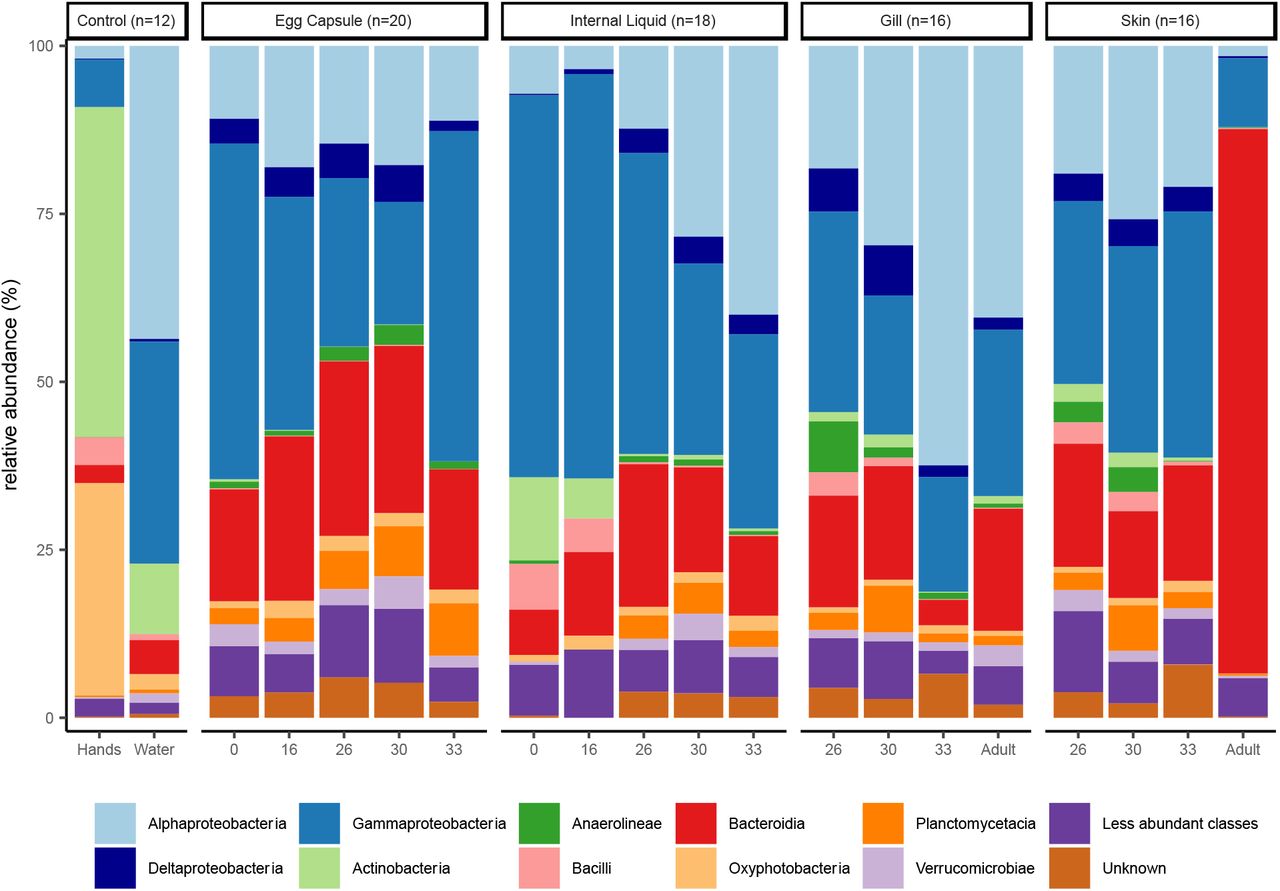
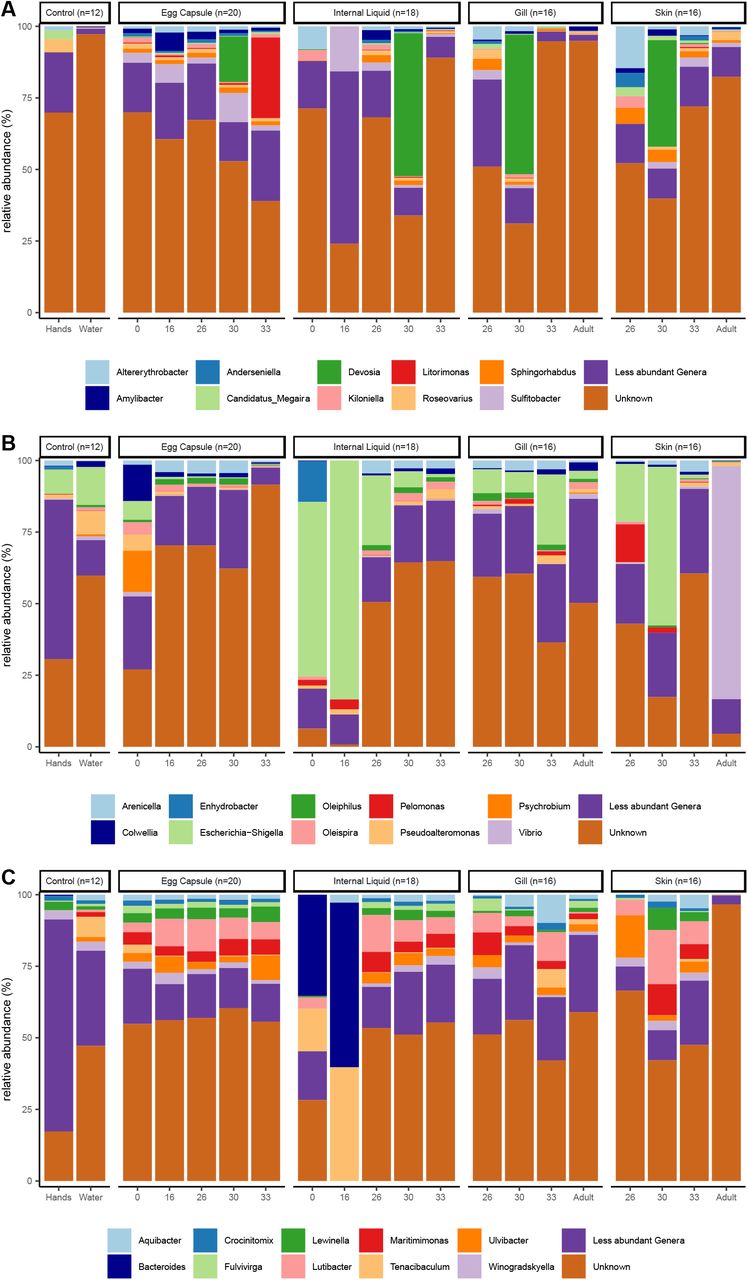
Skate and egg capsule samples are dominated by ASVs of the phyla Proteobacteria (58% of ASVs), Bacteroidetes (21%), and Planctomycetes (5%). The dominant classes identified are Gammaproteobacteria (33%), Alphaproteobacteria (21%), Bacteroidia (21%), and Planctomycetacea (4%) (Fig. 1). Adult skin samples are uniquely enriched for Bacteroidetes, which accounts for 81% of the bacterial community, while in all other samples Proteobacteria are most abundant. Within the control samples, the water samples are dominated by Proteobacteria (87%), followed by Bacteroidetes (5%), Cyanobacteria (2.6%), and Actinobacteria (2%), while the investigators’ hand samples are quite distinct from all other samples and are dominated by the phyla Actinobacteria (49%), Cyanobacteria (31%), Proteobacteria (9%) and Firmicutes (5%). Fewer ASVs can be categorized at increased phylogenetic resolutions, with <98% being classified at the phylum level, 97% at the class level, <83% at the order level, <70% at the family level and <60% at the genus level. The most common families identified are Rhodobacteraceae (8.1%), Flavobacteriaceae (6.9%), Enterobacteraceae (6.1%), Saprospiraceae (3.2%), and Devosiaceae (3.2%) (Supplementary Fig. 1). The most common genera are Escherichia-Shigella (6.1%), Cutibacterium (3.2%), Devosia (2.0%), and Lutibacter (0.9%) (Supplementary Fig. 2).
