

ABSTRACT
Systemic sclerosis (SSc; scleroderma) is a poorly understood autoimmune rheumatic disease that primarily affects women. The clinical hallmark is hardening of the skin, but internal organ dysfunction is the leading cause of death. Diagnosis and treatment are complicated by heterogeneity within the disease including variable lethality, fibrosis severity, serum autoantibody production, and internal organ involvement. Important gaps remain in our knowledge of the exact molecular and cellular pathways underlying distinct SSc subtypes. Herein, we identify genome-wide chromatin accessibility profiles of peripheral CD4+ T cells to distinguish and better understand the observed heterogeneity in SSc patients. We identify a link between serum anticentromere autoantibody (ACA) subtype and elevated levels of T helper 2 (Th2) cells and increased chromatin access at gene loci encoding fibrosis-driving Th2 cytokines IL4, IL13, and IL4 receptor. Biological sex followed by autoantibody subtype are the predominant variables associated with differences in CD4+ T cell epigenomic profiles, while mycophenolate mofetil treatment appeared to have no effect. These results suggest new mechanistic basis and therapeutic strategies to address SSc, especially the ACA+ subtype that is associated with pulmonary arterial hypertension.
INTRODUCTION
Systemic sclerosis (SSc; scleroderma) is a clinically heterogeneous systemic disease with the highest case-fatality rate of the autoimmune rheumatic diseases. Although disease pathogenesis is known to involve an early vascular phase followed by immune dysregulation and skin and internal organ fibrosis, there is a poor understanding of the deregulated molecular systems that drive disease. Two clinical SSc subtypes, limited cutaneous (lc) and diffuse cutaneous (dc) have been described based upon the pattern and extent of skin fibrosis (LeRoy et al., 1988). The role of serum autoantibodies as more than diagnostic indicators of autoimmune disease has increasingly come to light. In particular, anti-RNA polymerase III (RNAIII) autoantibodies are associated with an increased risk for scleroderma renal crisis and worse skin fibrosis while anticentromere antibodies (ACA) are associated with the development of pulmonary arterial hypertension (PAH) (Cepeda and Reveille, 2004; Ho and Reveille, 2003), but many gaps persist in connecting the autoantibody profiles and clinical disease traits.
Immune modulators, such as mycophenolate mofetil (MMF), that inhibits purine synthesis and reduces lymphocyte proliferation, are often prescribed for SSc lung and skin disease (Hinchcliff et al., 2013; Walker et al., 2012), yet response is variable (Herrick et al., 2010; Panopoulos et al., 2013). Study results suggest that innate immune system activation plays a critical role in SSc pathogenesis (Chia and Lu, 2015; Christmann et al., 2011; Christmann et al., 2014; Higashi-Kuwata et al., 2010; Johnson et al., 2015). Perivascular lymphocytes are a predominant feature of early SSc skin disease and many therapies used in treating other skin and autoimmune diseases, such as B-cell depletion with rituximab, and cyclophosphamide and MMF, have also been utilized in the treatment of SSc skin disease with varied success (Fernández-Codina et al., 2018; Jordan et al., 2015; Taroni et al., 2017). To date, no drugs have been identified that are uniformly useful in the treatment of SSc skin fibrosis. Thus, a better understanding of the molecular derangements that underlie SSc skin disease are required in order to permit identification of targeted effective therapy.
The majority of existing genomic profiling in SSc patients has focused on differential gene expression, but the role of chromatin accessibility contributes greatly to gene regulation in health and disease and is an important noncoding metric for cell and tissue identity and functionality. Assay for Transposase-Accessible Chromatin by sequencing (ATAC-seq) is capable of interrogating the chromatin landscape at high sensitivity in rare cell types and particularly optimized in blood cells (Buenrostro et al., 2013; Corces et al., 2016; Corces et al., 2017). Furthermore, ATAC-seq profiling of chromatin accessibility patterns in multiple human primary blood cell types reflected cell identity better than mRNA expression levels obtained through RNA-seq (Corces et al., 2016). ATAC-seq is, therefore, a powerful tool for assessing, at high sensitivity and accuracy, genomic profiles unique to SSc and SSc clinically-defined subtypes.
Herein, we characterize genome-wide chromatin accessibility state of circulating lymphocytes from patients with active SSc skin disease prior to initiation of MMF and at regular intervals during treatment and determine associations with clinical variables of treatment, biological sex, age, and serum autoantibody expression.
RESULTS
Clinical characteristics are shown in Table 1 and Supplemental Table 1 for 18 subjects with SSc (14 of 18 patients, 77.8% with dcSSc). The median (range: 0.5-142.5 months) SSc disease duration between first Raynaud symptom and the initial visit was 16 (stdev of 42.6) months. The median (range: 2-43) modified Rodnan skin score (mRSS) was 13.5 (stdev of 10.6). 3 subjects (16.7%) had anticentromere antibody (ACA), 9 (50%) had anti-RNA polymerase III (RNAIII), and only 1 (5.6%) had anti-topoisomerase I/Scl-70 serum autoantibodies as confirmed through serum autoantibody testing.
Metrics include the patient ID in 4 numerical digits, age at study baseline, biological sex, SSc disease subtype, anti-centromere antibody (ACA), RNA polymerase III (RNAIII) antibody, Scl70 antibody, presence of any SSc antibodies (ACA, RNAIII or Scl70), antinuclear antibody (ANA), treatment group, and response from baseline to study termination. (“+” indicates present, “NA” indicates results were not available because patient was not tested, blank indicates absence)
Longitudinal profiling of systemic sclerosis patients
Peripheral blood was collected through venipuncture from consenting patients followed by CD4+ Rosette separation and FACS selection of CD3+CD4+ T-lymphocytes for ATAC library preparation (Corces et al., 2017) (Figure 1A). A total of 18 patients comprised of 14 females and 4 males (Table 1) were analyzed across three separate timepoints, henceforth referred to as timepoint 1 (or, baseline if treatment naïve or falling within two months of treatment start date), timepoint 2 and timepoint 3 (Supplementary Table 1). ATAC-seq library quality checks and mitochondrial SNP profiling—to confirm consistent identity of each patient across each timepoint—were performed prior to analyses (Supplemental Table 1, Supplemental Figure 1).

A Diagram of sample collection from patients and ATAC-seq library preparation of CD4+ cells. B Spearman Correlation between PC1/PC2 and each contributing variable tested, color indicates spearman correlation value (from −1 red to 1 blue), size indicates strength of correlation (larger is stronger). C Differential peaks across all timepoints and samples for each of the main variables. D enrichment of GWAS SNPs in open chromatin regions from the SSc study samples to verified autoimmune disease-associated SNPs. Scale bar corresponds to fold enrichment of SNPs in diseases compared to the random sampling background (red=high enrichment, white = low enrichment), ** indicates FDR < 0.05 and fold enrichment > 2. E GO term and pathway analysis of SSc SNPs enriched ATAC peaks associated genes.
