

ABSTRACT
The 1000 Genomes Project (1kGP), launched in 2008, is the largest fully open resource of whole genome sequencing (WGS) data consented for public distribution of raw sequence data without access or use restrictions. The final (phase 3) 2015 release of 1kGP included 2,504 unrelated samples from 26 populations, representing five continental regions of the world and was based on a combination of technologies including low coverage WGS (mean depth 7.4X), high coverage whole exome sequencing (mean depth 65.7X), and microarray genotyping. Here, we present a new, high coverage WGS resource encompassing the original 2,504 1kGP samples, as well as an additional 698 related samples that result in 602 complete trios in the 1kGP cohort. We sequenced this expanded 1kGP cohort of 3,202 samples to a targeted depth of 30X using Illumina NovaSeq 6000 instruments. We performed SNV/INDEL calling against the GRCh38 reference using GATK’s HaplotypeCaller, and generated a comprehensive set of SVs by integrating multiple analytic methods through a sophisticated machine learning model, upgrading the 1kGP dataset to current state-of-the-art standards. Using this strategy, we defined over 111 million SNVs, 14 million INDELs, and ∼170 thousand SVs across the entire cohort of 3,202 samples with estimated false discovery rate (FDR) of 0.3%, 1.0%, and 1.8%, respectively. By comparison to the low-coverage phase 3 callset, we observed substantial improvements in variant discovery and estimated FDR that were facilitated by high coverage re-sequencing and expansion of the cohort. Specifically, we called 7% more SNVs, 59% more INDELs, and 170% more SVs per genome than the phase 3 callset. Moreover, we leveraged the presence of families in the cohort to achieve superior haplotype phasing accuracy and we demonstrate improvements that the high coverage panel brings especially for INDEL imputation. We make all the data generated as part of this project publicly available and we envision this updated version of the 1kGP callset to become the new de facto public resource for the worldwide scientific community working on genomics and genetics.
INTRODUCTION
The 1000 Genomes Project (1kGP) was the first large scale whole genome sequencing (WGS) effort to deliver a catalog of human genetic variation1–4. The project sampled participants from 26 populations across 5 continental regions of the world. Spanning seven years of data generation and analysis, it culminated in 2015 with a publication of the final, phase 3, variant callset3,4 consisting of 2,504 unrelated samples, a subset of which is from the HapMap collection5. The set of 2,504 samples was selected with the goal to maximize the discovery of single nucleotide variants (SNVs) at minor allele frequencies (MAF) of 1% or higher in diverse populations, hence related samples were not included. The phase 3 callset was generated based on the combination of low coverage WGS (mean depth 7.4X), high-coverage whole exome sequencing (WES, mean depth 65.7X), and microarray genotyping data. It included 84.7 million SNVs, and 3.6 million short insertions and deletions (INDELs), as well as a separate set of 60,000 structural variants (SVs; alterations ≥50bp). The 1kGP resources have been collectively cited over 15,000 times to date and have been utilized for foundational applications such as genotype imputation, eQTL mapping, variant pathogenicity prioritization, population history, and evolutionary genetics studies6–15. While the phase 3 dataset captured the vast majority of common variants (MAF > 1%) in the population (>99% of SNVs and >85 % INDELs)3, detection of rare variants (MAF <= 1%) was limited due to low sequencing coverage outside of the coding regions of the genome.
Here, we present high coverage WGS and comprehensive analyses of the original 2,504 1kGP samples, as well as additional 698 related samples. These related samples were not included as part of the phase 3 callset, but now provide complete WGS on 602 trios in the 1kGP cohort. A small subset of these pedigrees have been sequenced previously as part of various efforts, such as Platinum Genomes16, Complete Genomics3, and the Human Genome Structural Variant Consortium (HGSVC), which generated long-read WGS from Pacific Biosciences (PacBio), Bionano Genomics, and Strand-seq technology17,18; however, this is the first time that nearly all 1kGP trios have been sequenced at high coverage and jointly analyzed for the discovery and genotyping of genomic variation across the size and frequency spectrum, ranging from SNVs to large and complex SVs in a singular resource. We sequenced the expanded cohort of 3,202 samples to a targeted depth of 30X (minimum 27X, mean 34X) genome coverage using Illumina NovaSeq 6000 instruments. We aligned reads to the GRCh38 reference and performed SNV/INDEL calling using GATK’s HaplotypeCaller. Using this strategy, we called over 111 million SNVs and over 14 million INDELs with false discovery rate (FDR) of 0.3% and 1.0%, respectively, across the entire cohort of 3,202 samples. We also discovered and genotyped a comprehensive set of SVs, including insertions, deletions, duplications, inversions, and multiallelic copy number variants (CNVs), by integrating multiple algorithms and analytic pipelines19,20. Comparison with previous low coverage sequencing performed in phase 3 of the 1kGP demonstrated significant improvements in sensitivity and specificity in the SNV, INDEL and SV callsets, highlighting that the re-sequencing effort and expansion of the cohort to include trios brought significant value to the field.
One of the major applications of the phase 3 1kGP callset has been its widespread use as a reference panel for variant imputation in sparse, array-based genotyping data with a goal of improving the statistical power of downstream genome-wide association studies (GWAS) and facilitating fine-mapping of causal variants. We leveraged the presence of full trios in the expanded 1kGP cohort and performed haplotype phasing of SNVs and INDELs using a statistical phasing approach with pedigree-based correction. We demonstrate the power that inclusion of family members has on improving the accuracy of phasing and show how it compares to the phase 3 version. Finally, we evaluate the imputation performance of the high coverage panel and demonstrate improvements especially in INDEL imputation as compared to the phase 3 panel.
Over the past few years, the cost of high coverage WGS has decreased dramatically which, combined with substantial progress in analytics tools, has contributed to the emergence of several population-scale high coverage WGS panels, such as the Genome Aggregation Database (gnomAD; 15,708 WGS and 125,848 WES samples)21, Trans-Omics for Precision Medicine (TOPMed: https://www.nhlbiwgs.org/; 138,000 samples)22, or the UK Biobank (UKBB: https://www.ukbiobank.ac.uk/; goal to sequence 500,000 samples by 2023) to name a few. These growing resources, many fold larger in sample size than the 1kGP cohort, enable continuous expansion of the catalog of genetic variation in the human population and facilitate discoveries that improve human health. Unlike the 1kGP, most of the recent large scale WGS efforts have restrictions on public data sharing as they are often linked to clinical data, which amplifies privacy concerns. As a consequence, only aggregate population-level allele frequencies are available for public access in most cases. In contrast, samples within the 1kGP cohort have been consented for full public release of genetic information which allows for unrestricted sharing of the complete sample-level genotype (GT) data. This enables granting access to a downloadable reference imputation panel, as well as use of the dataset for methods development and benchmarking, among other applications. All the data generated as part of this project including BAM files, VCFs, and functional annotations, has been made publicly available (see the Data Access section). We envision this updated version of the 1kGP cohort to become the new de facto public resource for the worldwide scientific community working on genomics and genetics.
RESULTS
Overview of SNV and INDEL calls across the 3,202 1kGP samples
We performed WGS of the original 2,504 1kGP unrelated samples as well as additional 698 related samples, which completed 602 parent-child trios in the 1kGP cohort and brought the total number of sequenced samples to 3,202 (Figure 1A, Table S1). The final variant callset across the 3,202 samples contains 111,048,944 SNVs and 14,435,076 INDELs. The mean SNV density across the callable genome was 39.46 per 1kb of sequence. Chromosome X (30.16 SNVs per 1kb) displayed the lowest density across all chromosomes, followed by chromosome 1 (36.46 SNVs per 1kb) among the autosomes, whereas chromosome 19 (43.21 SNVs per 1kb) had the highest density overall (Table S2). Figure 1B summarizes variant counts by alternate allele frequency (AF), restricted to unrelated samples to prevent overestimation of AF in the population. We found 74,264,978 (59.2%) variants were singletons (allele count (AC) = 1) or doubletons (AC = 2); 33,008,491 (26.3%) variants to be rare (AC > 2 and AF <= 0.01), and 18,210,551 (14.5%) variants to be common (AF > 0.01). Overall, we found 19,239,888 (15.3%) total variants called were novel, defined as not reported in dbSNP build 151. Among the novel variants, 83.5% are singletons or doubletons, 11.8% are rare, and 4.6% are common variants. As expected, we see a higher percentage of novel variants among singleton and doubleton categories.