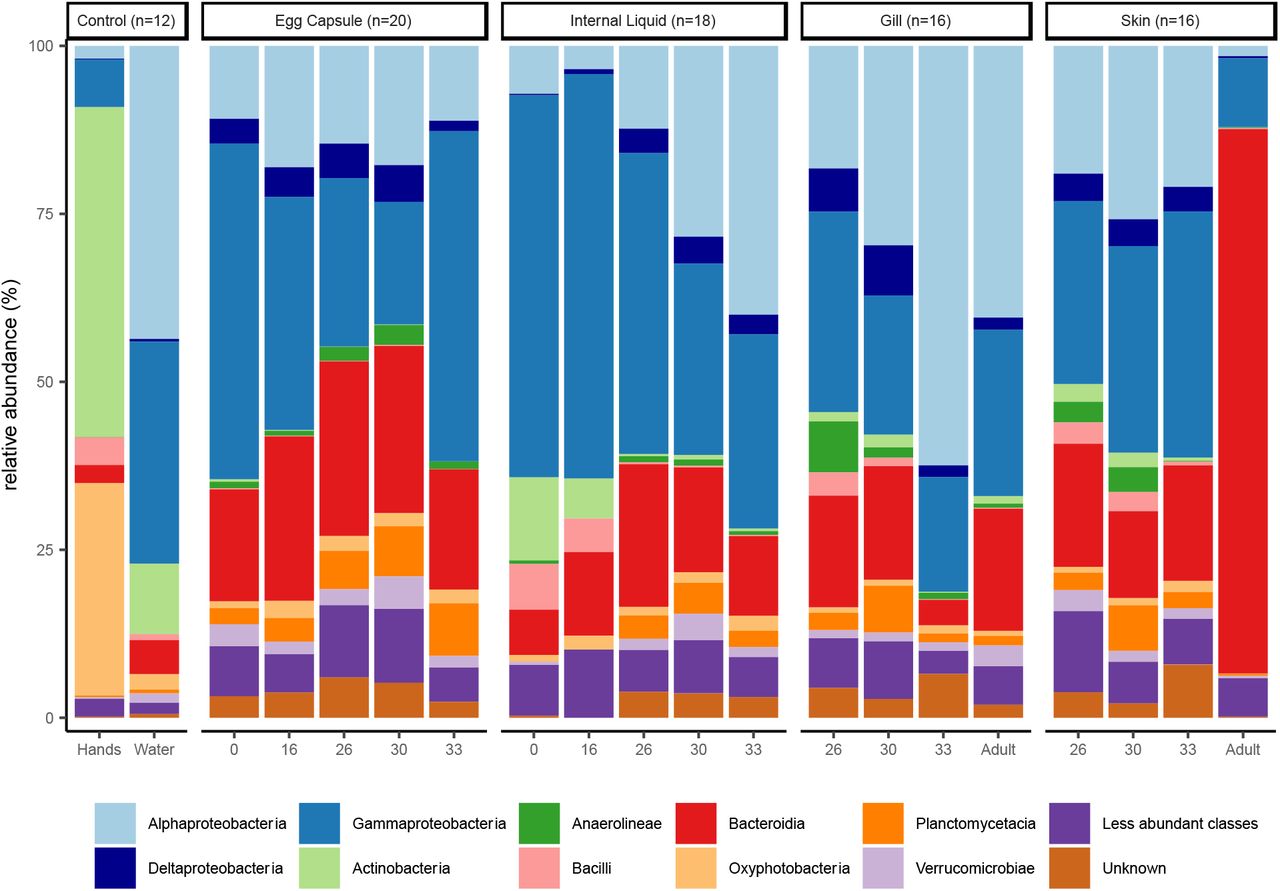
Relative abundance of the top ten bacterial classes in the dataset are shown for each site and timepoint as well as for water and hand controls. Classes of phylum Proteobacteria are shown in blue, other classes are ordered alphabetically.
Beta and alpha diversity of skate bacterial communities
Beta diversity by sample tissue and stage was assessed using Bray-Curtis dissimilarity (Fig. 2). Due to the low number of internal liquid samples at stage 16 which successfully amplified (n=2), these samples cannot be statistically differentiated from other samples but cluster tightly with stage 0 internal liquid. Water is likewise distinct from all skate tissues (q<0.05) except internal liquid from stage 33 embryos (q<0.08), at which time the internal liquid contains a significant amount of water due to the egg capsule opening. All egg capsule samples form a tight cluster distinct (q<0.08) from all other samples except mid-stage internal liquid (stages 16-30; q<0.50). Gill tissue samples split into two clusters (q <0.05): the external, embryonic gill stages 26-30, and the internal, later stages (33-Adult). Internal gill samples are distinct from egg capsule (q<0.05) and late skin samples (stage 33-Adult, q<0.05) while