

ABSTRACT
Microorganisms need to adapt to environmental changes, and genome plasticity can lead to adaptation by increasing genetic diversity.
The CTG (Ser1) clade of fungi is a diverse yeast group that can adapt remarkably well to hostile environments. Genome plasticity has emerged as a critical regulatory mechanism, in one member of the (Ser1) clade: the human fungal pathogen Candida albicans. However, in many aspects, C. albicans differs from other CTG (Ser1) members as it lacks a canonical sexual cycle and is an obligatory commensal.
It is still unknown whether environmental CTG (Ser1) fungi with a canonical sexual cycle utilise genome plasticity as a strategy for adaptation.
To address this question, we investigated genome plasticity in the CTG (Ser1) yeast Scheffersomyces stipitis. The non-pathogenic S. stipitis yeast does not live in the human host and has a canonical sexual cycle. We demonstrated that the S. stipitis genome is intrinsically unstable. Different natural isolates have a genome with a dissimilar chromosomal organisation, and extensive genomic changes are detected following in vitro evolution experiments. Hybrid MinION Nanopore and Illumina genome sequencing demonstrate that retrotransposons are major drivers of genome diversity and that variation in genes encoding adhesin-like proteins is linked to distinct phenotypes.
INTRODUCTION
Eukaryotic genomes are often described as stable structures with well-preserved chromosome organisation, and genome instability is viewed as harmful. However, the genome of eukaryotic microorganisms is plastic and can adopt different chromosomal organisations. Indeed, genome instability can be beneficial in microbial organisms that need to adapt rapidly and reversibly to changing environments (26–29). This is because genomic instability can increase genetic diversity, thereby allowing selection of genotype(s) better adapted in a new environment. For example, chromosome translocation can result in gene expression changes by disrupting the transcriptional regulatory networks or heterochromatin driven-gene silencing (30, 31). Selective environmental pressures coupled to genome instability can lead to intra-species genetic variation where an individual of the same species exhibits distinct genetic make-up and phenotypic properties (32, 33). Genome instability is not random; it occurs more frequently at specific hotspots that are often repetitive or enriched in DNA repeats (34). For this reason, transposable elements (TE) are one of the most common instability hotspots. TEs include a diverse group of DNA sequences present in multiple copies along eukaryotic chromosomes, which can jump to new sites in the genome (35). TEs can shape genome structure by jumping into regulatory regions and/or Open Reading Frames (ORFs) altering or disrupting gene expression and protein function. Alternatively, homologous recombination of nearly identical TE copies can generate complex chromosomal rearrangements such as translocations and gene deletion (36). TEs belong to two major classes: DNA transposons and retrotransposons. DNA transposons utilise a “cut and paste” mechanism in which the parental element excises from its original location before integrating elsewhere (37). In contrast, retrotransposons replicate through reverse transcription of their RNA and integrate the resulting cDNA into another locus. Retrotransposons can be further classified into Long Terminal Repeats (LTR) retrotransposon and non-LTR retrotransposons (22).
Genome instability can also drive adaptation to different environments by altering gene dosage and/or by allowing the accumulation of mutations that can change the duplicated gene’s structure and function(s) (38). Accordingly, gene families’ expansion and contraction are linked to functional diversification, and genetic variation in gene families is often observed between individual of the same species (39, 40). For example, in the yeast Saccharomyces cerevisiae and the human fungal pathogen Candida albicans, variation in the sequences and/or the number of cell surface adhesin proteins contribute to phenotypic plasticity by altering cell-to-cell and cell-to-environment interactions (41). Additionally, changes in the number of serine and threonine-rich repeats, a hallmark of adhesin proteins, is often observed between different S. cerevisiae and C. albicans isolates (41).
The CTG (Ser1) clade of fungi, in which the CTG codon is translated as serine rather than leucine, can adapt remarkably well to extreme environments (42). Members of the CTG (Ser1) clade can be highly tolerant to environmental changes such as changes in environmental osmolarity, and they can grow on a variety of carbon sources (43, 44). One key question is to understand how members of the CTG-clade can adapt so efficiently to different environments. Understanding these adaptive processes is particularly important for this group of ascomycetous yeasts as they feature dangerous human fungal pathogens, such as Candida albicans, and yeasts of high biotechnological potential, such as Scheffersomyces stipitis (43).