Variables considered included patient age at blood collection, sample timepoint, biological sex, SSc subtype (limited or diffuse cutaneous), skin disease over the study course (improved, stable, worsened), skin disease treatment (MMF or treatment naïve), presence of any of three SSc-specific serum autoantibodies (ACA, RNAIII, and/or Scl70), and specifically ACA and RNAIII autoantibody status (Scl70 was not studied due to low prevalence in the study cohort, n=1) (Table 1, Figure 1B). Because Principle Components Analysis (PCA) showed that ACA positivity individually had larger variation than the three SSc autoantibodies grouped together (i.e. SSc autoantibodies expressed vs. none) (Figure 1B), we focused our studies on the ACA and RNAIII individually.
PCA performed on all sample ATAC-seq data identified that sex followed by SSc autoantibody status (specifically ACA positivity) and, to a lesser extent, age and treatment was associated with the most data variance (Figure 1B, Supplemental Figure 1E). There was very little ATAC-seq peak overlap between the individual variables (Figure 1C), indicating that these differences are attributable to each variable. GWAS-enrichment analysis of ATAC-seq data demonstrated significant alignment to multiple autoimmune diseases including, as expected, SSc as well as other rheumatic diseases (i.e., rheumatoid arthritis) (Figure 1D). Furthermore, associated ontology from the GWAS SNPs enriched in our dataset highlighted autoimmune-related pathways and categories as well as SSc-specific disease-drivers, such as Th1/Th2 differentiation, IL-35 and IL-21 signaling, NFKB signaling, and IFN-alpha production (Figure 1E) (Dantas et al., 2015).
MMF treatment does not significantly alter CD4+ T-cells in SSc peripheral blood
Mycophenolate mofetil (MMF), also known as Cellcept, is a commonly used treatment for various autoimmune diseases that works through inhibiting lymphocyte proliferation to decrease elevated immune activity and fibrosis (Allison and Eugui, 2000; Hinchcliff et al., 2013; Taroni et al., 2017). In this study, patients with active skin disease in the opinion of one treating physician based upon physical exam findings (described in Materials and Methods) were prescribed MMF treatment and a control group of patients with stable, non-active disease remained untreated. In order to determine the potential impact of MMF treatment on T helper cells in SSc, we identified patients who donated a blood sample within two months of treatment initiation (baseline) as well as at two subsequent treatment timepoints. Using these criteria, we compared the CD4+ T-cell ATAC-seq profile of eight MMF-treated, and four untreated, patients (Supplemental Table 1).
Comparison of all peaks over the three timepoints showed a stable, unchanging pattern in MMF-treated patients and very few changes in untreated patients (Figure 2A, 2C). Similarly, CIBERSORT analysis using ATAC-seq data showed that the relative proportions of T-cell subsets within the CD4+ population are also not significantly impacted by MMF treatment or lack of treatment (Figure 2D, Supplemental Figure 2). The differential peaks identified between the MMF-treated and untreated groups at baseline (Figure 1C) remain consistent at subsequent timepoints and thus may be explained by differences in SSc disease severity and activity at study onset (Figure 2B). Based on these results, MMF treatment in SSc patients does not change the circulating CD4+ T-cell epigenome.

A Violin plot of differential ATAC peaks between MMF vs. healthy across all three timepoints in MMF-treated (red) and treatment naïve (blue) SSc patients. B Venn diagram of number of differential peaks in MMF-treated patients as compared to treatment naïve (untreated) across timepoints. C MA plots of differential peak comparisons between timepoints in both MMF-treatment and untreated groups. Red dots indicate significance. D CIBERSORT analysis of CD4+ T-cell subsets in MMF-treatment patients across time (composite of all patients by cell type, left, individual patient examples, right).
Sex differences contribute the greatest variance in CD4+ T-cell regulome in SSc patients
The prevalence of autoimmune diseases, in particular SSc, is higher in females than males (Peoples et al., 2016). This observation is corroborated with the PCA results of our study participants that show that the greatest variance arises from biological sex differences (Figure 1B, Figure 3A) with 82 of 203 (40.4%) differential peaks arising from the X chromosome in females with SSc, 4 and 80 (1.2% and 24.8%) of 323 mapping to the X and Y chromosomes, respectively, in males with SSc, and the remaining differential peaks to the autosomes (Supplementary Figure 3A). To focus the differential analyses on sex differences exclusively, we compared only SSc females and males at baseline (if treated, or first available timepoint if treatment naïve) and were able to find clear segregation of gene regions more accessible in female with SSc that were distinct from those in males with SSc (Figure 3B, 3D).

A PCA analysis of all patients and all timepoints grouped by sex. B Violin plots depicting peaks more accessible in females (red) and in males (blue). C GREAT predicted functions of cis-regulatory regions associated with peaks accessible in SSc females and males. D Heatmap of differential ATAC-seq peaks between male and female SSc study patients at treatment-naïve, baseline timepoint grouped by sex (red label = female, blue label = male), scale bar indicates z-score of accessibility (red is more accessible and blue is less accessible). E Heatmap of motif enrichment of the genomic region from overlay of significant ATAC-seq peaks with HiChIP anchors, scale bar indicate significance of enrichment (values correspond to −log10(p-value), darker blue is more significant). F Volcano plot of differential peaks with FDR < 0.2 and absolute fold change larger than 1.5 highlighted in red. G CIBERSORT analysis of CD4+ T-cell subsets in male and female SSc patients (composite of all patients by cell type, left, individual patient examples, right). H Percentage of Naïve T effectors grouped by sex in SSc study patients calculated by CIBERSORT. I Percentage of Naïve T effectors in general population control patients calculated by CIBERSORT.
General population (healthy control) data were obtained through published CD4+ T-cell ATAC data (Qu et al., 2015) and analyzed separately from our SSc data to provide a comparison basis for sex-related differences unrelated to disease. Interestingly, there were fewer differential peaks arising from sex differences in the general population (Females = 54, Males = 154) and a larger fraction of differential peaks arose from the sex chromosomes (Female X = 77.8%, Male Y = 39.0%) than in SSc males and females (Supplemental Figure 3B). This disparity suggests that factors other than the sex chromosomes may drive the sex differences observed in SSc.