A) Sample counts per super-population stratified by sex. Shaded areas represent counts coming from the newly added 698 related samples. B) SNV/INDEL counts, stratified by AF bins (unrelated samples only). Counts of variants with AC=1 or AC=2 (singletons and doubletons) are reported as a separate bin and were excluded from the ≤1% AF bin. C) Number of small variant loci per genome, stratified by population. From top to bottom: total count of variant loci, counts of single nucleotide variants (SNV), count of small deletions, count of small insertions. Complex variants and MNPs were not included in the breakdown. In all panels, counts are restricted to variants that passed VQSR. Color labels along the x axis correspond to the following super-populations: blue: EUR, green: EAS, purple: SAS, red: AMR, yellow: AFR.
The callset contains 117,175,809 variant sites of which 6.5% (7,676,044) are multiallelic. We divided the genome into easy, medium, and hard regions as defined in the CCDG functional equivalence paper23. Easy refers to parts of the genome that are mostly unique and where we can confidently call variants, hard is made up of low complexity and repetitive regions, and any region that did not fall into the two categories was classified as medium. We looked at the distribution of multiallelic sites across the three sets of regions and found that although hard regions only make up 8% of the genome they contain 38% of all the multiallelic sites, compared to easy regions that make up 70% and contain 40% of sites. About 50% of the sites in hard regions are multiallelic but that drops down to ∼15% in the filtered phased callset (described in the “Haplotype phasing of high quality SNVs and INDELs’’ section) suggesting that many of these variants might be of poor quality. This is not surprising as hard regions are made up of low complexity and repeat elements that make it harder to call variants confidently.
At a per sample level, we called an average of 5,038,683 small variants total (Figure 1C, top panel). This includes an average of 4,080,991 SNVs, 420,645 short insertions, and 451,276 short deletions per genome, across samples from all populations (Figure 1C, bottom 3 panels; mixed and complex variants and multi-nucleotide polymorphisms (MNPs) were not included in the breakdown). We observed an average transition to transversion ratio (Ti/Tv) of 2.01 and heterozygous to non-reference homozygous ratio (Het/Hom) of 1.70 (Figure S1), consistent with expectations for WGS data. As expected, the average number of variant sites was higher in the individuals from African populations, with 4,653,521 SNVs, 465,797 short insertions, and 503,995 short deletions per genome. In line with that, we also observed a higher Het/Hom ratio of 2.03 among the AFR samples (Figure S1). We also noticed higher variability in the number of variants in individuals belonging to the admixed American population. On average, we called 21,800 novel variant sites per sample across all populations, with African and South Asian populations containing more novel sites than Europeans, East Asians, and Admixed Americans (Figure S2).
Lastly, we determined the FDR of the high coverage callset by comparing genotype calls from sample NA12878 to the high confidence calls from the Genome in a Bottle (GIAB) NA12878 callset24 (see precision vs. recall plot in Figure S3). Using this approach, we calculated an FDR of 0.3% for SNVs with sensitivity of 99.7%, and an FDR of 1% for INDELs with sensitivity of 98%.
Overview of structural variation across the 3,202 1kGP samples
We generated an SV callset across all 3,202 1kGP samples with short read sequencing data. These SV genotypes were discovered and integrated from three analytic pipelines: GATK-SV19, svtools20 and Absinthe (see Methods, Table S5). This final ensemble callset included 170,242 loci, comprised of 89,269 deletions, 24,068 duplications, 674 multiallelic CNVs (mCNVs), 51,829 insertions, 956 inversions, 3,430 complex SVs (CPX) consisting of a combination of multiple SV signatures, and 16 inter-chromosomal translocations (CTX, Figure 2A). The size and allele frequency distribution of SVs followed expectations; Mobile element signatures were observed for ALU (200-300 bp), SVA (1-2 kb), and LINE1 (5-6 kb) variants. Most SVs were rare, and SV allele frequencies were inversely correlated with SV size (Figure 2B, C). On average, ∼9,266 SVs were discovered in each genome (see Figure 2D). The distribution of SVs observed per individual followed expectations for ancestry with the greatest number of SVs per genome derived from African populations (Figure 2E)25. The specificity of the SV callset was also quite high, with a de novo SV rate of 1.8%, which represents the combination of false positive SVs in children, false negative SVs in parents, and cell line artifacts in probands (Figure 2F).

A) The count, (B) size distribution, and (C) allele frequency distribution of each SV for each SV class is shown. The per sample counts of SVs by variant class (D) and ancestral population (E) is also provided, as well as (F) inheritance rates of all SVs.
Comparison of SNV and INDEL calls against the 1kGP phase 3 callset
We compared our SNV and INDEL calls against the phase 3 callset to quantify the improvements brought by high coverage sequencing and pipeline upgrades. A direct comparison to the original callset was not possible as the phase 3 dataset was aligned to the GRCh37 reference. To overcome this issue, we used the GRCh38 version of the phase 3 callset, which was generated by dbSNP, European Genome-phenome Archive (EGA), and European Variation Archive (EVA)26 by lifting over the coordinates of variant sites using dbSNP build 14927. Due to the high number of liftover failures on chromosomes X and Y, the comparison was limited to autosomes. On average, about 10,465 sites failed to liftover in autosomes, whereas the number of sites that failed to liftover in chromosome X and Y was 2,020,268 and 55,528, respectively. The liftover failures occur when the coordinate position has been moved to a different chromosome in the new build or due to inability to resolve the reference allele or strand changes. For consistency, we limited the comparison of the SNV/INDEL calls to the 2,504 samples that are common between the high coverage and the phase 3 datasets.
When restricted to autosomes, the 2,504-sample high coverage callset included 94.84 million SNVs and 9.08 million INDELs, as compared to 78.24 million SNVs and 3.15 million INDELs in the phase 3 callset. Figure 3A,B shows the breakdown of SNVs and INDELs from the two datasets into singleton (AC=1), rare (AC > 1 and AF <= 0.01), and common (AF > 0.01) bins based on non-reference AF. We called ∼15 million more singleton SNVs and ∼4 million more rare SNVs in high coverage as compared to phase 3, whereas the number of common variants remained similar. We called ∼3 times more INDELs in the high coverage callset, with increase in INDEL counts observed across the entire AF spectrum. The phase 3 callset contained only 4,377 singleton INDEL calls, as compared to 2,999,027 in the high coverage dataset. The low number of ultra-rare INDEL calls in the phase 3 set can be attributed to more stringent filtering applied to INDELs as compared to biallelic SNVs, as INDELs were harder to call with low coverage sequencing3. We also called significantly more longer INDELs. The lifted-over phase 3 set contains only 17 INDELs that are >50bp in size, whereas the high coverage callset contains 192,583 calls in this size range (Figure S4). Notably, the original phase 3 callset on GRCh37 contained 2,285 INDELs that are >50bp with the largest being 661bp insertion. We suspect the larger INDELs failed liftover and did not make it into the GRCh38 callset.

For consistency, the high coverage dataset was restricted to the 2,504 samples that are shared between the two callsets. Number of SNVs (A) and INDELs (B) across the 2,504 samples in phase 3 (aqua) and high coverage (purple) datasets, stratified by AF bins. Counts of variants with AC=1 (singletons) are reported as a separate bin and were excluded from the ≤1% AF bin. Multiallelic loci were split into separate lines and INDEL representation was normalized prior to counting, i.e. the reported counts are at the alternate allele (as opposed to locus) level. (C) Percent of phase 3 variants recalled in the high coverage dataset stratified by variant type (blue: all, orange: SNVs, green: INDELs) and regions of the genome (easy: regions where we can confidently call variants, medium: regions that did not fall in either easy or hard category, hard: centromeres, repetitive and low complexity regions). (D) Correlation of non-reference allele frequency of shared variants in the high coverage vs. phase 3 callsets, stratified by easy, medium, hard regions of the genome, as defined in C. Plotted on y-axis is the spearman correlation coefficient (rho). (E) Total number of SNV and INDEL loci per genome in the phase 3 (aqua) and the high coverage (purple) dataset, stratified by 1kGP super-populations. (F) Comparison of FDR between the high coverage (purple) and phase 3 (aqua) callsets across AF bins, stratified by variant type (star: SNV, plus sign: INDEL).
Overall, we recalled 94.7% of phase 3 variants in the high coverage callset. More than 95% of phase 3 variants in the easy genomic regions were recalled in the high coverage callset, compared to 88% and 73% in medium and hard regions, respectively (Figure 3C). The SNV recall rate was higher than the INDEL recall rate in the easy and medium regions whereas it was lower in the hard regions, suggesting there might be more false positives among SNV calls in the hard regions. We observed high correlation of AF among shared variants between the high coverage and phase 3 callsets, with Spearman correlation coefficient (rho) of >0.95 for both SNV and INDEL across all regions, except for INDELs in the hard region where it drops to 0.9 (Figure 3D).
At a per sample level, there are ∼4.3 million variant sites on average in the phase 3 dataset compared to ∼4.9 million in the high coverage set (Figure 3E). On average, 84% of variant loci called per sample in the high coverage callset were discovered in phase 3. We calculated FDR of the 2,504-sample high coverage and the phase 3 callsets by comparing the genotype calls from sample NA12878 to high confidence calls for the same sample from the GIAB24. We excluded any variants from both the callsets that fell into regions in GRCh38 that could not be lifted over from GRCh37 (see Table S3). Overall the FDR among SNVs was 0.3% in the high coverage, compared to 1.2% in the phase 3 callset. Among INDELs, FDR was 1.1% in the high coverage as compared to 12.6% in the phase 3 callset. Figure 3F shows FDR for the two sets in rare (AF <= 0.01), common (AF > 0.01 and AF <= 0.05), and very common (AF > 0.05) AF bins. As expected, in both callsets, FDR for SNVs decreases for high AF variants. We see the opposite trend for phase 3 INDELs because there are very few calls in the rare and common AF bins compared to very common.
Evaluation of the SV callset against the 1kGP phase 3 callset
This ensemble SV callset was benchmarked against the 1kGP phase 3 SVs (7.4X average coverage)4 on the 2,504 shared samples to assess the quality and unique value brought by high coverage sequencing and genotyping capabilities of new analytic pipelines. The current ensemble SV callset discovered over two-fold more SV sites than phase 3 (166,752 vs. 68,698), and encompassed 87.4% of the phase 3 SV calls (Figure 4A). This increased sensitivity and high overlap of phase 3 SVs was consistent across all SV classes (Figure 4A), and per genome, with an average of 9,266 SVs detected in the current ensemble callset compared to 3,431 SVs in the phase 3 callset (Figure 4B).