It is well established that genome plasticity is a critical adaptive mechanism in one member of the CTG (Ser1)-clade: the human fungal pathogens Candida albicans (29). C. albicans has a diploid genome organised in 8 chromosomes. However, clinical isolates exhibit karyotypic diversity that can confer antifungal drug resistance due to Copy Number Variation (CNV) of drug-resistance genes (45). Although transposable elements can be the source of this genetic variation, long repeat sequences with no homology to transposons are the major drivers of C. albicans genome instability (46–48).
In many respects, C. albicans is different from other members of the CTG-clade, and it is not always representative of CTG-clade biology. Indeed, C. albicans belong to a group of the CTG-clade that lacks a canonical sexual cycle. Instead, a parasexual cycle has been reported where mating of diploid cells is followed by mitosis and concerted chromosome loss (49). Although recombination can be associated with this parasexual cycle, mitotic events are major drivers of C. albicans genome diversity (50, 51). Consequently, genome plasticity may have evolved as a critical adaptive mechanism specifically in C. albicans as it can lead to an increase of genetic diversity in the absence of classical meiosis and associated recombination.
Notably, the C. albicans lifestyle is different from the lifestyle of other CTG (Ser1)-clade members as it is an opportunistic human fungal pathogen that lives almost exclusively in the human host (52). Consequently, C. albicans genome plasticity might be critical for adaptation to a wide range of host niches and for survival under the drastic environmental changes imposed by the host immune response (53, 54).
In this study, we aim to establish whether genome plasticity is a defining feature of the CTG-clade or is a mechanism that has emerged only in members of the CTG-clade yeast that lack a canonical sexual cycle and/or live in the human host. To address this question, we investigate genome plasticity in the homothallic CTG (Ser1)-clade yeast Scheffersomyces stipitis. S. stipitis a non-pathogenic haploid CTG-clade yeast that does not live in the human host, and that is found in the gut of wood-ingesting beetles, in hardwood forests or areas high in agricultural waste (55). S. stipitis has a canonical sexual cycle whereby mating of haploid cells generate diploid cells that undergo meiosis and produce haploid spores (56). S. stipitis holds great biotechnological potential because, contrary to S. cerevisiae, it can ferment xylose, a major component of lignocellulose biomass derived from forest and agriculture waste (57, 58). Consequently, S. stipitis is one of the most promising yeasts for the development of second-generation biofuel derived from green waste, an eco-friendly and ethical alternative to fossil-based biofuels (59).
Although several S. stipitis natural isolates are used for the optimisation of second-generation biofuels production, the genome of only one strain (Y-11545) has been sequenced and assembled to the chromosomal level (60). The Y-11545 strain has a genome organised in 8 chromosomes containing transposon-rich regional centromeres (11, 60, 61).
To investigate whether the S. stipitis genome is plastic, we have taken several complementary approaches. Firstly, we investigated the phenotypic and genotypic diversity of 27 different S. stipitis natural isolates collected from different environments. Secondly, we combined MinION Nanopore with Illumina genome sequencing to generate a high-quality chromosome-level sequence assembly of a second S. stipitis natural isolate (Y-7124). Lastly, we performed in vitro evolution experiments for eight weeks (~56 passages) and analysed changes in S. stipites genome organisation following laboratory passaging. Thanks to this combined approach, we discovered that the S. stipitis genome is highly plastic and that extensive genomic changes can appear quite rapidly in mitotic dividing cells. We demonstrated that different S. stipitis natural isolates have distinct chromosomal organisations and that this diversity in chromosome structure correlates with specific phenotypes. The Y-7124 genomic sequencing revealed that transposable elements drive this extensive intra-species genetic variation. Furthermore, we demonstrated that adhesin gene families’ genetic variation is linked with distinct sedimentation phenotypes, a trait that facilitates bioethanol purification following fermentation. In summary, results presented here demonstrate that genome plasticity is also associated with members of the CTG-clade that are not commensal in humans and have a canonical sexual cycle.