GREAT analysis of the differential peaks identified high prevalence of multiple immune-related pathways. In particular, T-lymphocyte-related programs associated to peaks more accessible in SSc males than SSc females (Figure 3C). Similarly, overlapping differential ATAC-seq peak regions to HiChIP anchor sites identified significant motif enrichment of genes involved in immune response and increased T-cell activation (AP-1, FOS, JUN, BATF) that are present in SSc males but not in any of the other groups (Figure 3E) and the Th2-activator GATA3 is significant in males in the volcano plot comparison of differentially accessible gene regions (Figure 3F). Moreover, enrichment of Th2-related activity in SSc males is supported by CIBERSORT analysis of the CD4+ T-cell subtypes which revealed a trend (p-value=0.23) of a higher proportion of pathogenic Th2 cells compared to SSc females (Supplemental Figure 3E). Conversely, SSc females do have a significantly higher proportion of the more undifferentiated naïve T effector cells (Figure 3G, 3H). These differences are not present between the sexes in the general population (Figure 3I, Supplemental Figure 3F), suggesting that divergences in differentiation and homeostasis of T effector cells may play a role in the sex differences observed in scleroderma.
In contrast, in separate analyses of healthy and SSc females, many of the same predicted cis-regulatory functions appeared in both groups and neither group showed motif enrichment of the SSc male-associated chromatin regions (Figure 3C, Supplementary Figure 3C, Figure 3E). Since biological females develop autoimmune diseases more commonly than males, this result is not surprising. Furthermore, males who develop SSc tend to have much more severe disease than females (Allison and Eugui, 2000; Peoples et al., 2016), supporting the hypothesis that more changes in immune-related expression are required to push males from stasis to disease.
Aging does not significantly alter chromatin accessibility in SSc peripheral T-cells
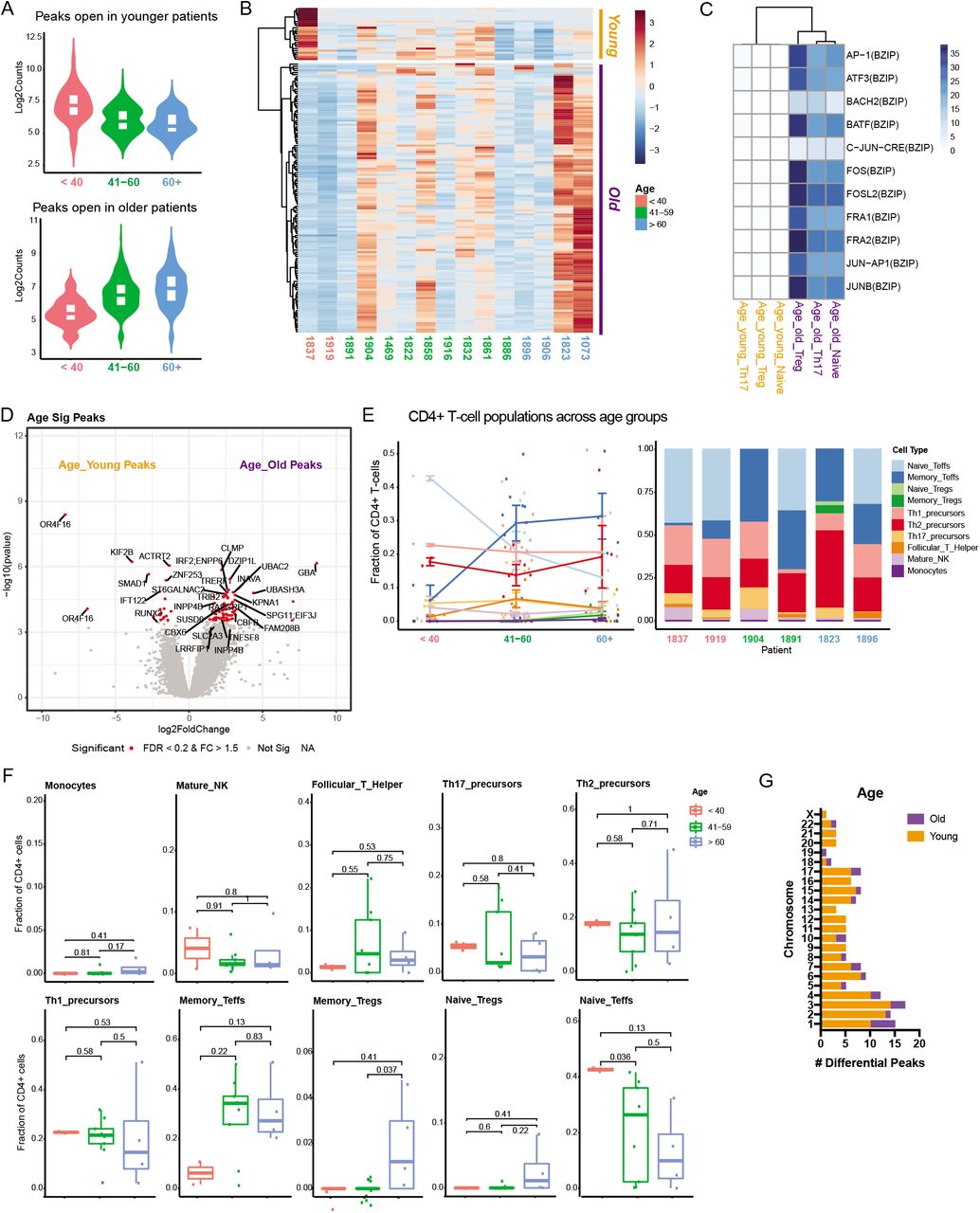
It is well-documented that aging changes the immune system and immune responses (Castelo-Branco and Soveral, 2014; Müller et al., 2019). SSc is primarily a disease of adulthood and our study did not have subjects <30 years old (Table 1). In our SSc study, age is a potential differential contributing factor that arises when sex-related differences are excluded from the comparison (Figure 1B, Supplemental Figure 1E). In contrast with sex, age-related differential peaks were more evenly distributed across both autosomes and sex chromosomes (Supplemental Figure 3G). For age-specific analyses we also included only patients treatment-naïve or at baseline to avoid potentially confounding factors (Supplemental Figure 4).
We observed a trend of increased chromatin accessibility in circulating CD4+ T-cells with advancing age (Supplemental Figure 4A). Heat maps of accessible peaks revealed two distinct clusters corresponding to genes more accessible in younger versus older patients (Supplemental Figure 4B). The accessible genes comprising the older patients’ cluster were related to immune activity pathways, such as the JUN-AP1 and BACH2-BATF motifs, interferon response factor IRF2 and T-cell proliferation regulator TNFSF8 (Supplemental Figure 4C, D). CIBSERSORT assignment of the CD4+ T-cell subsets based on ATAC-seq signatures did not display any statistically significant or consistent trends between the three age categories (Supplemental Figure 4F). These findings are in line with previous findings of immune system aging (Keenan and Allan, 2019) and indicate that the effects of aging on CD4+ T-cell chromatin accessibility is not a major mechanism of SSc pathobiology.