external gill samples (stage 26-30) cannot be distinguished from early skin samples (stages 26-30; q>0.20). Stage 30, 33 and adult skin are all significantly different (q<0.05) from each other. Adult skin is distinct from all other samples (q<0.05) and separates from all others along PC3 (Fig. 2B). Samples at stage 30 show no clear separation on open or closed status for any of the tissues examined.

PCoA analysis (Bray-Curtis) of skate and control samples. PC1 versus PC2 (A) and PC1 versus PC3 (B).
We next stratified the data into individual tissues and reran Bray-Curtis dissimilarity analyses on the reduced datasets (Fig. 3). Egg capsule samples, which have indistinct subclustering in Fig. 2, show more substructure when analyzed independently, with stages 0, 30, and 33 forming unique subclusters (q<0.05; Fig 3A). Internal liquid has no statistically significant subclusters (q>0.09; Fig. 3B). Bray-Curtis dissimilarity analysis on all gill samples supports the internal and external gill subclusters seen in the full dataset (Fig 3C; q<0.05). Adult skin is distinct from all other skin (Fig. 3D; q<0.05) along PC1 which explains 36.5% of the variance within skin samples. Due to high variability in stage 26 and 30 skin, stage 33 skin is statistically differentiated (q<0.05) but clusters tightly with the majority of these samples.