MATERIAL AND METHODS
Yeast strains and Growth Conditions
Strains were obtained from the Agricultural Research Service (ARS) Collection, run by the Northern Regional Research Laboratory (NRRL) (Peoria, Illinois, USA), or the National Collection of Yeast Cultures (NCYC) (Norwich, United Kingdom) (Table S1) and confirmed by sequencing (primers AB798 and AB799 of the 26S rDNA (D1/D2 domain) (1) (Table S2). Routine culturing was performed at 30 °C with 200 rpm agitation on Yeast Extract-Peptone-D-Glucose (YPD) media. Phenotypic analyses were conducted on SC glucose (SC-G), xylose (SC-X), or a mixture of 60% glucose and 40% xylose (SC-Mix). Uridine (0.08 g/L in YPD and SC) and adenine hemisulfate (0.05 g/L in YPD) were added as growth supplements. Solid media were prepared by adding 2% agar.
Contour-clamped homogeneous electric field (CHEF) electrophoresis
Intact yeast chromosomal DNA was prepared as previously described (2): Cells were grown overnight, and volume equivalent to an OD600=7 was washed suspend in 20 μl of 10 mg/ml Zymolyase 100T (Amsbio #120493-1) and 300 μl of 1% Low Melt agarose (Biorad® # 1613112) in 100 mM EDTA. Chromosomes were separated on a 1% Megabase agarose gel (Bio-Rad) in 0.5X TBE using a CHEF DRII apparatus. Run conditions as follows: 60-120s switch at 6 V/cm for 12 hours followed by a 120-300s switch at 4.5 V/cm for 12 hours, 14 °C. The gel was stained in 0.5x TBE with ethidium bromide (0.5 μg/ml) for 30 minutes and destained in water for 30 minutes. Chromosomes were visualised using a Syngene GBox Chemi XX6 gel imaging system.
Southern Blotting
DNA from CHEF gel was transferred overnight to a Zeta-Probe GT Membrane (Biorad®, #162-0196) in 20x SSC and crosslinked using UV (150 mJ). Probing and detection of the DNA were conducted as previously described (3). Briefly, probes were generated by PCR incorporation of DIG-11-dUTP into target sequences following manufacturer’s instructions (Roche). Primer pairs used in probe design are AB1028 and AB1029 (Table S4). The membrane was hybridised overnight at 42 °C with DIG Easy Hyb (Roche®, 11603558001). The DNA was detected with anti-digoxigenin-Alkaline Phosphatase antibody (Roche®, #11093274910) and CDP Star ready to use (Roche®, #12041677001) according to manufacturer instructions.
Genome sequencing
The genome of S. stipitis NRRL Y-7124 isolate was sequenced by Illumina short-read and MinION long-read technologies. To this end, DNA was extracted from an overnight culture using the QIAGEN genomic tip 100/G kit (Qiagen®, #10243) according to manufacturing protocol. For long-read sequencing, MinION (Oxford Nanopore, Oxford UK) was performed on a DNA library prepared from size selected gDNA. DNA fragments greater than 30 Kb were selected using a Blue Pippin (Sage Science) and concentrated using Ampure beads. From this, a DNA library was prepared using a Ligation Sequencing Kit 1D (SQK-LSK108) and run on the Oxford Nanopore MinION flowcell FLOMIN 106D. The same gDNA extract was also used for the preparation of Illumina libraries. In this case, the DNA was sheared using the Covaris M220 with microTUBE-50 (Covaris 520166) and size selected using the Blue Pippin (Sage Science). The library was constructed using a PCR-free kit with NEBNext End Repair (E6050S), NEBNext dA-tailing (E6053S) and Blunt T/A ligase (M0367S) New England Biolabs modules. Sequencing was performed on a MiSeq Benchtop Analyzer (Illumina) using a 2×300bp PE (MS-102-3003) flow cell.
Genome assembly
Base-calling and demultiplexing were conducted with Albacore v2.3.3 (available at https://community.nanoporetech.com). Adapters and low-quality data were trimmed using the eautils package fastq-mcf 1.04.636 (https://expressionanalysis.github.io/ea-utils/). On nanopore sequence data, adapter trimming was performed with Porechop v.0.1.0 (https://github.com/rrwick/Porechop). Genome assembly was completed using long reads, with read correction performed with Canu v1.8(4) followed by assembly in SmartDenovo github commit id 61cf13d (5)). The draft assembly was corrected using the corrected nanopore reads through five rounds of Racon github commit 24e30a9 (6), and then by raw fast5 files using 10 rounds of Nanopolish v0.9.0 (7). Illumina sequencing reads were then used to polish the resulting assembly through 10 rounds of Pilon v1.17 (8). Following assembly, BUSCO v3 was run to assess gene space (9), identifying the presence of 1683 conserved genes from the Saccharomycetales_odb9 gene database. Assembly size and contiguity statistics were assessed using QUAST v4.5 (10) The final assembly in contigs was further corrected and assembled to chromosome level by identification of centromeric sequences and overlapping regions between the contigs (11).