Serum autoantibody profiles are correlated with Th2-related pathways in SSc
Serum autoantibodies in SSc patients were the most significant non-sex related factor associated with CD4+ T-cell ATAC-seq peaks (Figure 1B, Supplemental Figure 1E). When patients were grouped by autoantibody profile, the most pronounced peak differences were observed at the latest time point collected (data from 3rd timepoint shown), potentially because of prolonged disease progression. Thus, we utilized the third timepoint data for subsequent autoantibody-related analyses. Additionally, we included all patients with available ACA and RNAIII antibody data since there was very little overlap of total differential peaks across timepoints between each of the comparison factors (i.e., age, sex, treatment, etc., Figure 1C), Table 1).
ACA and RNAIII antibodies are mutually exclusive in > 95% of patients (Domsic and Medsger, 2016) and are associated with different clinical outcomes (e.g., pulmonary arterial hypertension and scleroderma renal crisis most commonly present in ACA and RNAIII-positive SSc patients, respectively) (Nguyen et al., 2010; Odler et al., 2018; Steen et al., 2007). Given these clinical attributes, the lack of a significant number of Scl70 patients, and the consistent presence of ANA in nearly every patient, we focused our autoantibody analyses on differences arising from ACA and RNAIII expression (Table 1). A point of interest comes from patient 1837, who was not included in our ACA analysis due to lack of an ACA confirmatory test. However, since patient 1837 has confirmed expression of RNAIII autoantibodies, we can reasonably infer the absence of ACA (Domsic and Medsger, 2016). Furthermore, the ATAC-seq profile of subject 1837 clustered independently with the ACA negative patients, suggesting that chromatin accessibility profiles may predict autoantibody expression.
There was a clear distribution pattern of accessible DNA elements in CD4+ T-cells in positive-compared to negative-ACA patients (Figure 4A,C). GREAT analysis of cis-regulatory functions of differential peaks revealed that more accessible peaks corresponded to Th2, cell-mediated immune responses, and T-cell differentiation in ACA+ patients (Figure 4B). Overlap of differential peaks with Hi-ChIP anchor sites in CD4+ T-cell subsets showed enrichment of multiple motifs corresponding to immune cell function and autoimmunity (ETS family) (Garrett-Sinha et al., 2016), T-cell development (FLI1), Th2 differentiation (GATA3) as well as other immune cell programs (PU.1) that are significantly present in ACA+ T-cells but not ACA-T-cells (Figure 4D). Regions of genomic accessibility corresponding to the pro-inflammatory transcription regulator, NFKB1, and Th2 genes, IL4, IL4R, and IL21R, associated with ACA+ (Figure 4E). Additionally, CIBERSORT analysis showed that CD4+ T-cell subset composition was significantly different between ACA+ and ACA− patients (Figure 4F), with a much higher percentage of Th2 cells in ACA+ compared to ACA-patients (Figure 4G).

A Violin plots depicting peaks more accessible in ACA-positive (red) and in ACA-negative (blue) patients. B GREAT predicted functions of cis-regulatory regions associated with peaks accessible in ACA-positive patients (GREAT analysis for ACA-negative patients not significant). C Heatmap of differential ATAC-seq peaks of SSc study patients at latest timepoint available grouped by ACA-positive (red) and negative (blue). Grey indicates status not reported. Scale bar indicates z-score of accessibility (red is more accessible and blue is less accessible) D Heatmap of motif enrichment of the genomic region from overlay of differential ATAC-seq peaks with HiChIP anchors, scale bar indicate significance of enrichment (values correspond to −log10(p-value), darker blue is more enriched and significant). E Volcano plot of differential peaks with FDR < 0.3 and absolute fold change larger than 1.5 highlighted in red. F CIBERSORT analysis of CD4+ T-cell subsets in ACA-positive and negative patients (composite of all patients by cell type, left, individual patient examples, right). G Percentage of Th2 precursor cells grouped by ACA-status in SSc study patients calculated by CIBERSORT.
Next, we sought to determine the relationship between peaks in patients with different serum autoantibodies. As in the ACA comparison group, there was a clear distribution pattern distinguishing RNAIII+ and RNAIII-genomic accessibility in CD4+ T-cells (Figure 5A, C). Upon closer examination of genomic regions and associated pathways, there were clear similarities shared between ACA- and RNAIII+ patients even though not all ACA-patients are also RNAIII+, as some patients were negative for both autoantibodies (ACA-/RNAIII-). Similar to the ACA+ group, a subset of RNAIII- patient cells showed greater and more differential peaks at the genomic accessibility level as analyzed by ATAC-seq compared to RNAIII+ associated peaks (Figure 5A). These two different peak sets (RNAIII+ and RNAIII-) gave rise to two distinct clusters on the Z-score heatmap (Figure 5C).

A Violin plots depicting peaks more accessible in RNAIII-positive (red) and in RNAIII-negative (blue) patients. B GREAT predicted functions of cis-regulatory regions associated with peaks accessible in RNAIII-negative patients (GREAT analysis for RNAIII-positive patients not significant). C Heatmap of differential ATAC-seq peaks of SSc study patients at latest timepoint available grouped by RNAIII-positive (red) and negative (blue). Grey indicates status not reported. Scale bar indicates z-score of accessibility (red is more accessible and blue is less accessible) D Volcano plot of differential peaks with FDR < 0.3 and absolute fold change larger than 1.5 highlighted in red. E CIBERSORT analysis of CD4+ T-cell subsets in RNAIII-positive and negative patients (composite of all patients by cell type, left, individual patient examples, right). F Percentage of Th2 precursor cells grouped by RNAIII-status in SSc study patients calculated by CIBERSORT.
Furthermore, GREAT analysis revealed that CD4+ T-cells from ACA+ and RNAIII-patients shared many immunity and T-cell related pathways, particularly Th2 differentiation (Figure 5B) and the volcano plot of significant RNAIII-differential peak regions highlighted Th2-related genes such as IL4, IL4R, and IL21R (Figure 5D). These attributes were corroborated with CIBERSORT breakdown of the CD4+ T-cell subset that once again highlighted the significant difference in Th2 composition in RNAIII+ and RNAIII-cohorts. Both ACA+ and RNAIII-groups contained higher Th2 percentages when compared to their respective counterparts (Figure E, F). Additionally, the majority of differential peaks came from the ACA+ and RNAIII-groups while ACA- and RNAIII+ contributed fewer differential peaks (Supplemental Figure 5A, B), suggesting that major autoantibody-related differences primarily arise from ACA+ and/or RNAIII-carriage status.