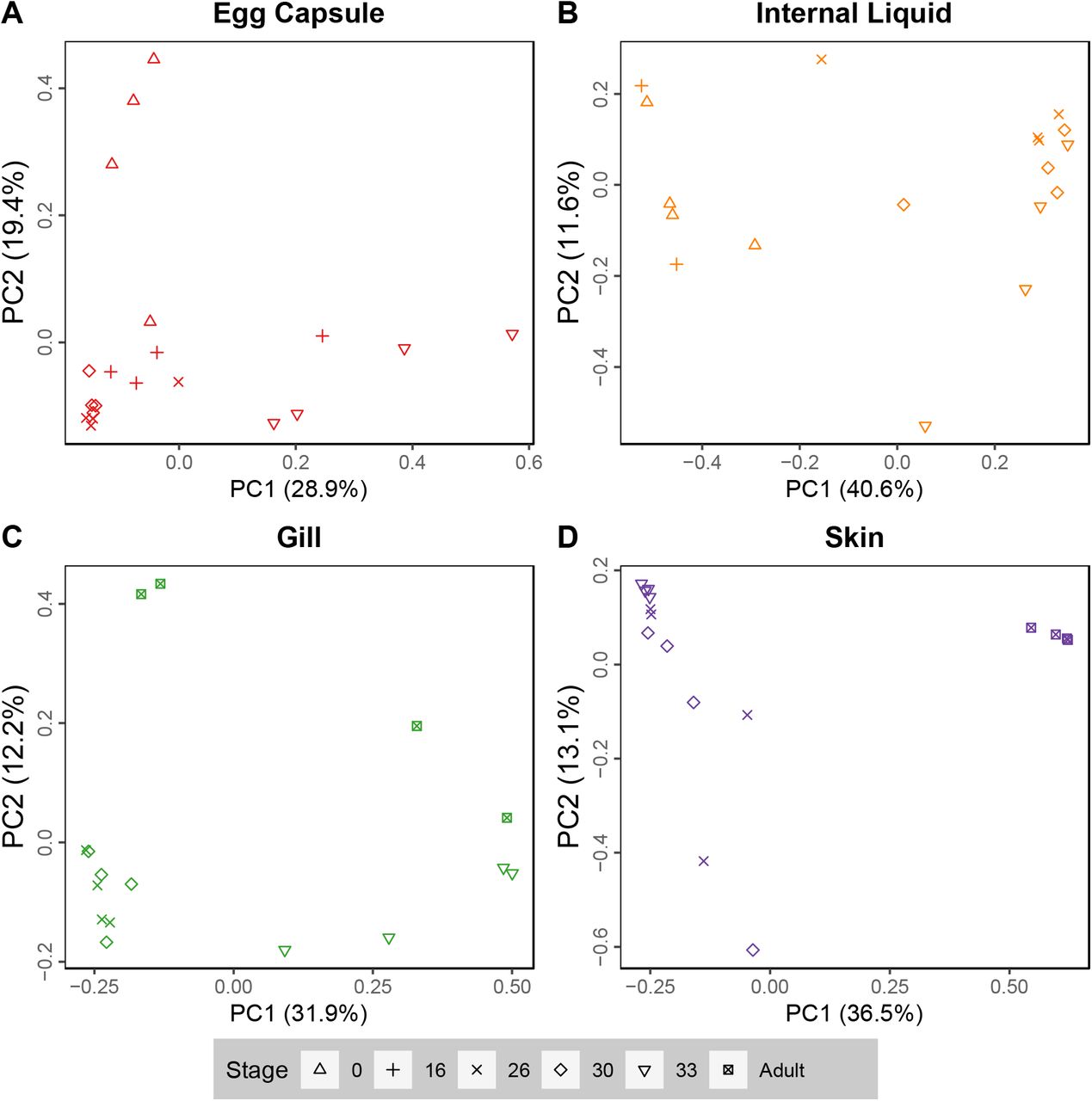
PCoA analysis (Bray-Curtis) plots of PC1 versus PC2 for (A) egg capsule, (B) Internal liquid, (C) gill, and (D) skin samples.
Shannon and Chao1 alpha diversity indices were calculated for all samples to assess species evenness and richness, splitting gills into external (stages 26-30) and internal (stages 33-Adult), as well as skin into embryonic (stages 26-33) and adult (Fig. 4). Egg capsule samples are significantly more diverse than skin, internal gill, internal liquid, hands, and water by Shannon and Chao1 (q<0.05, pairwise Kruskal-Wallis), but only significantly different from external gill by Chao1 (q<0.05). Adult skin was significantly less diverse than embryonic skin, external gills, and internal liquid (q<0.05) using the Shannon index but not Chao1. No other pairwise comparisons were significant using either metric.

Boxplots of Shannon (A) and Chao1 (B) alpha diversity metrics for each sample site. Datapoints shown in black. Colored points are statistical outliers. Stage is indicated by shape. Grey: experimenters’ hands, blue: water, red: egg capsule, orange: internal liquid, green: gill, and purple: skin.
Microbial source identification for each stage
We performed FEAST source tracking on each stage to evaluate the potential for intergenerational microbial transfer in the little skate. Samples from the preceding stage were used as source pools, except at stage 0 for which adult tissues were used as the source pools (Fig. 5). Water samples were included as an environmental source pool, and the experimenters’ hands were also considered as a source of potential contamination. However, hand samples are a negligible source for all samples except stage 0 internal liquid (hand contribution 21.6±5.5%, mean±standard deviation, Fig. 5A). Environmental water rarely contributes to the little skate microbiome; the exceptions are stage 0 (egg capsule 18.7±24.4%, internal liquid 16.0±18.8%), and stage 33 internal liquid (37.1±38.5%). In addition to water, the microbiome at stage 0 is primarily derived from an adult gill-like community (egg capsule 47.4±16.2%, internal liquid 5.6±7.2%) or an unknown source (egg capsule 32.7.4±14.2%, internal liquid 51.1±31.4%, Fig. 5A). At stage 16, the egg capsule and internal liquid microbiomes are both largely conserved from the same tissue of the previous stage (50.6±10.7% and 35.9±34.8% continuity, respectively, Fig. 5B). At stage 26, egg capsule from the previous stage is the dominant bacterial source for all tissues (60.8±16.7%, Fig. 5C). At stage 30, egg capsule and internal liquid are sourced primarily from stage 26 egg capsule (57.1±8.5%), but gill and skin are sourced from a combination of the egg capsule, internal liquid, and gill of the previous stage (egg capsule 20.8±13.1%, internal liquid, 14.7±12.8%, gill 12.0±6.0%, Fig. 5D). At stage 33, egg capsule remains contiguous (36.7±21.3%) while the internal liquid draws from egg capsule (24.3±21.4%) and internal liquid (13.1±9.1%) sources along with environmental water (Fig. 5E). The stage 33 gill microbiome, comprised of the earliest internal gill samples, is largely of unknown source (75.5±12.0%) while the skin is primarily derived from egg capsule bacteria (36.3±7.0%). Adult gill has small contributions from all stage 33 embryonic tissue types (egg capsule 9.1±5.6%, gill 2.4±2.7%, internal liquid 15.0±9.3%, skin 10.1±10.1%) but most sources are unknown (53.3±22.0%). The adult skin bacterial community is primarily derived from that of the late embryonic egg capsule (84.1±11.4%) (Fig. 5F). When stage 0 is used as the source pool for adult tissues instead, egg capsule is the dominant source for the skin (54.8±42.6%) and gills (29.7±17.0%), highlighting the continuity of sourcing from the egg capsule.