Genome annotation
Genome annotation was performed using FUNGAP v1.0.1 (12) with fastq reads from NCBI SRA accession SRR8420582 used as RNA-Seq training data and protein sequences taken from NCBI assembly accession GCA_000209165.1 for S. stipitis NRRL Y-11545 (CBS6054) used for example proteins. Protein fasta files were extracted from predicted gene models using the yeast mitochondrial code (code 3) and the alternative yeast nuclear code (code 12). Functional annotation of gene models was performed through BLASTp searches vs all proteins from the NCBI reference fungal genomes (downloaded 11th April 2020), retrieving the top-scoring blast hit with an E-value < 1×10−30. These annotations were supplemented with domain annotations from Interproscan v5.42-78.0 (13). The annotated genome was submitted to NCBI, with submission files prepared using GAG v2.0.1 (http://genomeannotation.github.io/GAG.), Annie github commit 4bb3980 (http://genomeannotation.github.io/annie) and table2asn_GFF v1.23.377 (available from https://ftp.ncbi.nih.gov/toolbox/ncbi_tools/converters/by_program/tbl2asn/).
Comparative genomics
Whole-genome alignment between NRRL Y-7124 and NRRL Y-11545 (CBS6054) was performed using the nucmer tool from the MUMmer package v4.0 (14) with results visualised using Circos v0.6 (15). Orthology analysis was performed between predicted proteins from these isolates using OrthoFinder v2.3.11 (16), with results visualised using the package VennDiagram in R (17).
Sequence variants were identified in NRRL Y-7124 through comparison to the NRRL Y-11545 assembly. Short read sequence data for NRRL Y-7124 were aligned to the reference genome using BWA v 0.7.15-r1140 (18), before filtering using using picardtools v2.5.0 to remove optical duplicates (http://broadinstitute.github.io/picard/). SNP and insertion/deletion (InDel) calling was performed using GATK4 (19). Low confidence variants were then filtered using VCFtools v0.1.15 (20) using minimum mapping quality of 40, phred quality of 30, read depth of 10 and genotype quality of 30. Effect of variants on NRRL Y-11545 gene models was determined using SnpEff v4.2 (21).
Identification of Transposons and adhesin genes
Long sequences (>100 nucleotides) present more than once in the NRRL Y-7124 and NRRL Y-11545 genomes were identified by aligning the NRRL Y-7124 and NRRL Y-11545 genomes to themselves using BLASTN. The genomic position of repeats were manually verified using IGV/SNAPGene, and clustered repeats were combined. These repeats datasets were interrogated to identify non-centromeric transposons and genes encoding putative adhesin proteins. Transposons were classified using established guidelines (22). Briefly, LTR-transposons were identified by detecting two Long-terminal Repeat sequences (size 260-430 nt) flanking an internal coding region. These potential LTR-transposons were further annotated for the presence of the following marks: LTR flanked by a TG and CA di-nucleotides, presence of a Primer Binding Site (PBS) with homology to S. stipitis tRNAs (GtRNAdb (http://gtrnadb.ucsc.edu/index.html), presence of a coding region with homology to pol gene and containing an Integrase (INT), Reverse Transcriptase (RT) and RNAse H (RH) domain. Additional coding regions were interrogated for the presence of known PFAM domains using Simple Modular Architecture Research Tool (SMART(http://smart.embl.de) (23). Non-LTR transposons were identified by detection of coding regions homologous to LINE retrotransposons ORF1 (containing a Zn-finger), ORF2 (containing an Endonuclease and a Reverse Transcriptase domain) and terminal Poly-A sequence. Retrotransposons were classified into different families based on sequence similarity with a 90% cut-off. Repetitive coding regions containing serine and threonine-rich repeats (a hallmark of Adhesin proteins) were extracted from the NRRL Y-7124 and NRRL Y-11545 repeats datasets and PFAM domains associated with these proteins were identified with SMART(http://smart.embl.de) (23). Sequence alignment was performed with Clustal Omega and visualised with Jalview v2.11.1.0 (24).