Anti-centromere antibodies may be predictive of Th2-mediated fibrosis in SSc
We have shown that CD4+ T-lymphocytes in SSc patients that are ACA+ and/or RNAIII- evinced significantly higher proportions of Th2 cells and related motifs and regulatory pathways. There were 44 total genes in the overlap of differential peaks associated with both ACA+ and RNAIII-, of which 19 were shared with SSc males, and featured multiple Th2-related genes, such as IL4 and IL21R (ACA+ and RNAIII-only) and GATA3 and IL4R (shared with SSc males) (Figure 6A). Here, we focus on the IL-4-KIF3A and IL4R-IL21R regions, which were previously identified as associating with ACA+ and RNAIII-in the volcano plots (Figure 4E, Figure 5D).

A Venn diagram of accessible differential peaks in ACA-positive and RNA-negative SSc study patients. The genes within the intersection of the two autoantibody groups are listed and the intersection with male peaks are in blue. Bold italic indicates Th2-related genes. Representative ATAC-seq tracks for B the IL4;KIF3A and C the IL4R;IL21R genomic regions. Representative tracks for females at baseline (red), males at baseline (blue), ACA-positive at timepoint 3 (orange), RNA-III positive at timepoint 3 (purple), and ACA/RNAIII dual-negative at timepoint 3 (green) with HiChIP interaction loops from Naïve T-cell, Th17 and Tregs (black loops). The predicted enhancer for IL-13, IL-4, and KIF3A is highlighted in yellow. D Proposed model of relation between autoantibody expression and Th2 activation.
Th2 cells produce both IL-4 and IL-13 to stimulate fibrosis. The IL-4 gene locus is in close genomic proximity to both IL-13 and KIF3A (Figure 6B), another gene shared by the Th2-high groups (ACA+, RNAIII-). KIF3A is a ciliary protein important in myofibroblast development in SSc (Rozycki et al., 2014; Teves et al., 2019). The expected trends for accessibility in the IL-4-KIF3A genomic interval was reflected across all three comparison groups (females vs. males, ACA+ vs ACA- and RNAIII+ vs. RNAIII-) in relation to predicted Th2 cell proportions: peaks were larger in males compared to females, and significantly larger in ACA+ compared to RNAIII+. We also identified an active enhancer located between IL-13 and KIF3A that is predicted through HiChIP to interact with IL-4, IL-13, and KIF3A in Th17 and Treg cells (Figure 6B). The peak levels of this putative enhancer also reflect predicted Th2 activity levels.
Examination of IL4R locus in the IL4R-IL21R genomic region showed a similar accessibility pattern as the IL4 region with higher accessibility peaks in samples with higher proportions of Th2 (Figure 6C). IL21R neighbors IL4R and also displays more chromatin access in the Th2-high ACA+ CD4+ T-cells compared to the Th2-low RNAIII+ CD4+ T-cells (Figure 6C). IL21R is the receptor for IL-21, a potent cytokine secreted by both pathogenic Th2 and Th17 cells that promotes Th17 differentiation, inhibits Th1 differentiation, and drives fibrosis in diseases of chronic skin inflammation as well as pulmonary fibrosis in SSc (Brodeur et al., 2015; Costanzo et al., 2010; Kastirr et al., 2014; Lei et al., 2015; Sarra et al., 2011; Wurster et al., 2002). Interestingly, patients without either ACA or RNAIII antibodies (double negative) possessed accessibility levels falling at an intermediate level between the higher ACA+ and lower RNAIII+ peak levels in both the IL13-KIF3A and IL4R-IL21R regions (Figure 6B, C), suggesting that expression of either autoantibody influences Th2-cytokine genomic accessibility, and potentially expression, patterns.
Our results suggest an association between ACA+ and/or RNAIII-autoantibody status and higher Th2 prevalence and Th2-cytokine activity in SSc patients and suggest autoantibody profiles impact circulating cellular pathway activation in patients with SSc. Furthermore, previous studies have identified Th2 cells as a driving force behind fibrosis in SSc (Chizzolini et al., 2003; Mavalia et al., 1997). Taken together, the higher Th2 cell proportions and activity signatures detected in ACA+ and RNAIII-patients in this study may explain differences in observed clinical phenotypes.
DISCUSSION
Herein, we have comprehensively analyzed the chromatin landscape of CD4+ T-lymphocytes in a longitudinal study of 18 SSc patients across multiple clinical variables. While patient age did not significantly correlate with differences in CD4+ T-cell chromatin accessibility, we did find the well-established SSc biological sex bias to underly significant differences in the SSc chromatin landscape. We included the whole genome in our study, expanding from existing studies on SSc sex biases focusing exclusively on X-linked SNPs and X-linked epigenetic modifications (Saveria Fioretto et al., 2020). Importantly, we have identified a novel correlation of serum ACA positivity to fibrosis-driving Th2 cells, including a prospective pathway of cytokine involvement connected with PAH development.
ACA and RNAIII are autoantibodies highly specific for SSc rarely expressed in healthy individuals with low prevalence in other diseases. Serum autoantibodies are currently used as predictive tools for disease outcome as ACA patients tend to have lower early mortality but greater likelihood for developing PAH while RNAIII patients are more likely to experience earlier lethality and renal crisis (Cepeda and Reveille, 2004; Ho and Reveille, 2003). However, mechanisms to explain the association between the presence of serum autoantibodies in SSc patients with specific outcomes remains murky. We have therefore advanced a critical missing link in connecting ACA positivity to increased Th2 fractions and fibrotic cytokine activity.
The Th2 cytokines IL-4 and IL-13 are both detected at higher serum levels in SSc patients (Allanore et al., 2020), and have been implicated as drivers of PAH (Christmann et al., 2011; Kumar et al., 2015; Soon et al., 2010; Sweatt et al., 2019). Combined with the clinically-documented association between ACA+ and PAH development, these results support our proposed model that ACA carriage is connected to the Th2 fibrosis-promoting pathway whereas RNAIII carriage is associated with an alternative pathway involving less Th2 activity (Figure 6D). Additionally, in the tight skin scleroderma mouse model, IL-4 has been strongly linked to dermal fibrosis: IL-4 mice display less skin fibrosis (Ong et al., 1999) and addition of anti-IL-4 antibody reduces collagen production in IL-4 stimulated tight skin dermal cells (Ong et al., 1998). CD8+ production of IL-13 is directly correlated with dermal fibrosis in SSc patients (Fuschiotti et al., 2013; Fuschiotti et al., 2009). Together with our data correlating ACA+ but not RNAIII+ to increased chromatin accessibility to the IL13-IL4 genomic loci, autoantibody profiles may, thus, not only be used as a predictive diagnostic tool for internal organ involvement such as PAH or renal crisis, but also fibrotic severity in SSc patients when combined with tracking of Th2-cytokines and cellular fractions.