Boxplots showing source contributions to the bacterial community of the skate microbiome at stage 0 (A), stage 16 (B), stage 26 (C), stage 30 (D), stage 33 (E) and adult (F) estimated using FEAST. Stage 0 used adult tissues as the source pools. Source contributions to adults (F) are shown for stage 33 (top) and stage 0 (bottom) source pools. Sources are colored as in Fig. 2. Letter codes refer to source pools from the previous stage or controls. H: Experimenters’ hands, W: water; EC: egg capsule, IL: egg capsule internal liquid, G: gill, S: skin, U: unknown source.
Identification of the skate core microbiome
For subsequent analyses, we grouped samples of interest into four statistically significant and biological relevant groups: (1) egg capsule (all stages), (2) external gills (stages 26-30) and embryonic skin (stages 26-33) together, (3) internal gill (stage 33-Adult), and (4) adult skin. Due to heterogeneity in the composition of the internal liquid throughout ontogeny and lack of large source contributions to other tissues, these samples are not considered in further analyses.
To extract the core microbiome of the little skate from our dataset, we identified taxonomic groups classified to the most specific level possible which were present in 75% or more of the samples comprising the four groups specified above (Supplementary Table S2). Egg capsule samples have a rich core microbiome with 212 identified taxonomic groups. Skate samples have smaller core microbiomes: combined external gill and embryonic skin has ten, internal gills have twenty-two, and adult skin has fifty-six groups. There is high overlap between the core microbiomes of these tissues (Table 1) and only egg capsule and adult skin house unique taxa (Supplementary Table S2).
List of taxa identified as part of the common core microbiome at a 75% presence cut-off in a least two of the following tissues: egg capsule, combined external gill (stages 26-30) and embryonic skin, internal gill (stage 33-adult), and adult skin.
We cross-referenced the genera identified as core to only the adult skin with the full taxa list for all samples in order to identify taxa exclusively present on adult tissues. This comparison identified six exclusively adult taxa. Undibacterium is the only genus found in all adult skin and gill samples that does not appear in any egg capsule or embryonic sample. Spiroplasma, Salimicrobium, and Proprionivibrio were each found in a single adult gill sample and in the adult skin core microbiome. Sulfurospirillum sp. SM-5 and Aeromonas were unique to adult skin tissues.
Differential abundance of bacterial taxa and predicted functions
For each of the four sample groups in Table 1, we used LEfSe analysis to identify uniquely enriched taxa (P<0.05, LDA>2; Supplementary Fig. 3). Adult skin is enriched in Bacteroidia, Vibrio, and Mycoplasma agassizii. Embryonic skin and the external gills are enriched in Sphingomondales, Flavobacteriaceae, and Escheria-Shigella. Internal gills are enriched in Rhodobacterales and Alphaproteobacteria of SAR11 clade 1. Finally, the egg capsule is characterized by higher abundance of Alteromonadales, Pirellulales, Saprospiraceae, and Verrucomicrobia.