Phenotypic characterisation
Growth analyses were performed using a plate reader (SpectrostarNano, BMG labtech) in 96 well plate format at 30 °C for 48 hours in SC-G, SC-X or SC-Mix. The growth rate (μ, hours−1) was calculated using: μ = (ln(X2)-ln(X1))/(t2-t1), where: (i) X1 is the biomass concentration (OD600) at time point one (t1) (ii) X2 is the biomass concentration (OD600) at time point two (t2). The maximum OD (OD units) was determined with the MAX() from Excel (Microsoft®). The lag time (minutes) was determined visually as the time in which the exponential growth starts. Experiments were performed in 3 technical and 3 biological replicates. Sedimentation analysis was conducted as previously described (25) with modifications. Strains were grown overnight in SC-Mix and were standardised to a starting OD600 = 1.0. Absorbance was measured every hour, with no agitation between measurements, for 4 hours. The percentage of sedimentation was calculated using: Sedimentation (%) = 100 – (Atx/At0·100), where: (i) Atx is the absorbance (OD600) recorded at time x (ii) At0 is the absorbance (OD600) recorded at time 0. The experiment was conducted in three biological replicates.
Adaptive Laboratory Evolution
A single colony of the S. stipitis strain NRRL Y-7124 was grown overnight in 5 ml of YPD at 30 °C, plated in YPD at a cell density of 100 and grown 48 hours at 30 °C. 36 single colonies were streaked in two SC-Mix plates and grown at 30 °C and 37 °C, respectively and streaked daily for a total of 56 passages (8 weeks). The karyotype variability of the colonies was assessed by CHEF electrophoresis.
Statistical analyses
Growth parameters and sedimentation percentages were compared by ANOVA. The equality of variances presumption was checked by the Levene’s test, whereas the normality of the data was checked by Shapiro-Wilk. When both presumptions were obeyed the Tukey’s honest significant difference test was used to determine were the differences stated by the ANOVA test lied. When the variances were not statistically the same, the one-way test was used to indicate significance. In case of equal variances, but no normal distribution of the data, the Kruskal-Wallis rank sum test was used to indicate statistical differences. When significant, the pairwise testing was used to determine were those differenced lied. A p-value lower than 0.05 was considered significant for all the statistic tests.
RESULTS
S. stipitis natural isolates are characterised by distinct genomic organisation