(A) Count of SV sites in the current ensemble SV callset and low-coverage phase 3 SV callset, and their overlap. Numbers next to each bar represent the counts of SV sites in each dataset. (B) The distribution of SVs counts per sample in both callsets, and their average overlap displayed in the venn diagram. (C) Count of genes altered by SVs in both datasets. pLoF: probable loss of function, CG: complete copy gain, IED: intragenic exon duplication. (D) Count of genes altered by pLoF SVs across ancestral populations.
The high coverage SV callset provided significant added value in terms of the discovery of SVs that alter gene function by comparison to the phase 3 low-coverage SV dataset. Consistent with previous large population studies19, we observed that biallelic SVs in each genome resulted in probable loss of function (pLoF) of 119 protein coding genes, complete copy gain (CG) of 24 genes, and duplications of intragenic exons (IED) of 6 genes. The same analyses of the low-coverage phase 3 callset predicted an average of 32 genes disrupted by SVs per genome (30 pLoF, 1 CG and 1 IEDs; Figure 4C). The 1kGP dataset also offered an estimate in the population diversity of functional SV variation, where African populations had the highest number of pLoF SVs per genome, (Figure 4D), with similar patterns observed for CG and IED SVs that altered protein coding gene sequences (Figure S11).
Haplotype phasing of high quality SNVs and INDELs
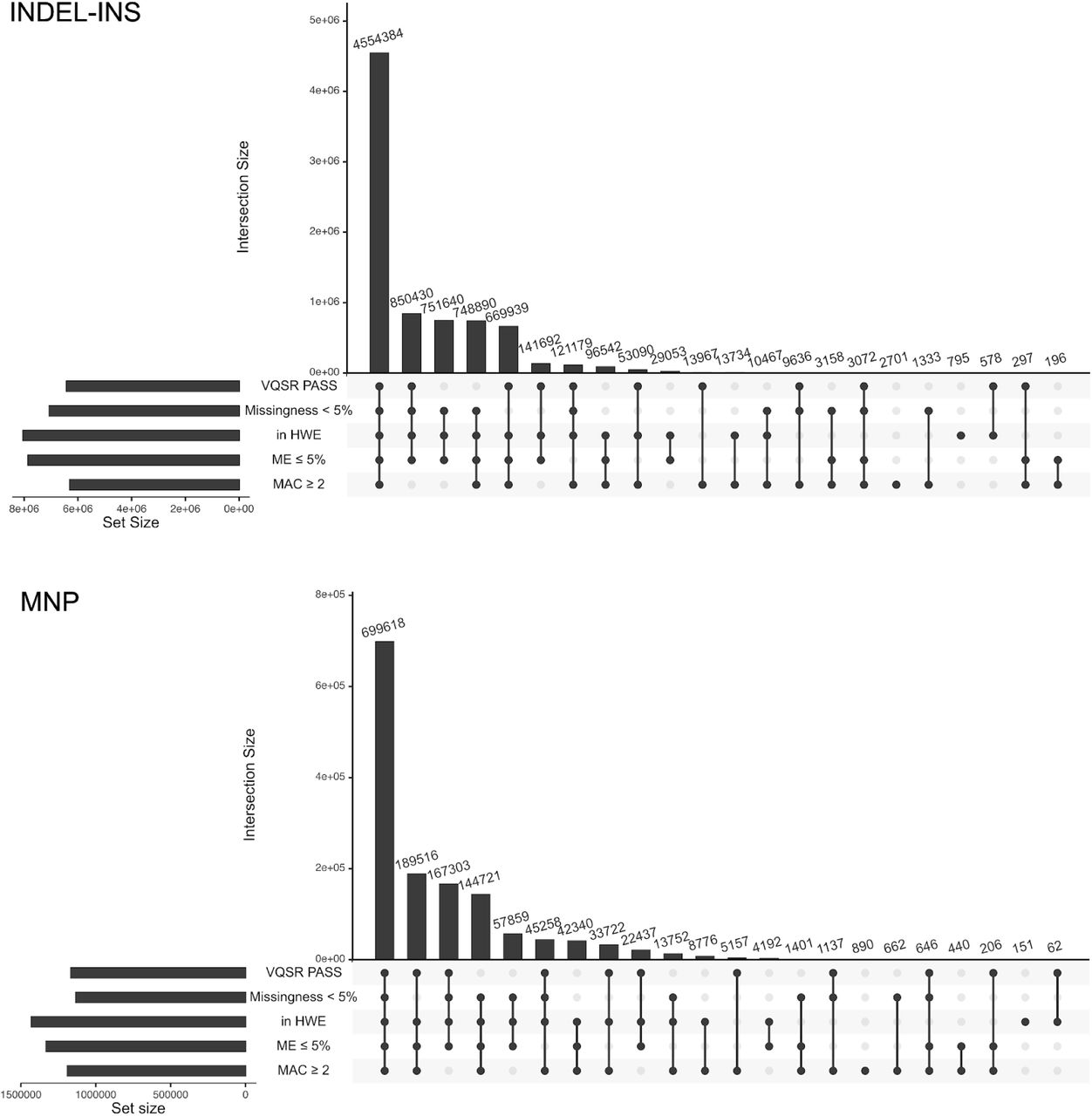
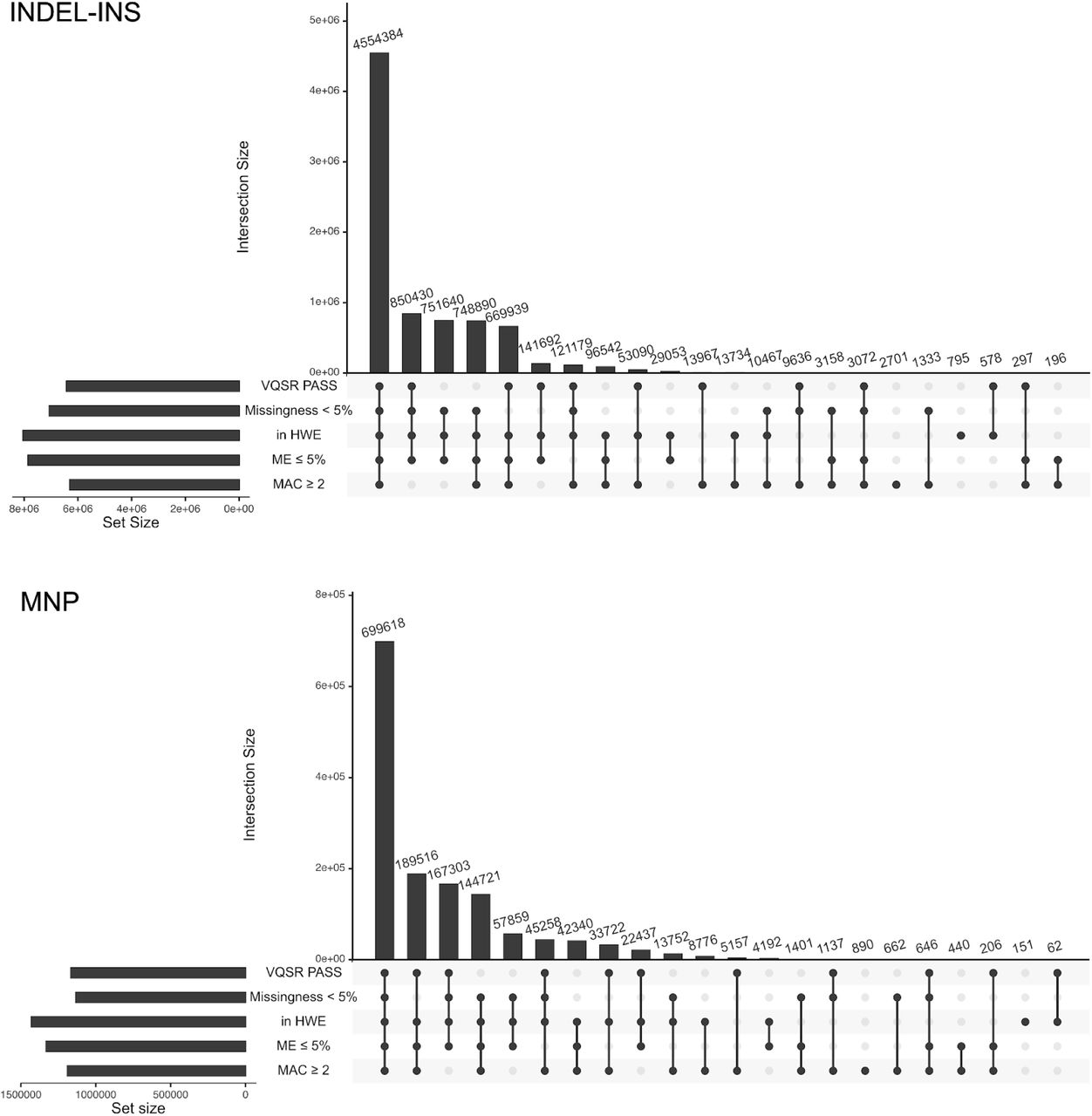
We performed haplotype phasing of high quality autosomal SNVs and INDELs across the 3,202-sample 1kGP cohort using statistical phasing with pedigree-based correction, as implemented in the SHAPEIT2-duohmm software28,29. Phasing of the high quality SNVs and INDELs on chromosome X was performed using statistical phasing, as implemented in the Eagle2 software30, which, unlike SHAPEIT2-duohmm, supports ploidy-aware phasing. We defined high quality SNVs and INDELs by applying the following set of filtering criteria: 1) VQSR PASS; 2) GT missingness rate < 5%; 2) Hardy Weinberg Equilibrium (HWE) exact test31 p-value > 1e-10 in at least one of the 5 super-populations (EUR, EAS, SAS, AMR, AFR); and 3) mendelian error rate (MER) for sites with complete trio calls ≤ 5%. Additionally, we excluded all sites with minor allele count (MAC) < 2, as singletons are not informative for phasing. The resulting set of high quality variants that went into phasing consisted of 72,065,314 sites on chromosome 1-22 and chromosome X (Figure 5A), which included 61,411,215 SNVs, 9,954,481 INDELs, and 699,618 MNPs (counts at the ALT allele-level) (Figure S5).