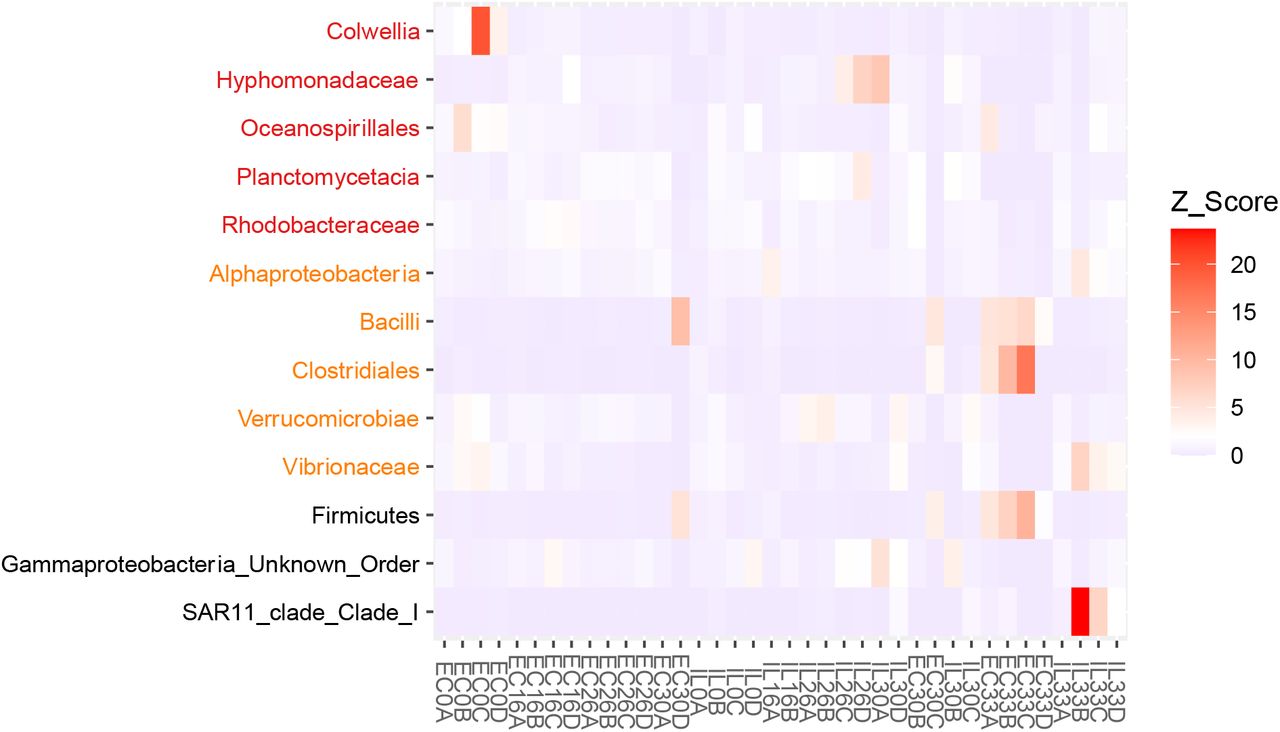
Additionally, we used LEfSe to identify shifts in taxa abundance associated with the opening of the egg capsule slits. We compared open and closed samples of the egg capsule, internal liquid, and a combined set of the two tissues. While a few taxa were identified as statistically significant, closer inspection of abundances in each sample did not exhibit the expected pattern of similar levels of abundance across all samples in one condition compared to the other. Instead, significance was driven by differences in group means due to a few samples with high abundances in either the open or closed condition (Supplementary Fig. 4).
We predicted functional differences between tissue types using PiCRUSt2 to generate KEGG Orthology (KO) gene predictions for each sample. Egg capsule, adult skin, internal gill, and combined external gill and embryonic skin bacterial communities have significantly different predicted functional properties (FDR-adjusted, P<0.01 for all, pairwise PERMANOVA). The most differentially abundant predicted genes for each of these groups were identified using LEfSe. Enrichments for each tissue group mapped predominantly to metabolic pathways using KEGG Mapper. Complete lists of predicted genes and enriched pathways for each group can be found in Supplementary Table 3.
Discussion
Our results show that the phyla Proteobacteria and Bacteroidetes comprise most of the bacterial communities associated with the little skate, as has been shown for other chondrichthyans (7,8,10,11). Adult skate skin within our study has a uniquely high proportion of Bacteroidetes (>50%) compared to all other batoids (10,11). Within Proteobacteria, relative proportions of each Class vary between chondrichthyan species (7,8,10,11). Below the phylum-level, there is evidence of unique site-specific communities in the little skate. While all skate samples included Gammaproteobacteria, Flavobacteriaceae, Pirellulaceae, Rhodobacteraceae, and Saprospiraceae, we are unable to identify a common core microbiome below the family level for all skate samples across all developmental time points. Instead, our data suggest that early embryonic tissues support similar bacterial communities which differentiate into distinct internal gill and adult skin microbiomes later in development.
We sampled two parts of the egg capsule: the inner surface of the capsule and the internal liquid, which fills the space in the egg capsule not occupied by the developing embryo and yolk. The microbiome of the egg capsule has the highest taxonomic richness of any tissue sampled and has a complex core microbiome. This provides evidence that the egg capsule is a rich reservoir of bacteria for the developing embryo, similar to the dense microbiome observed in squid egg capsules (53,54). This diversity likely takes the form of a biofilm lining the inside of the egg capsule, a hypothesis that is supported by an enrichment of processes related to biofilm formation such as pilus assembly and fimbrial biosynthesis (55,56). While the microbiome of the internal liquid fluid is generally similar to egg capsule samples, there appears to be a collapse of Actinobacteria and Bacilli after stage 16, both rare in the egg capsule, with relative replacement by Bacteriodia and Planctomycetacia, both more abundant in the egg capsule. Given the relatively limited quantity of internal liquid, many bacterial taxa present initially are likely outcompeted by more abundant strains living on the substrates provided by the egg capsule, yolk, and embryo. These initially abundant strains are likely able to endure despite opening of the egg capsule to the environment. Indeed, upon closer analysis, we find no taxa which undergo consistent shifts in abundance in either the egg capsule or internal liquid upon opening.