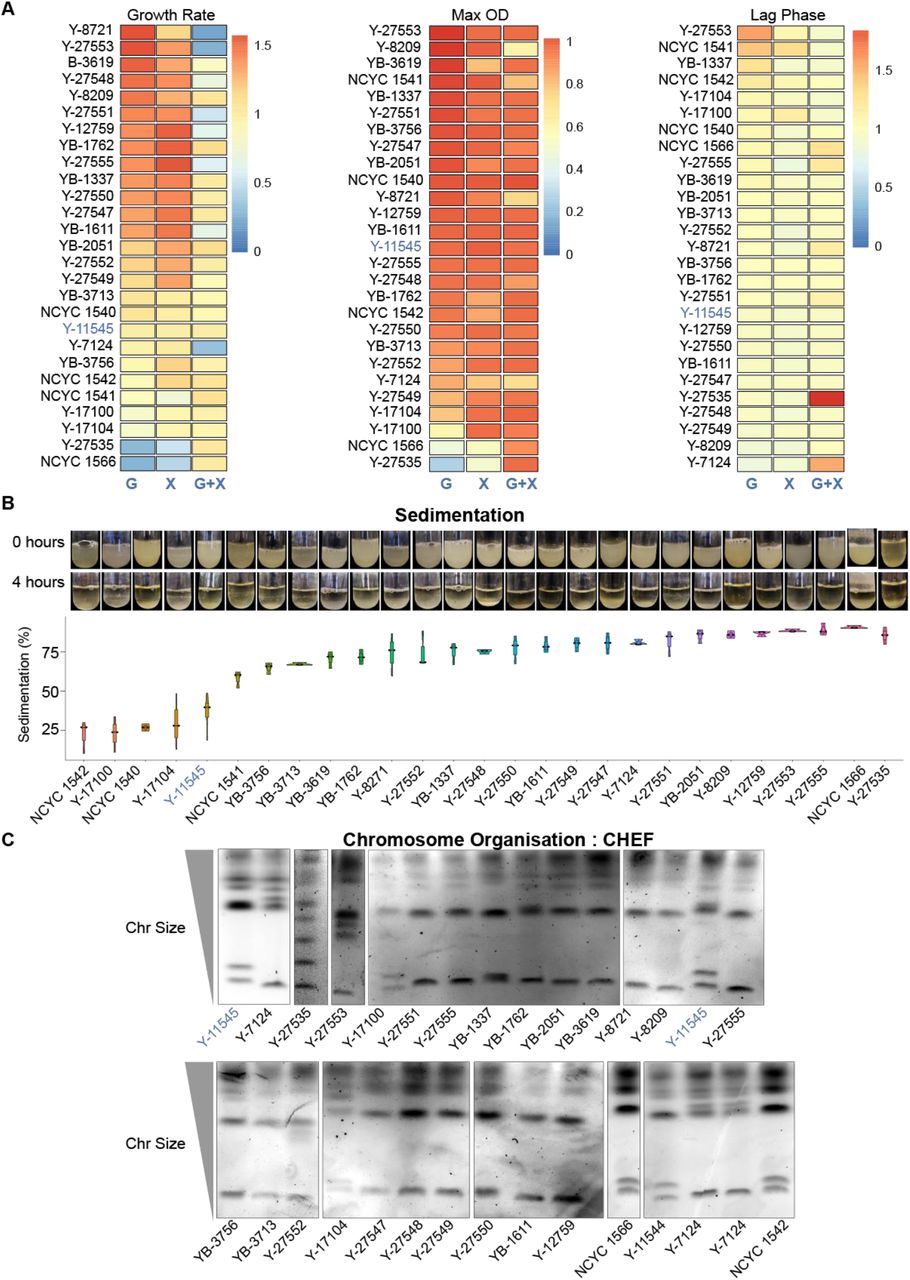
To examine S. stipitis diversity, we selected a geographically diverse set of strains (n=27) that were collected in different habitats (Table S1 source NRRL and NCYC collection), and that includes the sequenced NRRL-Y11545 strain (60). rDNA fingerprinting confirm that all isolates belong to the S. stipitis species (D1/D2 domain of the S26S rDNA similarity >99 %) (Table S2). Phenotypic analyses established that the natural isolates vary in their ability to utilise and grow on different carbon sources. Indeed, the growth rate, maximum culture density and the lag phase are different among natural isolates when cultured in Synthetic Complete media containing the hexose sugar Glucose (SC-G), Xylose (SC-X) or a mixture of both sugars (SC-Mix) (Fig 1A). The ability of yeast cells to aggregate and sediment, is a desirable trait for the bioethanol industry as it provides an environmental-friendly and cost-effective strategy to remove yeast cells at the end of fermentation (62). Sedimentation analysis demonstrated that the different natural isolates differ in their ability to settle out of suspension (Fig 1B).

Phenotypic and Genetic Diversity in S. stipites
A) Heatmaps comparing growth rate (Left), maximum OD (Middle) and lag time (Right) of each S. stipitis natural isolate in comparison to the reference NRRL Y-11545 strain (blue). Analyses were performed in Glucose (G), Xylose (X) and Glucose/Xylose (G+X) media. The heatmap data are the average of 3 biological replicates. B) Top: Photos of sedimentation experiment for each S. stipitis natural isolate at time 0 and 4 hours. Bottom: Sedimentation profile (% compared to t= 0) of S. stipits natural isolates. The reference NRRL Y-11545 strain is highlighted in blue. Violin plots= average of 3 biological replicate; error bars= Standard Deviation. C) Karyotyping of S. stipits natural isolates by CHEF electrophoresis. NRRL Y-11545 strain is highlighted in blue
To determine whether these different phenotypes are linked to distinct genomic organisations, we analysed the natural isolates’ karyotype by performing chromosomes Contour-clamped Homogenous Electric Field (CHEF) gel electrophoresis. This technique allows chromosome separation according to size. The CHEF electrophoresis analysis reveals a clear difference in the S. stipitis natural isolates (Figure 1C). Therefore, we concluded that intra-species phenotypic and genotypic variation is a common feature of S. stipitis.