A) Counts of SNVs/INDELs meeting specified filtering criteria. Multiallelic variants were split into separate rows prior to counting. HWE: HWE exact test p-value > 1e-10 in at least one of the 5 super-populations (EUR, AFR, EAS, SAS, AMR); ME: mendelian error among complete trios (i.e. trios where all family members have a called GT at a given site); MAC: minor allele count. The set of SNV/INDELs meeting all 5 QC criteria (first bar from the left) was selected for haplotype phasing. See Figure S5 for a similar plot stratified by variant type. B) Haplotype phasing accuracy evaluation of the high coverage (purple) vs. the phase 3 (aqua) 1kGP SNV/INDEL panel. Switch error rate (SER) was computed across all pairs of consecutive heterozygous SNV/INDELs in specified MAF bins for sample NA12878, using the Platinum Genome NA12878 callset as gold standard truth set (autosomes only). Three phasing conditions applied to the high coverage dataset (purple): (1) solid line with circles: 3,202-sample panel phased using statistical phasing with pedigree-based correction; (2) dashed line with triangles: 3,202-sample panel phased using statistical phasing without pedigree-based correction; (3) dashed line with diamonds: 2,504-sample panel phased using statistical phasing (unrelated samples only). The 2 panels represented with dashed lines were created for evaluation purposes only. Aqua solid line with squares: 2,504-sample phase 3 SNV/INDEL panel phased using statistical phasing with family-based scaffold. C) Imputation accuracy of SNV (solid lines with circles) and INDEL (dashed lines with triangles) genotypes as a function of non-reference allele frequency in a given 1kGP super-population (blue: EUR, green: EAS, purple: SAS, red: AMR, yellow: AFR), achieved using the complete high coverage panel. Imputation accuracy is measured by the aggregate squared correlation (R2) between imputed dosages and WGS dosages of the Simons Genome Diversity Project samples (average across 22 samples per super-population). Comparison of the imputation performance between the high coverage (solid lines with circles) and phase 3 (dashed lines with triangles) panels for SNVs (D) and INDELs (E), stratified by super-population (the same colors as in C). Comparison in (D) and (E) was restricted to sites that are shared between the phase 3 and the high coverage panels (unlike panel C which shows performance across all sites for the high coverage panel).
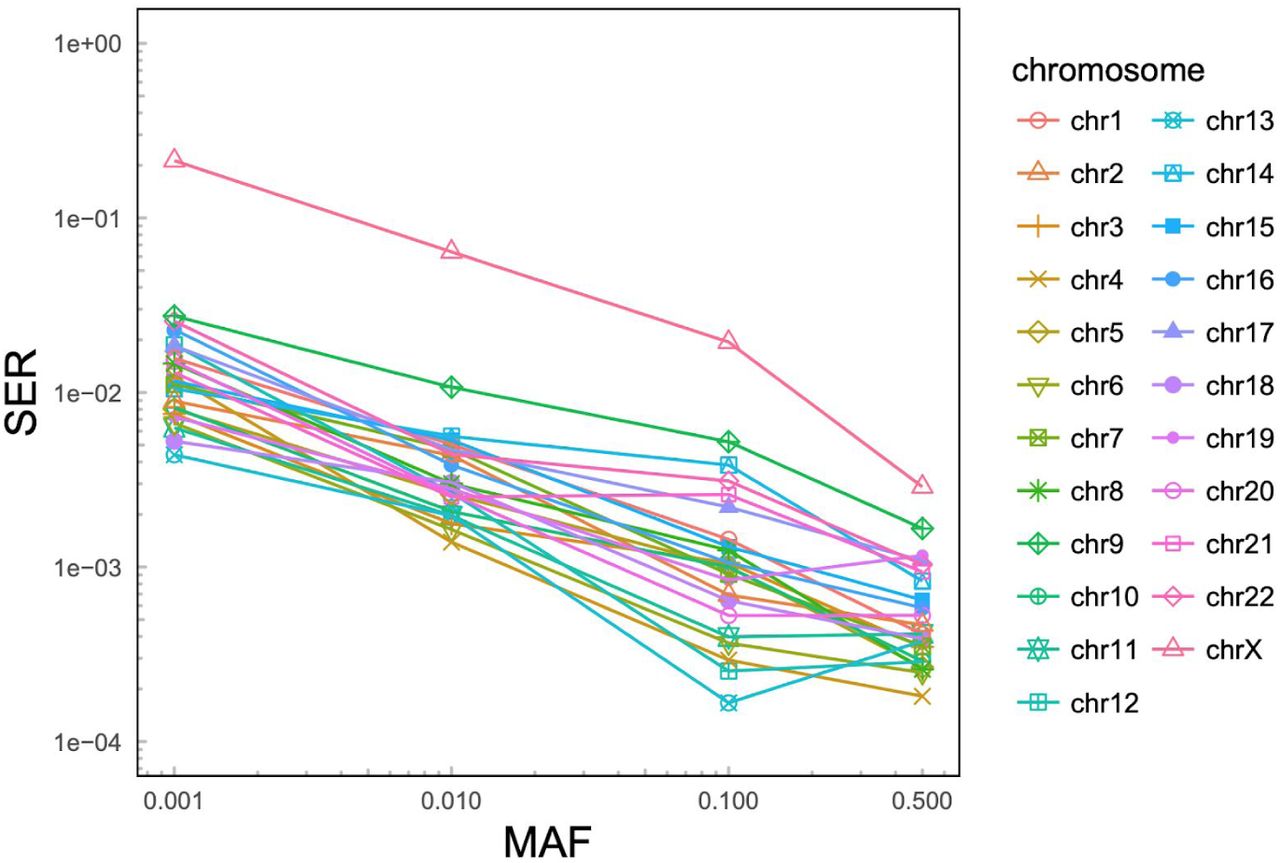
We evaluated phasing accuracy of the phased panel by computing switch error rate (SER) among pairs of consecutive heterozygous sites in sample NA12878 (child in a full trio in the expanded 3,202-sample cohort). As the phasing truth set against which the evaluation was performed, we used the extensively validated, haplotype-resolved Platinum Genome NA12878 callset generated by Illumina16. The SER among SNVs and INDELs across all autosomes was 0.074% (1,754 switches among 2,338,955 assessed SNV/INDEL heterozygous pairs), indicating high accuracy of phasing. As expected, chromosome X, which was phased without pedigree-based correction, displayed higher SER as compared to autosomes (SER=0.491%; 362 switches among 73,794 assessed SNV/INDEL heterozygous pairs total; Figure S6; Table S4). We did not observe a significant difference in phasing accuracy between SNVs and INDELs, other than on chromosome X, where SER for INDELs was 2.01% (187 switches among 9,298 assessed INDEL heterozygous pairs total) as compared to 0.51% for SNVs (328 switches among 64,583 assessed SNV heterozygous pairs total) (Figure S7). In addition to assessing SER genome-wide, we also assessed it at the following four MAF bins: 1) (0%, 0.1%]; 2) (0.1%, 1%]; 3) (1%, 10%]; and 4) (10%, 50%]. While we noticed an expected increase in SER with decrease in MAF, the SER remained low throughout the entire MAF spectrum, reaching a maximum of 1.2% in the (0, 0.1%] MAF bin across autosomes (Figure 5B, violet solid line; see Figure S6 for per chromosome breakdown). Such high phasing accuracy at the low end of the MAF spectrum can be attributed to both the presence of family members in the expanded 1kGP cohort (Figure 5B, dashed violet line with open triangles vs. dashed violet line with open diamonds) as well as to pedigree-based correction applied after statistical phasing (Figure 5B, solid violet line vs. dashed violet line with open triangles).
Finally, we compared phasing accuracy of the high coverage family-based panel against the phase 3 panel. The phase 3 panel was phased using statistical phasing with family-based scaffold (built from chip array data on the entire 1kGP cohort including related samples), without pedigree-based correction, as no trios were sequenced as part of the phase 3 panel3. We observed an order of magnitude lower SER in the high coverage as compared to the phase 3 panel, across the entire MAF spectrum (Figure 5B, solid violet vs. solid aqua line). This significant improvement in SER underscores the power that inclusion of trios has on the quality of haplotype phasing in the upgraded panel. It is worth noting that the phasing accuracy of the 2,504-sample phase 3 dataset was slightly better than that of the 2,504-sample high coverage dataset (Figure 5B, solid aqua line vs. dashed violet line with open diamonds) due to the fact that the latter dataset was phased using statistical phasing alone, without the family-based scaffold.
Imputation performance of the high coverage 3,202-sample SNV/INDEL panel
To assess imputation performance, we imputed a set of 279 diverse samples from the Simons Genome Diversity Project (SGDP)32 using IMPUTE2 software33 with the high coverage and the phase 3 panels separately as the reference. We evaluated the accuracy of imputed genotypes by computing the squared correlation (R2) between imputed allele dosages and dosages from WGS data across 110 samples, 22 from each of the five super-populations (the maximum number of samples in all populations). For the high coverage panel, the aggregate R2 between imputed SNV genotypes and WGS genotypes for EUR and SAS samples reaches 0.88 at an AF of 0.2%, and increases at more common variant frequencies (Figure 5C). Imputation of AFR, EAS, and AMR samples attains an R2 ≥0.8 at AF of 3%, 5%, and 5%, respectively. In general, INDELs are imputed with lower accuracy than SNVs, reaching an R2 >0.8 at an alternate allele frequency of 5% in EUR and SAS samples, and 20% in AFR, EAS, and AMR samples. Figures 5D and 5E compare the imputation accuracy of the phase 3 and high coverage panels for SNVs and INDELs across sites shared between the panels. Using the high coverage reference panel leads to a significant increase in imputation performance compared to the phase 3 panel for SNVs and INDELs across all super-populations. These differences are largest for EUR (high coverage panel SNP R2=0.98 at 0.1% AF; phase 3 panel SNP R2=0.49 at 0.1% AF) and SAS samples (high coverage panel SNP R2=0.99 at 0.2% AF; phase 3 panel SNP R2=0.85 at 0.2% AF). Differences in imputation performance between the panels are most pronounced for INDELs which see a 9.6-fold increase in the accuracy of imputing EUR samples (AF=0.1%), 4.2-fold increase in SAS (AF=0.2%), 8.9-fold increase in AFR (AF=0.1%), 0.29-fold increase in EAS (AF=0.1%), and 9-fold increase in AMR (AF=0.2%) samples when using the high coverage panel.
DISCUSSION
We present results from high coverage WGS of the expanded 1kGP cohort, consisting of 2,504 original samples as well as additional 698 related samples, completing 602 trios in the cohort. We called >111 million SNVs, >14 million INDELs, and ∼170,000 SVs across the 3,202 samples, using state-of-the-art methods. When compared to the low coverage phase 3 1kGP dataset published in 2015, the variant counts in the high coverage callset reflect an estimated increase of 258,505 SNVs (1.07-fold), 321,965 INDELs (1.59-fold), and 5,835 (2.7-fold) SVs per genome, and a cohort-level increase of 16.6 million SNV, 5.9 million INDEL, and ∼100 thousand SV loci, across the original 2,504 unrelated samples. As expected, given that the phase 3 dataset identified nearly all common SNVs (MAF > 1%) in the population, the vast majority of the new SNVs identified here were in the rare MAF spectrum (AC ≤ 2). Since limitations of the low coverage sequencing had a greater negative impact on INDEL as compared to SNV calling in the phase 3 dataset, we observed gains in INDEL counts across the entire MAF spectrum, with gains in the rare end of the spectrum being the most pronounced.