Gills are the primary site of gas and waste exchange with the environment, offering a unique habitat for microbial life. Little skate embryos develop transient external gill filaments between stages 25 and 32, which later regress into the body to form the adult internal gills (32). During early stages, we find that the gill microbiome is undifferentiated from that of the embryonic skin. The mature gills, however, harbor a distinct microbiome, which is enriched for Rhodobacterales and Alphaproteobacteria of the SAR11 clade. These taxa are also enriched in the gills of reef fish, suggesting that marine vertebrate gills provide similar microbial environments (57). Additionally, gills are the largest site of urea loss in skates (58). This makes adult gill tissue particularly well-suited for a commensal relationship with Nitrosopumilus, which is known to use urea as an energy source (59), and is identified as a core bacterial genus in the gills starting at stage 33. Overall, functional predictions for the internal gill microbiome show an abundance of molecular transportation pathways consistent with the biology of the gills as an active site of gas and waste exchange.
Adult skin has the lowest Shannon diversity of any tissue and unlike all other sites its microbiome is primarily composed of Bacteroidetes ASVs assigned to class Bacteroidia (81.2%). Bacteroidetes are dominant in many niches, are adapted to life on marine surfaces, play roles in polymer degradation, and contribute to immune function, suggesting a potential fitness benefit to the host (60,61). This low diversity may be due to the distinct properties of chondrichthyan skin, which is characterized by dermal denticles and a thin mucus layer (62). This structure is hypothesized to offer a selective microbial environment due to micropatterning of the dermal denticles, reduced laminar flow, and antimicrobial compounds (8,63). In support of this hypothesis, shark skin micropatterning has been shown to hinder microbial colonization and migration (64,65). The biophysical properties of chondrichthyan skin likely select for a distinct microbial community only after embryonic development is complete as skin denticles do not develop until around the time of hatching (66). Our data show that the adult skate skin bacterial community is distinct from that of embryonic skin, which clusters closely with the other embryonic tissues (Fig. 2). We hypothesize that the development of mature denticles creates a distinct selective pressure on the bacterial community of adult chondrichthyan skin that is not present in the embryo, though this hypothesis requires explicit testing.
An alternate possibility for the low-diversity skin microbiome is that the taxa present possess antimicrobial properties and suppress competition. While stingray skin microbiota show some antibiotic potential, skate skin microbes exhibit lower levels of antibiotic activity and are unlikely to be particularly potent (67,68). Predicted gene enrichment among the bacterial communities present in adult skin in this study identify some antimicrobial properties. Future studies are needed to validate this finding. In addition, multiple genes were identified relating to iron utilization, including iron complex outer membrane receptor protein, outer membrane receptor for ferrienterochelin and colicins, and ferrous iron transport protein B. A similar enrichment in iron acquisition and metabolism was detected in the skin microbiome of black-tip reef sharks (8). Iron is scarce in marine environments and required for growth by the majority of bacteria (69). The taxa that are able to persist on the challenging substrate created by the denticles may be those most able to metabolically outcompete rivals for limited resources, such as iron, rather than those which compete using antimicrobial secretions to reduce competition.
We tracked community continuity between different developmental stages and tissues. Despite large contributions from unknown sources, we find that vertical transmission accounts for at least 25% of the adult gill and 50% of the adult skin microbiome in the little skate. This is the first evidence of vertical transmission in an elasmobranch, and is significant as widespread vertical transmission offers a potential explanation for the pattern of phylosymbiosis observed in this group (10). In contrast, teleosts do not exhibit phylosymbiosis (10) and there is only limited evidence of vertical transmission in that clade (19). The prevalence of vertical transmission in chondrichthyans with alternate life histories awaits further research, which will expand our current understanding of the mechanisms underlying the signature of phylosymbiosis in elasmobranchs.
On average, unknown sources contributed about 50% of the bacterial community. Horizontal transmission likely plays a role in shaping the skin and gill microbiomes of adult skates and may account for many of the unknown sources. The one environmental source included in this study, the surrounding water, was not found to contribute meaningfully, however, deeper sampling may be necessary to detect low abundance taxa. Other potential environmental sources, such as diet and benthic substrates, were not included. If adult skates acquire large portions of their microbiome from environmental sources, we would expect large numbers of shared taxa between adults housed together. However, we identify only six genera uniquely present on adult tissues. Of these six, only a single genus, Undibacterium, was present in all adult samples of both skin and gill. Undibacterium species have been isolated from other fishes and may play a role in biofilm degradation (70–73). Given that they were not detected in embryonic or water samples, skates likely acquire Undibacterium through horizontal transfer from an unknown source or drastically enrich this genus from starting levels below the limit of detection.
The large proportion of unknown sources can also be explained by our experimental design. Since individual egg capsules were not associated with particular adult females, direct pairwise comparisons between parent and offspring were not possible. Furthermore, embryonic sampling is lethal, so each developmental timepoint is comprised of different, unrelated embryos. Thus, inter-individual variation limits our ability to accurately source all ASVs between timepoints. Furthermore, while we did sample adult female skin at the cloaca, these samples are unlikely to capture the extent of diversity housed in the reproductive tract where the egg capsules form (24). Like most elasmobranchs, little skates are polyandrous and multiple paternity is likely (74). Similar promiscuity has been associated with higher microbial diversity in the female reproductive tract in other vertebrate groups (75–77), a pattern that may hold for skates. We hypothesize that the microbiome of the reproductive tract is highly diverse, potentially providing a richer source of microbiota to the egg capsule than is captured by the adult tissues sampled in this study. Nonetheless, the precise mechanisms by which the egg capsule is seeded with its rich bacterial community, and how transmission of pathogenic bacteria is minimized, await further investigation.