MMF is an immunosuppressive drug that is used in autoimmune diseases for its immunomodulatory effects. However, we showed here that MMF treatment does not confer any significant changes to the chromatin state of circulating T-cells over the time course of our study (Figure 2). It is possible that MMF works on a different level and/or cell type(s) than the CD4+ T-cell epigenome or requires a longer time period to show an effect. Our study results suggest that targeted therapies to modulate Th2 activity can potentially be a specific and effective treatment option for ACA+ and/or for male patients with high levels of Th2-cytokines. In particular, IL-4 and IL-13 have emerged as promising therapeutic targets in SSc (Gasparini et al., 2020), and two drugs are in clinical trials: (1) Dupilumab (DB12159), an anti-IL-4 and IL-13 monoclonal antibody used to treat atopic dermatitis, is currently in Phase II trials for localized SSc (trial identifier: NCT04200755, 2019-002036-90, Uni-Koeln-3815) and (2) romilkimab (SAR156597), an IL-4 and IL-13 neutralizing IgG bi-specific antibody that appears to significantly improve skin fibrosis based upon the results of a Phase II trial in patients with early dcSSc (Allanore et al., 2020). It is important to note that neither trial published the autoantibody profile of enrolled patients and thus, it is unclear if targeted IL-4/IL-13 therapy was more effective in ACA+ than RNAIII+ patients or whether study outcomes were affected by a preponderance of ACA+ or RNAIII+ subjects in these Phase II trials. And given the recent trend towards repurposing medication, such as rituximab, to address PAH specifically in SSc-PAH (Clinicaltrials.gov identifier: NCT01086540) (Prins et al., 2019; Zamanian et al., 2019), it will be useful to explore if targeting IL-4/IL-13 in SSc-PAH patients is a more direct approach than adjunctive therapy. Moving forward, future trials of targeted therapies should include SSc autoantibody expression for consideration.
Herein, we have demonstrated that ATAC-seq is a powerful and sensitive tool capable of high-fidelity detection of distinct open chromatin signatures that are associated with individual SSc-specific serum autoantibodies. ATAC-seq analysis of CD4+ lymphocytes was sufficient to consistently group each patient both by sex and by autoantibody profile. From our pathway analysis, we predict that future clinical studies involving ACA+ SSc patients and anti-IL-4 and anti-IL13 therapy will be promising avenues to explore. Future experiments will further expand investigation into other SSc autoantibodies, such as pathways related to RNAIII+ and Scl70+, and the involvement of other immune cell types, such as B-cells and myeloid cells.
The expansion of ATAC-seq datasets into larger sample sizes, more cell types, additional SSc autoantibodies and clinical disease traits (i.e. mRSS, localized or diffuse manifestation, organ involvement, and etc.) will power the development of robust machine learning models that may be used to analyze a single sequencing sample at high confidence. These dataset models will not only provide pathway and predictive disease information, but also replace multiple invasive and expensive clinical visits and tests. In a chronic and painful disease such as SSc, where epidermal and vascular stiffness can complicate even standard venipunctures, reducing the amount of blood and biopsies needed for evaluation and diagnosis will immediately improve standard of care. Furthermore, while this particular study focused on SSc patients, the technological applications and analysis used here and advancements discovered can be applied to the investigation of other autoimmune diseases.
AUTHOR CONTRIBUTIONS
Conceptualization: D.R. Dou and H.Y. Chang; Methodology and Investigation: D.R. Dou, Y. Zhao, K. Aren, M. Carns, and R. Li; Data Analysis: D.R. Dou, Y. Zhao, and B. Abe; Writing: D.R. Dou, Y. Zhao, B. Abe, L.C. Zaba, L.S. Chung, M. Hinchcliff, and H.Y. Chang; Funding Acquisition: H.Y. Chang; Resources: M. Hinchcliff and H.Y. Chang; Supervision: M. Hinchcliff, and H.Y. Chang.
DISCLOSURE
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, and an advisor to 10x Genomics, Arsenal Biosciences, and Spring Discovery. M.H. has received consulting fees from AbbVie and Boehringer Ingelheim.
MATERIALS AND METHODS
Clinical Cohort
The Northwestern University Institutional Review Board approved the study (IRB# STU00080199), and SSc patient participants provided informed consent in accordance with the Declaration of Helsinki. Patient participants fulfilled American College of Rheumatology SSc, or three out of five CREST (calcinosis, Raynaud, esophageal dysmotility, sclerodactyly, telangiectasias), criteria (1980). Research participants’ blood samples were collected in green-topped tubes containing sodium heparin. A subset of SSc patients were commencing mycophenolate mofetil (MMF) for a clinical indication at 250 mg PO BID with dose escalation to 1000-1500mg PO BID as tolerated. One physician performed clinical exams including mRSS (LeRoy et al., 1988). Serum autoantibodies were measured by indirect immunofluorescence at Specialty Laboratories, Valencia, CA. Subsequent blood collection for research was performed at the time of clinically indicated testing.
CD4+ T-cell isolation and ATAC-seq library preparation
Lymphocyte isolation for ATAC-seq library preparation was performed as previously described (Qu et al., 2015). 5mL of whole blood was enriched for CD4+ cells using RosetteSep Human CD4+ T Cell Enrichment Cocktail (StemCell Technology). CD4+ cells were frozen in 10% DMSO/FBS and stored in liquid nitrogen. Frozen cells were thawed and washed twice in 5% FBS in PBS1x, filtered through 100 uM cell strainers, stained for viability with 7-AAD (BD Biosciences, 559925), and T-cell markers mouse anti-human CD3 (Thermo Fisher Scientific, 11-0037-41) and mouse anti-human CD4 (Tonbo Biosciences, 20-0048-T025), and sorted for live 7-AAD- CD3+ CD4+ T-lymphocytes using the BDFACS ARIA II with a 100 uM nozzle. After CD4+ cells were sorted, 50,000 CD4+ T cells were used for Omni-ATAC library prep (Corces et al., 2017). Sequencing library quality metrics are included in Supplemental Table 2 and visualized in Supplemental Fig. 1.