We acknowledge that a direct comparison of the high coverage 1kGP SNV/INDEL dataset against the phase 3 set was impossible due to differences in genomic builds that were used for variant calling during generation of the two callsets. To address this, we used a version of the phase 3 dataset that was lifted over from the GRCh37 to the GRCh38 reference. As a result, over 2 million variants (99% of which are on chromosomes X and Y) that failed the lift-over had to be excluded from the comparative analysis. Importantly, we were not positioned to dissect the impact of various factors that likely contributed to differences in variant calls between the high coverage and phase 3 datasets. Differences in sequencing platforms and read length (phase 3: Illumina HiSeq 2000 and HiSeq 2500, 76 bp or 101 bp paired-end reads; high coverage: Illumina NovaSeq 6000, 150 bp paired-end reads), library preparation (phase 3: PCR-based; high coverage: PCR-free), sequencing coverage (phase 3: mean depth 7.4X; high coverage: mean depth 34X), reference genome (phase 3: GRCh37; high coverage: GRCh38), alignment software (phase 3: BWA 0.5.9; high coverage: BWA-MEM 0.7.15), as well as in downstream bioinformatics pipeline most likely all contributed to various degrees to the differences in variant calls that we described here. Overall, despite major differences in data generation and analysis, we found high concordance between the high coverage and the phase 3 SNV/INDEL callset. Nearly 95% of small variants from the phase 3 callset were recalled in the high coverage dataset with AF correlation of 0.90-0.975, depending on the region of the genome.
The SVs presented here provide a significant increase over the phase 3 callset, both in the number of SVs detected per genome (9,266 vs. 3,431) and in the number of SVs predicted to directly alter gene function (149 vs. 32 per genome) across these populations. Notably, one limitation of the 1kGP dataset is the use of cell line DNA, which can include somatic mutations that arise during cell proliferation. In agreement with the Polaris project34, we observed aneuploidy of allosomes in 0.94% of the samples, and sub-chromosomal level mosaic CNVs on multiple autosomes (Figure S12). We further performed manual inspection of all large CNVs (>50Kb, n = 3717), as well as benchmarked large inversions against Strand-seq (n = 256), and the variants that lack support were labeled as ‘LowQual’ in the SV callset so that they can be easily excluded from future analyses. This data, coupled with the availability of inheritance information from 602 complete trios and the independent availability of long read WGS, Strand-seq, and optical mapping data on 34 of these samples18, provides an unprecedented open access SV resource for methods development and genomic studies.
Inclusion of 602 trios in the expanded 1kGP cohort led to high accuracy of SNV/INDEL haplotype phasing due to both an increase in long-range haplotype sharing between related samples which facilitates phasing, and pedigree-based correction applied after statistical phasing to ensure consistency of phased haplotypes with the pedigree structure. We also demonstrate that the phased high coverage SNV/INDEL panel exhibits an order of magnitude higher phasing accuracy as compared to the phase 3 dataset across the entire MAF spectrum. Importantly, improvements in small variant calling, coupled with higher phasing accuracy of the high coverage panel, translated into significantly better imputation accuracy, especially for INDELs, across all of the 1kGP super-populations when the high coverage panel was used as the reference for imputation as compared to the phase 3 panel.
For more than a decade, the 1000 Genomes collection has been a key resource in the field of genomics. These datasets have produced scientific insights into population genetics and genome biology, as well as provided an openly sharable resource that has been widely used in methods development and testing as well as for technical validation. By generating high coverage sequencing data for the complete phase 3 set of 2,504 unrelated individuals and completing 602 trios with 698 additional samples, we have updated this critical resource with benchmarks and standards for a new generation of large-scale international whole genome sequencing initiatives. Our state of the art SNV, INDEL, and SV callsets, freely released, provide the most accurate and comprehensive catalog of variation compiled to date across this diverse genomic resource, particularly in rare SNVs and all classes of indels and SVs that were challenging to detect using earlier sequencing and analysis methods on low coverage data. We also present an improved phasing and imputation panel leveraging full sequence from trios that outperforms the existing imputation panels. Importantly, this panel is fully public and can be freely downloaded and used in combination with other panels and for use with any target dataset. Although many larger sequencing projects have now been conducted, the open nature of the 1000 Genomes samples will continue to make this a foundational resource for the community in the years to come.
METHODS
Data collection, WGS library preparation and sequencing
DNA extracted from lymphoblastoid cell lines (LCL) was ordered from the Coriell Institute for Medical Research for each of the 3,202 1kGP samples. Whole genome sequencing (WGS) libraries were prepared using the Truseq DNA PCR-free (450bp) Library Preparation Kit in accordance with the manufacturer’s instructions. Briefly, 1ug of DNA was sheared using a Covaris LE220 sonicator (adaptive focused acoustics). DNA fragments underwent bead-based size selection and were subsequently end-repaired, adenylated, and ligated to Illumina sequencing adapters. Final libraries were evaluated using fluorescent-based assays including qPCR with the Universal KAPA Library Quantification Kit and Fragment Analyzer (Advanced Analytics) or BioAnalyzer (Agilent 2100). Libraries were sequenced on an Illumina Novaseq 6000 sequencer using 2 x 150bp cycles.
Alignment and SNV/INDEL calling
Read alignment to the human reference genome GRCh38, duplicate marking, and Base Quality Score Recalibration (BQSR) were performed according to the functional equivalence pipeline standard developed for the Centers for Common Disease Genomics project 23. SNV and INDEL calling was performed using GATK version 3.5, as described below. For variant discovery we used HaplotypeCaller in GVCF mode35 with sex-dependent ploidy settings on chromosome X and Y. Specifically, variant discovery on chromosome X was performed using diploid settings in females, diploid settings on PAR regions in males, and haploid settings on non-PAR regions in males. Variant discovery on chromosome Y was performed with haploid settings in males and was skipped entirely in females. We combined GVCFs in batches of ∼200 samples using CombineGVCFs and joint-genotyped all 3,202 samples with GenotypeGVCFs. We then used VariantRecalibrator to train the Variant Quality Score Recalibration (VQSR) model using “maxGaussians 8” and “maxGaussians 4” parameters for SNVs and INDELs, respectively. We applied the VQSR model to the joint callset using ApplyRecalibration with truth sensitivity levels of 99.8% for SNVs and 99.0% for INDELs. Variant annotations were performed using SnpEff 36 and BCFtools37. SnpEff was used to annotate variant effect prediction and BCFtools was used to annotate membership and allele frequency from various variant databases like 1kGP phase 33, cosmic (v79) (https://cancer.sanger.ac.uk/cosmic)38, dbNSFP (v3.2a)39,40, dbSNP (v151)27 and ExAC (v0.3)41.
Quality control of sequence data
We ran a number of quality control (QC) tools to look for quality issues, sample swaps, and contamination issues. We ran FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) on the raw sequence data to assess yield and raw base qualities. We ran Picard CollectMultipleMetrics and CollectWGSMetrics (http://broadinstitute.github.io/picard/) on the aligned BAM to collect alignment and insert size metrics. Picard CollectGcBiasMetrics was run to compute normalized coverage across multiple GC bins. Reads duplication metrics were quantified by running Picard MarkDuplicates on the BAM.
All the samples had at least 27X mean coverage across the genome (average per sample coverage: 34X, range: 27X-71X) and at least 91% of the bases at base quality score 30 or higher (Figure S8A, B). The mean duplicate rate across the samples was 9% but there were 5 samples that had a duplicate rate greater than 20 (Figure S8C). The median per sample insert size was 433 bp (Figure S8D). Higher duplication rate is a known issue with Illumina’s patterned flow cell that uses exclusion amplification clustering method to increase data output, but this chemistry is very sensitive to library loading concentrations. Higher loading concentrations can lead to low throughput because of polyclonal clusters being formed in the nanowells of the patterned flow cell, whereas low concentration can lead to pad hopping which increases the duplication rate. VerifyBamID42 was run in chip-free mode to estimate the likelihood of sample contamination. We use a cutoff of 2% to flag any sample for contamination and none of the samples reached the cutoff.