Transposable elements are drivers of S. stipitis genome plasticity
To date, only one S. stipitis isolate (Y-11545) has been sequenced and assembled at chromosome level (60). To determine the cause of S. stipitis genetic diversity, we generated a chromosome-level sequence assembly of a second S. stipitis natural isolate (Y-7124) by combining MinION Nanopore with Illumina genome sequencing. This Y-7124 isolate was chosen because (i) karyotypic analysis reveals that its genomic organisation is distinct from the genomic organisation of the reference strain Y-11545, (ii) phenotypic analysis reveals that Y-7124 sediments faster than Y-11545 (Fig 1) and (iii) Y-7124 is widely used both for industrial applications and for basic research (63).
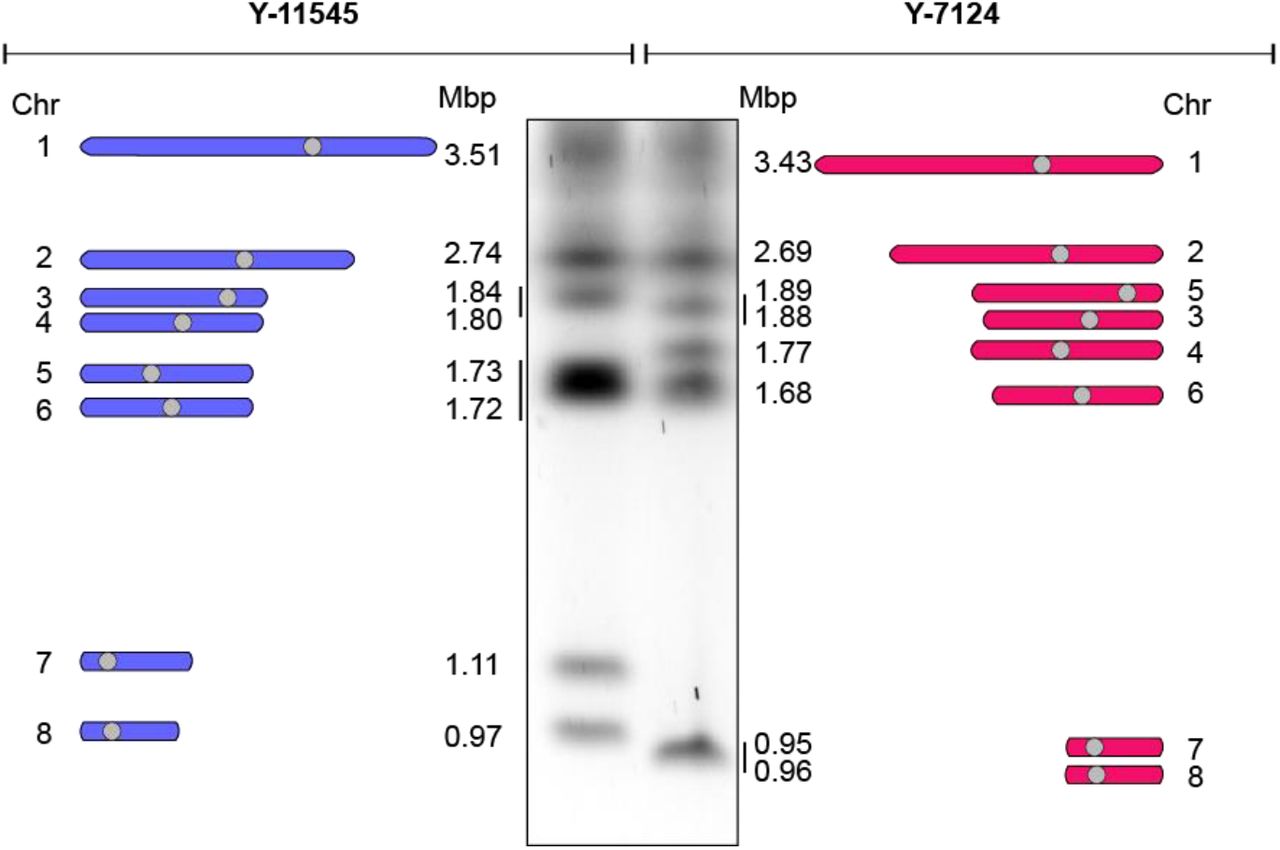
The NRRL Y-7124 genome was sequenced to 186.88x coverage resulting in a 15.69 Mb assembly arranged in 11 contigs (Table S3). High accuracy reads from Illumina-sequencing enabled correction of errors that are associated with the MinION technology. Identification of unique centromeric sequences (present once in each chromosome) supported identification of chromosomes in the final assembly. We found that Y-7124 has a genome organised in eight different chromosomes with different sizes and organisations than Y-11545 (Fig 2).