Mitochondrial mutation calling and patient samples consistency confirmation
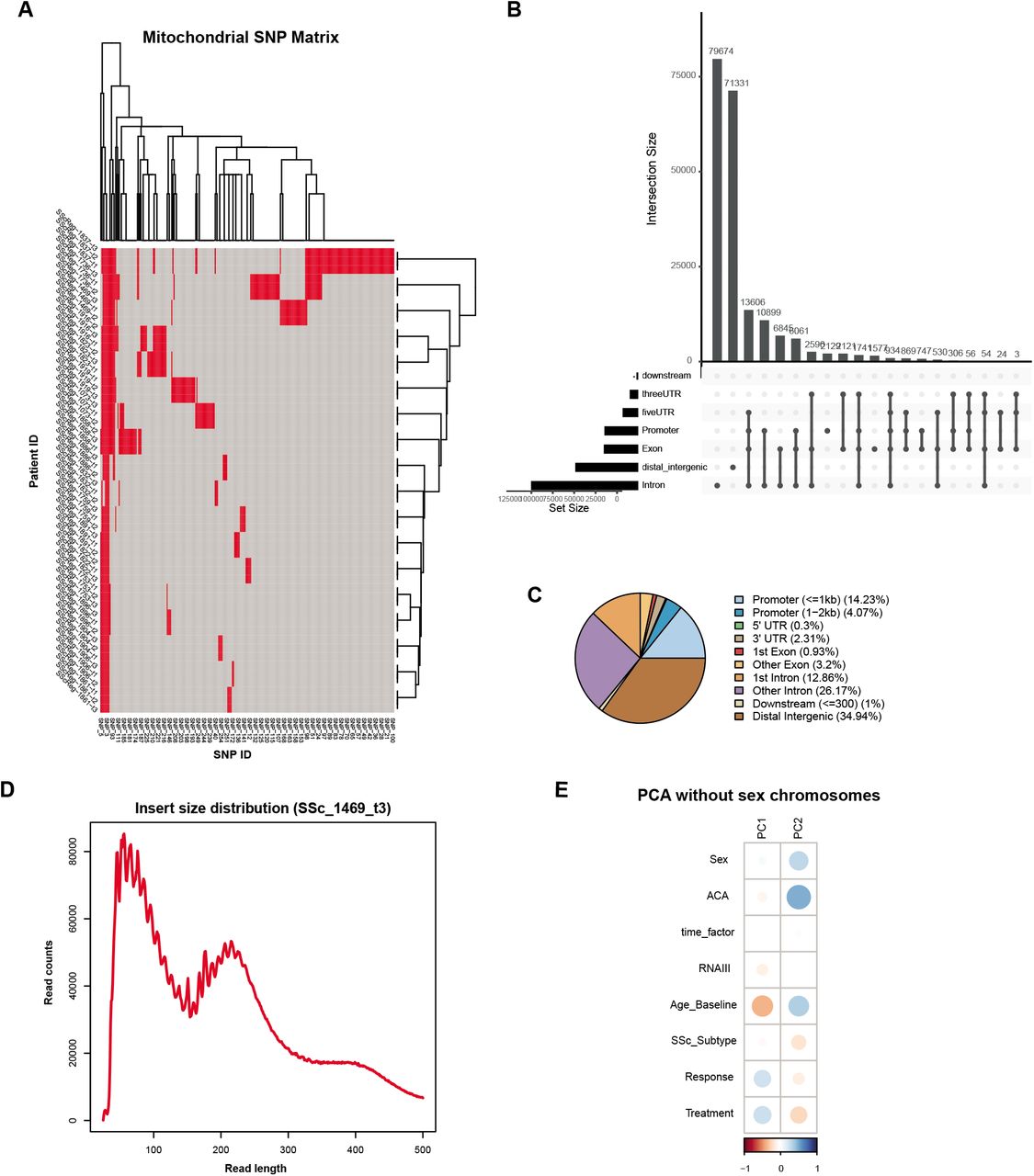
We used the mitochondrial mutation information to confirm consistent identity of patients across timepoints using the previously described mitochondrial SNP pipeline (Xu et al., 2019). The GRCh37 reference from the 1000 Genomes Project and the mtDNA sequence rCRS (revised Cambridge reference sequence) were used for mitochondrial mutation calling pipeline. Paired-end ATAC-seq fastq files were first aligned to the reference genome using BWA (Li and Durbin, 2009). Reads aligned to the mitochondria reference genome were then extracted. Samtools (Li et al., 2009) was used to convert mitochondrial sam files to bam files, sort bam files, and remove duplicated reads. Samtools mpileup was used to generate the pileup file for each sample with option “-q 30 -Q 30”. A custom perl script was used to get mutation information from pileup file and filter low quality reads. SNPs with allele frequency larger than 0.9 and support reads larger than 3 were selected. A heatmap was used to check whether the mitochondrial mutation is consistent across samples from three timepoints for each patient (Supplemental Fig. 1A).
ATAC-Seq data Analysis
The adaptor of paired-end ATAC-seq data were first trimmed by an in-house software, and then aligned to hg38 genome using bowtie2 (Langmead and Salzberg, 2012). The mitochondrial reads and reads with low alignment score (<10) were removed. The aligned sam files were converted to bam files and sorted by Samtools. Picard (http://broadinstitute.github.io/picard/) was used to remove duplicate reads and Macs2 was used to call peaks (Zhang et al., 2008). Each ATAC-seq peak was annotated by its nearby genes using GREAT (McLean et al., 2010) under the basal plus extension default setting. Bam files were converted to bedGraph format using the Bedtools genomeCoverageBed module (Quinlan and Hall, 2010). After normalization by total reads, bedGraph files were converted to BigWig format using the bedGraphToBigWig module from ucscTools (Kent et al., 2010) for visualization purpose. The bedtools MultiBamCov module was used to generate read count matrix from bam files.
Differential ATAC-seq peaks were identified using the negative binomial models from the R package DESeq2 (Love et al., 2014). The Benjamini hochberg procedure (Benjamini and Hochberg, 1995) was used to adjust for multiple hypothesis testing. Peaks with FDR < 0.2 and absolute fold change larger than 1.5 were selected as significant for Sex and Age factors. Peaks with FDR < 0.3 and absolute fold change larger than 1.5 were selected as significant for ACA and RNAIII comparisons.
Healthy control CD4 T cell ATAC-seq data were downloaded from GEO (GSE85853) (Qu et al., 2017). When comparing males with females, we used the same methods to analyze the healthy control samples as we did in the SSc sample data set. However, due to concern for batch effects, downloaded healthy control data were compared only with each other and not directly with the SSc group samples from our study.