To make sure there were no sample mix-ups we ran genotype concordance against genotyping chip data. For that, we used the chip data that was released with phase 3. We did not find chip data for 15 samples in phase 3 so for those we ran Infinium CoreExome-24 v1.3 chip and performed genotype concordance. All the samples had >97% genotype concordance.
BCFTools was used to split multiallelic variants into multiple rows and normalize INDELs before performing cumulative variant counts. Per sample variant metrics were collected using GATK VariantEval. The reference genome sequence was divided into three categories: Easy, Medium, and Hard, as defined in the functional equivalence of genome sequencing analysis pipelines paper23. The Easy genomic regions consist of GIAB gold standard high confidence regions. These are regions with mostly unique sequences where variant calling can be performed confidently. The Hard regions consist of centromeres, microsatellite repeats, low complexity regions, and windows determined to have high copy number. Any sequence that did not fall into either Easy or Hard regions was classified as Medium. FDR was calculated by comparing variant calls on sample NA12878 from the 3,202 callset to the high confidence variant calls from the GIAB version 3.3.2 on the same sample. The VCF files were compared using hap.py (v0.3.12; github.com/Illumina/hap.py) using the rtg-tools (v3.8.2)43 vcfeval comparison engine. For FDR calculation the variant comparison was restricted to the confident regions as defined by the GIAB.
Comparison of the high coverage SNV/INDEL calls against the phase 3 dataset
To compare the high coverage against the phase 3 callset, we used the GRCh38 lifted over version of the phase 3 callset, as described in https://www.internationalgenome.org/announcements/updated-GRCh38-liftover/. Due to the high number of liftover failures on chromosomes X and Y we restricted the comparison to autosomes. Additionally, for FDR comparative analysis against the phase 3 callset, we excluded regions in GRCh38 that could not be lifted over from GRCh37 prior to computing FDR. This was done by going over each interval in GRCh38 GIAB confident regions and trying liftover to GRCh37 using CrossMap44. Any interval or part of the interval that either failed to liftover or lifted over to a different chromosome were part of the exclusion criteria. Overall only 0.1% of bases were excluded from the GIAB confident regions due to liftover issues. When we applied the exclusion list to the 2,504 high coverage callset, 7,486 variants were filtered out and 526 were filtered out from the phase 3 callset. See Table S3 for the percentage of bases in GRCh38 that were part of the exclusion list broken down by chromosome.
SV discovery using GATK-SV
GATK-SV involved an ensemble SV discovery and refinement pipeline for WGS data. The technical details of the method were previously described in Collins et al19 for application to the genome aggregation database (gnomAD) for SV discovery, and further described in analyses from the HGSVC18. In this study, the same methods were applied to all 3,202 samples for SV discovery. In brief, SVs discovered by Manta, Wham, MELT, cn.MOPS and GATK-gCNV from Ebert et al. were integrated, genotyped across all samples, resolved for complex SVs, and annotated for variant class and functional impact. The FDR was previously assessed from analyses in quartet families, which yielded a 97% molecular validation rate for de novo SV predictions45, as well as a 94% validation rate compared to long-read sequencing19.
SV discovery using svtools
The svtools46 method was previously described in Abel et al20 and applied for SV discovery across 17,795 genomes from the Centers for Common Disease Genomics (CCDG) program.20 The workflow combines per-sample variant discovery with lumpy47 and manta48 with resolution-aware cross-sample merging. The set of merged variants is then genotyped with svtyper49, followed by copy-number annotation with cnvnator50 and reclassification of variants based on concordance of read-depth with breakpoint orientation. All parameter settings and versions are as implemented in the wdl-based work (https://github.com/hall-lab/sv-pipeline).
Large insertion discovery using Absinthe
On a per-sample basis, insertions with a minimum length of 100bp were discovered through de novo assembly of unmapped and discordant read pairs using Absinthe (github.com/nygenome/absinthe), and then genotyped using Paragraph51, respecting sex-specific ploidies. Insertion calls from all 3,202 samples that were positively genotyped with a PASS filter flag were then clustered by genomic location and aligned using MAFFT52. For each locus, the most consensual allele was selected. Variants from the resulting merged callset were then re-genotyped on all 3,202 individuals. To produce the final callset only variants with 1) genotyping PASS filter rate >= 80%; 2) Mendelian Error Rate <= 5% for complete trio calls; and 3) HWE Chi-square test p-value > 1e−6 in at least one of the 5 super-populations were kept.
Integration of GATK-SV, svtools, and Absinthe callsets
We conducted a series of analyses to benchmark SVs from each of the three methods described above, including their false discovery rate (FDR) as indicated by inheritance rates and support from orthogonal technologies, as well as their breakpoint precision estimated by the deviation of their SV breakpoints from long read assemblies in three genomes from analyses in the HGSVC17. We also compared the three callsets to decide on the optimal integration strategy to maximize sensitivity and minimize FDR in the final ensemble callset (Table S6). Details of the comparison and integration strategies are described separately for insertions and all other variant classes below.
Integration of insertions from GATK-SV and Absinthe
We compared the de novo rate of variant calls from each pipeline for insertions, yielding results of 4.1% for GATK-SV, 25.8% for svtools, and 2.4% for Absinthe. Given these results we restricted integration of insertions to GATK-SV and Absinthe. Each insertion pair was considered concordant if the insertion points were within 100 bp. The FDR of each insertion callset was estimated from three measurements: 1. de novo rate of SVs observed in the 602 trios; 2. proportion of SVs that were not validated by VaPoR53, an algorithm that evaluates SV quality by directly comparing raw PacBio reads against the reference genome, and 3. proportion of SVs that were not overlapped by SVs from PacBio assemblies in the same genome (Figure S9D). Precision of an insertion call was estimated by the distance of the insertion point to the closest PacBio insertion and the difference between the length of inserted sequence versus the length of the closest PacBio insertion calculated as an odds ratio. Both insertion callsets display less than 5% FDR based on inheritance and PacBio support, and the callsets were thus merged for all subsequent analyses (Figure S9D). Notably, as Absinthe showed higher precision than GATK-SV, as measured from both the coordinates of the insertion point and the length of inserted sequences (Figure S9H, I), we retained the Absinthe record for insertions that were shared by both methods.
Integration of deletions, duplications, and inversions from GATK-SV and svtools
To consider a pair of SVs of the same variant class other than insertions as concordant, 50% reciprocal overlap was required for SVs larger than 5 kb and 10% reciprocal overlap was required for variants under 5 kb respectively. The FDR across variant calls was evaluated using the same measurements as described above. For deletions, duplications, and inversions, we observed low FDR (<5%) among variants that were shared by GATK-SV and svtools, but significantly higher FDR in the subset that were uniquely discovered by either algorithm (Supp Figure S9E-G). To restrict the final callset to high-quality variants, a machine learning model (lightGBM54) was trained on each SV class. Three samples that were previously analyzed in the HGSVC studies (HG00514, HG00733, NA19240)17,18 were selected to train the model. The truth data was defined by SVs that were uni-parentally inherited, shared by GATK-SV and svtools, supported by VaPoR, and overlapped by PacBio callsets. The false training subset was selected as SVs that appeared as de novo in offspring genomes, specifically discovered by either GATK-SV or svtools, not supported by VaPoR, and not overlapped by PacBio callsets. Multiple features were included in the model, including the sequencing depth of each SV, the depth of the 1kb region around each SV, the count of aberrant pair ends (PE) within 150 bp of each SV, the count of split reads (SR) within 100 bp of each breakpoints, the size, allele fraction and genomic location (split into short repeats, segmental duplications, all remaining repeat masked regions, and the remaining unique sequences) of each SV, and the fraction of offspring harbor a de novo variant among trios in which the SV is observed. Each SV per genome was assigned a ‘boost score’ by the lightGBM model, and SVs with >0.448 boost score were labeled as ‘PASS’ in the model (Figure S9M, N). This threshold was specifically selected to retain an estimated FDR < 5%. Callset specific SVs that failed the lightGBM model in less than 48% of all examined samples were included in the final integrated callst (Figure S9N).