This study provides the first exploration of the bacterial communities associated with the little skate throughout ontogeny and offers many intriguing possibilities for future microbiome research using this model chondrichthyan. Some potential avenues for future research include the reproductive tract microbiome as a source of vertically transmitted microbes to the embryo, the role of the host genome in modulating host-microbe interactions, and the effects of environmental variables, such as temperature and pH, on the little skate microbiome.
Funding
This project was funded by a Microbiome Pilot Project Grant from the University of Chicago Microbiome Center to D.M.W and N.H.S. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship to A.S.O. (DGE1745303).
Author Contributions
K.M. and A.S.O. conceived of and designed the project in consultation with N.H.S and D.M.W. K.M. and A.S.O. collected the data, performed the computational analyses, and wrote the manuscript. All authors approved the final paper.
Figures

Relative abundance of the top ten bacterial families in the classes Alphaproteobacteria (A), Gammaproteobacteria (B), and Bacteroidia (C) in the dataset are shown for each site and timepoint as well as for water and hand controls.

Relative abundance of the top ten bacterial genera in the classes Alphaproteobacteria (A), Gammaproteobacteria (B), and Bacteroidia (C) in the dataset are shown for each site and timepoint as well as for water and hand controls.
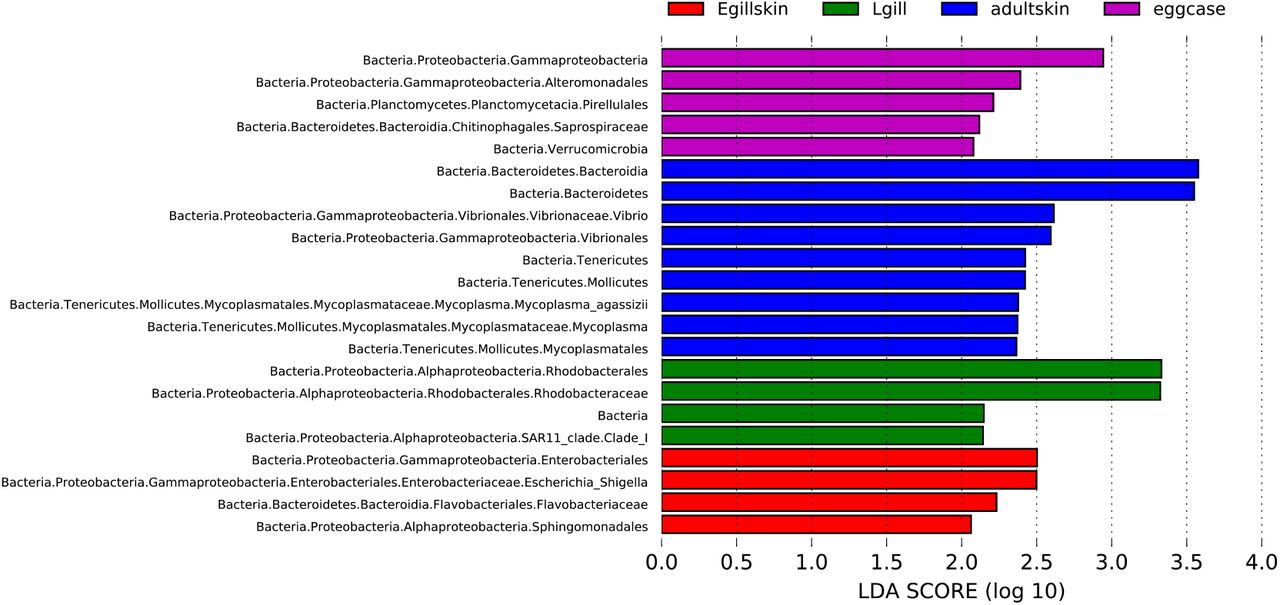
LEfSe analysis at P <0.05 and LDA>2. Only differentially abundant tree branches are shown. eggcase: egg capsule, Egillskin: embryonic external gills (stages 16–30) and embryonic skin (stages 16–33), Lgill: internal gill (stage 33-Adult), adultskin: adult skin.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Heatmap of the taxa identified by LEfSe (P<0.05, LDA>2) at the lowest categorized taxonomic level for comparisons of open versus closed egg capsule (red), internal liquid (orange) and combined egg capsule and internal liquid (black). Abundances of each sample are shown as Z-scores. Open samples include stage 30 replicates A & D, as well as all stage 33 samples. All other samples come from closed egg capsules.
Supplementary Table 1: Sample reads summary statistics
Supplementary Table 2: Complete core microbiome results
Supplementary Table 3: KO gene and pathways enrichments
Acknowledgements
The authors thank David Remsen, Dan Calzarette, and the MRC staff for assistance sampling the skates; Andrew Gillis for inspiration and staging the skate embryos, Tetsuya Nakamura for assistance with our IACUC protocol, Emily Davenport for directing us to computational resources, and Nipam H. Patel for use of lab space. We are deeply grateful to Tom A. Stewart, Terence D. Capellini, and the Carmody Lab for helpful comments on the manuscript.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References