CIBERSORT analysis
CIBERSORT (Newman et al., 2015) was used to estimate the abundance of CD4 T cell subtypes in bulk ATAC-seq data. First, the reference CD4 T cell ATAC-seq peaks and according read count matrix were downloaded from GEO (GSE118189 (Calderon et al., 2019)). The Bedtools intersect module was then used to identify the overlap peak set between the SSc ATAC-seq peaks and reference peaks. The overlap peak set, mixture file, reference sample file, and phenotype files were generated according to the CIBERSORT manual. The R package edgeR (Robinson et al., 2010) was used to normalize both Mixture count matrix and reference count matrix, and the subsequent matrices converted to log2CPM value. The signature file and the final CD4 T cell subtypes fraction matrix were then generated using CIBERSORT.
GWAS enrichment analysis and GO term analysis
We gathered known GWAS SNPs from the European Bioinformatics Institute GWAS catalog (https://www.ebi.ac.uk/gwas/docs/file-downloads), and retrieved index SNPs associated with autoimmune and control diseases. We then retrieve linked SNPs with Linkage Disequilibrium (LD) r2 > 0.8 to the index SNPs. This LD information was obtained from the haploreg website (http://archive.broadinstitute.org/mammals/haploreg/data/). The uscsTools liftOver module was used to lift SNP positions over to the hg38 genome. The numbers of SNPs within the SSc ATAC-seq peak regions were calculated for each disease. Random peaks were sampled from genome using the bedtools shuffle module with the same size of SSc ATAC-seq peaks and the number of SNPs within the random peak regions were subsequently recorded. The random shuffle procedure was conducted 1000 times to construct a null background, and the empirical p values were then respectively computed. The fold enrichment of GWAS analysis was calculated as the observed number of overlapping SNPs versus the mean random shuffled background.
SSc GWAS SNPs within our SSc ATAC-seq data were recorded as SSc ATAC-specific GWAS SNPs. Identities of these specific associated SNPs were assigned in accordance to nearby genes and then were passed to Gprofiler (Raudvere et al., 2019) for functional gene set enrichment analysis.
HiChIP data Analysis
HiChIP data for all valid pair matrices for Naïve T cells, Th17 cells and Treg were downloaded from GEO (GSE101498 (Mumbach et al., 2017)). H3K27ac ChIP-seq peaks for Naïve T cells, Tregs and T helper cells were downloaded from ENCODE as 1d peak sets. The FitHiChIP (Bhattacharyya et al., 2019) pipeline was used to call loops with 5kb bin, peak-to-all interaction type, loose background, and FDR < 0.01. LiftOver was used to convert the merged significant interaction files generated from the FitHiChIP pipeline from hg19 to hg38, the hg38-aligned files were then converted to BigBed format as described in the FitHiChIP online manual (https://ay-lab.github.io/FitHiChIP/usage/output.html) and visualized in UCSC web browser.
Motif analysis
Homer (Heinz et al., 2010) was used to conduct motif enrichment analysis using the intersecting regions between SSc ATAC-seq peaks and significant 5kb binned loops anchors called by FitHiChIP pipeline. Motifs with FDR < 0.05 were considered significant for each comparison.
Data Availability
All ATAC-seq files and processed files generated for this study have been deposited in the Gene Expression Omnibus (GEO) and are available under the accession identifier GSE163066.
SUPPLEMENTAL FIGURES

A Matrix of mitochondrial SNPs for every patient at every timepoint to confirm veracity of patient ID across the study. B Graph of ATAC-seq peak distribution by number of peaks. C Venn diagram of ATAC-seq peak distribution by percentages. D Representative plot of ATAC-seq library fragment length distribution (SSc_1469_t3). E PCA plots of contributing variables in the study, excluding peaks from the sex chromosomes (autosomes only).

CIBERSORT analysis of CD4+ t-cell subsets in treatment naive SSc patients (composite of all patients by cell type, left, individual patient examples, right)

A Differential peaks from each chromosome grouped by sex (female = red, male = blue) in SSc study patients. B Differential peaks from each chromosome grouped by sex (female = yellow, male = purple) in general population healthy control samples. C GREAT predicted functions of cis-regulatory regions associated with peaks accessible in healthy control females and males. D CIBERSORT analysis of CD4+ T-cell subsets in general population control males and females (composite of all patients by cell type, left, individual patient examples, right). Percentages of each CD4+ T-cell subset as calculated using CIBERSORT compared by sex in E SSc patients and F general population controls.

A Violin plots depicting peaks more accessible in younger than 40 years (red), 41-60 years (green), and older than 60 years (blue) at baseline. B Heatmap of z-score for accessibility of differential ATAC-seq peaks of SSc study patients at treatment-naïve, baseline timepoint grouped by age (red label = younger than 40, green label = 41-60 years, blue label = older than 60) and clusters for peaks more accessible in young patients (orange) and old patients (purple). C Heatmap of motif enrichment of the genomic region from overlay of differential ATAC-seq peaks with HiChIP anchors. D Volcano plot of differential peaks with FDR < 0.2 and absolute fold change larger than 1.5 highlighted in red. “Young” (orange) and “Old” (purple) refer to the two clusters from B. E CIBERSORT analysis of CD4+ T-cell subsets grouped by age of SSc patients (composite of all patients by cell type, left, individual patient examples, right). F Percentages of each CD4+ T-cell subset as calculated by CIBERSORT compared by age group in SSc patients. G Differential peaks from each chromosome grouped by age in SSc study patients.

Number of differential peaks at each chromosome grouped by absence and presence of A ACA or B RNAIII. Percentages of each CD4+ T-cell subset as calculated by CIBERSORT compared by positivity or absence of C ACA or D RNAIII in SSc study patients.
SUPPLEMENTAL TABLES
Supplemental Table 1: Patient Timepoint Table. Timepoint information (calculated in 0.5 month intervals) and baseline MRSS information for each patient.
Supplemental Table 2: QC Table. Quality Check metrics of ATAC-seq libraries for SSc study.
ACKNOWLEDGEMENTS
We thank members of the Chang lab and Dr. Oliver Distler for discussion. This work was supported by Scleroderma Research Foundation (H.Y.C.), NIAMS T32 AR007422 (D.R.D.), NIAMS T32 AR050942 (B.T.A.), NICHD K12 HD055884 (M.H.), NIAMS K23 AR059763 (M.H.), NIAMS R01 AR073270 (M.H.). H.Y.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
↵6 Co-senior authors





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}