To design strategies to merge SVs shared by GATK-SV and svtools, the precision of SV calls was evaluated by examining the distance between breakpoint coordinates of SVs to matched calls in the PacBio callset. Comparable breakpoint precision was observed for GATK-SV and svtools (Figure S9J-L). Thus, for SVs in each sample, the variant with the greatest number of split reads for each breakpoint was selected, or if equivalent then the variant with the higher boost score was retained, then for each locus the SV observed in the greatest number of samples was retained as final.
Inclusion of mCNV, CPX and CTX variants from GATK-SV
Other minority SVs types, including mCNVs, CPX and CTX, were specifically detected by GATK-SV, so we performed in-depth manual inspection to ensure their quality before including them in the final integration callset. The depth profile across all 3,202 samples around each mCNV was plotted for manual review, and mCNVs that did not show clear stratification among samples were labeled as ‘Manual_LQ’ in the filter column even if they showed clear deviation from the normal copy number of 2 (Figure S10). For CTX, the aberrantly aligned read-pairs across each breakpoint was manually examined, and variants that lacked sufficient support were labeled as ‘Manual_LQ’ in the final callset.
Haplotype phasing
For haplotype phasing we used statistical phasing with pedigree-based correction, as implemented in the SHAPEIT2-duohmm software28,29. Phasing with SHAPEIT2-duohmm was performed per chromosome using the default settings, except for the window size “-W” which was increased from 2Mb (default) to 5Mb to account for increased amounts of shared IBD due to pedigrees being present in the dataset (as recommended in the SHAPEIT2 manual). SHAPEIT2 does not handle multiallelic variant phasing. To phase both biallelic and multiallelic variants, we first split the multiallelic sites into separate rows, while left-aligning and normalizing INDELs, using the bcftools norm tool 37. We then shifted the position of multiallelic variants (2nd, 3rd, etc ALT alleles) by 1 or more bp (depending on how many ALT alleles there are at a given position) to ensure a unique start position for all variants, which is required for SHAPEIT2. The positions were shifted back to the original ones after phasing. SHAPEIT2 duohmm supports phasing of autosomal variants only. Therefore, to phase variants on chromosome X we used statistical phasing as implemented in the Eagle2 software30. Phasing with Eagle2 was performed using default parameters. No shifting of positions for multiallelic sites was needed as Eagle2 supports phasing of variants with the same start site. Phasing accuracy evaluation was performed using the WhatsHap compare tool55. As a measure of phasing accuracy we used switch error rate (SER), which is defined as:
 In all of the evaluations, SER was computed for sample NA12878 relative to the Platinum Genome NA12878 gold standard truth set 16.
In all of the evaluations, SER was computed for sample NA12878 relative to the Platinum Genome NA12878 gold standard truth set 16.
Imputation performance evaluation
We performed imputation on 279 samples from 130 diverse populations using WGS data from the Simons Genome Diversity Project (SGDP) 32. To create a pseudo-GWAS dataset, we extracted the genotypes at all sites included on an Illumina Infinium Omni2.5-8 v1.4 array. We performed quality control (QC) of the dataset using standard pre-imputation filters, removing sites which did not meet at least one of the following criteria: genotype call rate of >= 95%, MAF > 1%, and HWE p-value >= 1e−4. We used plink software 56 for all QC steps, and analysis was restricted to the autosomes. We imputed the data passing quality control with the phase 3 and the high coverage panels, separately. For the phase 3 reference panel, we used the low coverage 1000 Genomes phased SNV set called directly against GRCh38 by EBI. SHAPEIT228 was used to perform a strand check of the dataset and remove any problematic sites as determined by aligning with the respective panel. We pre-phased the data using SHAPEIT2 and an input reference panel. We imputed the pre-phased data using the IMPUTE233 software with default parameters. Following imputation, we concatenated the imputed intervals to create an autosome-wide imputed dataset with each of the panels. We evaluated imputation using 22 samples from each of the five super-populations (EUR, AFR, SAS, EAS, and AMR) and compared the held out imputed genotypes with the WGS genotypes stratified by MAF. For this evaluation, we converted the posterior genotype probabilities produced by IMPUTE2 to dosages using QCTOOL version (www.well.ox.ac.uk/∼gav/qctool_v2/), and the WGS genotypes to dosages using BCFtools37. For the data imputed with the high coverage panel, we computed the squared correlation (R2) between the imputed dosages and those from the WGS data for all non-missing sites. To compare imputation accuracy between the phase 3 and the high coverage panels, we restricted the evaluations to only sites shared between the two panels.
DATA ACCESS
To download the bam files, SNV/INDEL VCF, SV VCF, as well as the filtered haplotype-resolved SNV/INDEL callset, please visit: https://www.internationalgenome.org/data-portal/data-collection/30x-grch38. Sequence data was deposited in ENA under projects PRJEB31736 and PRJEB36890.
Competing interests
M.C.Z. is a shareholder in Merck & Co and Thermo Fisher Scientific. P.F. is a member of the scientific advisory boards of Fabric Genomics, Inc., and Eagle Genomics, Ltd. J.L. is an employee and shareholder of Bionano Genomics.
AUTHOR CONTRIBUTIONS
Writing of the manuscript and figure generation: M.B., U.S.E., X.Z., A.O.B.
SNV/INDEL calling: U.S.E., M.B.
SNV/INDEL analysis: U.S.E., M.B., A.O.B., R.M.
SV calling: X.Z., H.B., H.A., A.A.R., A.C., W.E.C., The Human Genome Structural Variant Consortium.
SV integration and analysis: X.Z., H.B.
Production and quality control of the WGS data: L.W., A.R., U.S.E., M.B., K.N.
Data coordination, data sharing, and user support: S.F., E.L., P.F.
These authors jointly supervised this work: S.G., I.M.H., M.E.T, G.N., M.C.Z.
CONSORTIA
The Human Genome Structural Variation Consortium
Evan E. Eichler1,2, Jan O. Korbel3, Charles Lee4,5, Scott E. Devine6, William T. Harvey1, Weichen Zhou7, Ryan E. Mills7, Tobias Rausch3, Sushant Kumar8, Can Alkan9,10, Fereydoun Hormozdiari11, Zechen Chong12, Xiaofei Yang4,13,14, Jiadong Lin15, Mark B. Gerstein8, Ye Kai15, Qihui Zhu4, Feyza Yilmaz4.
1. Department of Genome Sciences, University of Washington School of Medicine, 3720 15th Ave NE, Seattle, WA 98195-5065, USA.
2. Howard Hughes Medical Institute, University of Washington, Seattle, WA 98195, USA.
3. European Molecular Biology Laboratory (EMBL), Genome Biology Unit, Meyerhofstr. 1, 69117 Heidelberg, Germany.
4. The Jackson Laboratory for Genomic Medicine, 10 Discovery Dr, Farmington, CT 06030, USA.
5. Precision Medicine Center, The First Affiliated Hospital of Xi’an Jiaotong University, 277 West Yanta Rd., Xi’an, 710061, Shaanxi, China.
6. Institute for Genome Sciences, University of Maryland School of Medicine, 670 W Baltimore Street, Baltimore, MD 21201, USA.
7. Department of Computational Medicine & Bioinformatics, University of Michigan, 500 S. State Street, Ann Arbor, MI 48109, USA.
8. Program in Computational Biology and Bioinformatics, Yale University, BASS 432&437, 266 Whitney Avenue, New Haven, CT 06520, USA.
9. Department of Computer Engineering, Bilkent University, Ankara, 06800, Turkey.
10. Bilkent-Hacettepe Health Sciences and Technologies Program, Bilkent University, Ankara, 06800, Turkey.
11. Department of Biochemistry and Molecular Medicine, MIND Institute and Genome Center, University of California, Davis, CA 95616, USA.
12. Department of Genetics and Informatics Institute, School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
13. Department of Computer Science and Technology, School of Electronic and Information Engineering, Xi’an Jiaotong University, Xi’an, 710049, China.
14. MOE Key Lab for Intelligent Networks & Networks Security, School of Electronics and Information Engineering, Xi’an Jiaotong University, Xi’an, 710049, China.
15. School of Automation Science and Engineering, Faculty of Electronic and Information Engineering, Xi’an Jiaotong University, Xi’an, Shaanxi, 710049, China.
SUPPLEMENTARY INFORMATION
Supplementary Figures

Distribution of per sample heterozygous to homozygous ratio (Het/Hom) based on SNVs and INDELs across the 3,202 samples.

Novel variants are defined as variants that are not reported in dbSNP build 151.

Sensitivity vs. precision of SNV/INDEL calls in the high coverage 3,202-sample callset, based on comparison against the GIAB NA12878 truth set.

Length distribution of INDEL calls in the 2,504-sample high coverage and phase 3 callsets.

Same as Figure 5A, but stratified by variant type.

Analogous to Figure 5B (solid purple line), but stratified by chromosome.

Chromosome X is shown separately as it was phased using a different strategy as compared to autosomes (statistical phasing vs. statistical phasing with pedigree-based correction, respectively).

A) Mean coverage distribution. B) % bases with quality >= 30. C) % duplication. D) Insert size distribution.

(A) Overlap of insertion sites between GAKT-SV and Absinthe callsets. (B) Overlap of SV other than insertions between the GATK-SV and svtools callset. (C) Overlap of each SV type between GATK-SV, svtools and Absinthe. (D) Overlap of insertions in each genome between GATK-SV and Absinthe. (E-G) Overlap of deletions (E), duplications (F), inversion and complex SVs (G) in each genome between GATK-SV and svtools. The integers in (D-G) represent count of SVs per sample, followed by proportion of SVs validated by VaPoR / proportion of SVs assessable by VaPoR in the second row, proportion of SVs supported by PacBio SVs in Ebert et al. 2020 / proportion of SVs supported by PacBio SVs in Chaisson et al. 2019 in the third row, and transmission rate /rate of bi-parentally inherited SVs in the fourth row. (H-I) Precision of the insertion breakpoint (H) and length (I) assessed against PacBio assemblies. (J-K) Precision of the SV breakpoints in GATK-SV (J) and svtools (K) callsets assessed against PacBio assemblies. (L) Breakpoint distance of SVs shared by GATK-SV and svtools. (M-N) de novo rate of SVs in GATK-SV (M) and svtools (N) callset when filtered at different boost score cutoffs. (O) False positives and false negatives in the GATK-SV and svtools callsets when filtered at different boost score cutoffs.

Read depth distribution of large deletions (red), duplications (blue) and multiallelic CNVs (green) across the 3,202 samples.

Count of genes interrupted by copy gain (GC) and interagenic exon duplications (IED) SVs in the current ensemble callset and 1kGP Phase 3 SV callset.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A). Ploidy of allosomes. (B). Copy number of each chromosome. Each dot represents a copy number of the 1Mbp bin in a sample. Blue dots are samples with copy gain and red dots represent copy loss.
Supplementary tables
Sample counts broken down by ancestry, sex, cohort, and presence within pedigrees.
Mean SNV density per 1kb of sequence in 3,202-sample high coverage callset.
Percentage of bases in GRCh38 that could not be lifted over from GRCh37 within the GIAB confident regions.
Summary of the high coverage panel phasing accuracy evaluation stratified by chromosome.
Count of SV sites across 3,202 samples and SVs per sample.
Quality of SVs evaluated by PacBio support and inheritance.
ACKNOWLEDGEMENTS
The following cell lines/DNA samples were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research: [NA06984, NA06985, NA06986, NA06989, NA06991, NA06993, NA06994, NA06995, NA06997, NA07000, NA07014, NA07019, NA07022, NA07029, NA07031, NA07034, NA07037, NA07045, NA07048, NA07051, NA07055, NA07056, NA07340, NA07345, NA07346, NA07347, NA07348, NA07349, NA07357, NA07435, NA10830, NA10831, NA10835, NA10836, NA10837, NA10838, NA10839, NA10840, NA10842, NA10843, NA10845, NA10846, NA10847, NA10850, NA10851, NA10852, NA10853, NA10854, NA10855, NA10856, NA10857, NA10859, NA10860, NA10861, NA10863, NA10864, NA10865, NA11829, NA11830, NA11831, NA11832, NA11839, NA11840, NA11843, NA11881, NA11882, NA11891, NA11892, NA11893, NA11894, NA11917, NA11918, NA11919, NA11920, NA11930, NA11931, NA11932, NA11933, NA11992, NA11993, NA11994, NA11995, NA12003, NA12004, NA12005, NA12006, NA12043, NA12044, NA12045, NA12046, NA12056, NA12057, NA12058, NA12144, NA12145, NA12146, NA12154, NA12155, NA12156, NA12234, NA12239, NA12248, NA12249, NA12264, NA12272, NA12273, NA12274, NA12275, NA12282, NA12283, NA12286, NA12287, NA12329, NA12335, NA12336, NA12340, NA12341, NA12342, NA12343, NA12344, NA12347, NA12348, NA12375, NA12376, NA12383, NA12386, NA12399, NA12400, NA12413, NA12414, NA12485, NA12489, NA12546, NA12707, NA12708, NA12716, NA12717, NA12718, NA12739, NA12740, NA12748, NA12749, NA12750, NA12751, NA12752, NA12753, NA12760, NA12761, NA12762, NA12763, NA12766, NA12767, NA12775, NA12776, NA12777, NA12778, NA12801, NA12802, NA12812, NA12813, NA12814, NA12815, NA12817, NA12818, NA12827, NA12828, NA12829, NA12830, NA12832, NA12842, NA12843, NA12864, NA12865, NA12872, NA12873, NA12874, NA12875, NA12877, NA12878, NA12889, NA12890, NA12891, NA12892].
These data were generated at the New York Genome Center with funds provided by NHGRI Grants 3UM1HG008901-03S1 and 3UM1HG008901-04S1. A.C., W.E.C., and M.C.Z. were partially supported by NHGRI grant UM1HG008901. S.F., E.L., and P.F. were partially supported by the Wellcome Trust (WT104947/Z/14/Z) and the European Molecular Biology Laboratory. Support for analyses by X.Z. and M.E.T. was provided by NIMH MH115957-02.
REFERENCES