The genomic organisation of Y-11545 and Y-7124 is distinct
Left: Schematics of Y-11545 chromosome organisation. Chromosome (Chr) number and size (Mbp) is indicated. Middle: Karyotyping of S. stipits Y-11545 and Y-7124 strains by CHEF electrophoresis. Right: Schematics of Y-7124 chromosome organisation. Chromosome (Chr) number and size (Mbp) is indicated
In many organisms, transposons are instability hotspots (36). To test whether S. stipitis genome diversity originated at transposable elements, we scanned the Y-11545 and Y-7124 genomes for the presence and organisation of coding and non-coding regions with similarity to known transposable elements (Material and Methods). This analysis identifies six novel retrotransposons families that are not clustered at centromeres, but they are present in multiple copies along the chromosome arms of both natural isolates (Fig 3A and 3B). These are 3 Copia LTR-retrotransposons families, that we named Ava, Bea and Caia, and three novel non-LTR LINE families, that we named Ace, Bri and Can (Fig 3A). Our analysis did not identify any DNA transposons.

Transposable elements are major driver S. stipitis genomic diversity
A) Schematics of non-centromeric retrotraspons identified in this study. For each transposon, subclass and family is indicated. The structure displays coding and non-coding sequences of each transposon. Size in kilobases (kb) is indicated. B) Schematics of non-centromeric transposon family distribution in Y-11545 (left) and Y-7124 (right). C) Copy Number of full-length transposons (Left) and transposon-associated repeats associated with the Y-11545 (dark grey) and Y-7124 (light grey) genome. D) Left: Schematics of transposons Ancestral, Deletion and Insertion sites Right: Percentage (%) of Ancestral, Deletion and Insertion sites associated with the Y-11545 and Y-7124 genomes
Comparison of the Y-7124 and Y-11545 genomes establishes that non-centromeric retrotransposons are significant drivers of S. stipitis genome diversity as one of the most prominent differences between the two genomes is the abundance and localisation of these retrotransposons (Fig 3B). Indeed, the number of LTR and LINE non-centromeric retrotransposons and transposons-derived repeats is greater in the Y-11545 reference genome compared to the Y-7124 genome: retrotransposons, solo LTR and truncated LINE elements account for approximately 2% of the reference Y-11545 genome and only for ~1% of the Y-7124 genome (Fig 3C). We classified retrotransposons loci present in both isolates (ancestral loci), present in the reference Y-11545 genome but absent in Y-7124 (deletion loci) and not present in the reference genome but present in a given strain (insertion loci) (Fig 3D).
Out of 69 transposons loci, only ten ancestral loci (~15%) were detected in the two isolates. These sites are likely to be inactive transposons or transposons that rarely transpose. In addition, we detected 42 deletion loci (60 %) and 17 (24%) insertion loci (Fig 3D). The presence of deletion and insertion loci suggests that S. stipitis LTR transposons and LINE elements are active and competent of transposition. Active transposons can insert into genes to cause functional consequences (36).
Comparison of the Y-11545 and Y-7124 genome reveals that TE insertions do not disrupt any protein-coding genes.
One major genomic rearrangement differentiates the reference strain Y-11545 from the Y-7124 strain: a reciprocal translocation between chromosome 5 and chromosome 7 (Fig 4A). This translocation causes the size change in chromosome 5 Y-7124 and chromosome 7 Y-7124 detected by CHEF karyotyping (Fig 4B) Southern analyses with a probe specific for chromosome 5 Y-11545 confirms this finding (Fig 4B). The evolutionary history of Y-11545 and Y-7124 is unknown, and therefore it is difficult to predict the molecular events underlying these genomic changes. However, sequence analysis of the rearrangement breakpoint reveals that this structural variation occurs in a genomic region that (i) on chromosome 7 is a transposon-rich and contains two inverted repeats (ii) contains homologous sequences between chromosome 5 and 7 and (iii) contains the viral-related NUPAV sequence (64) (Fig 4C). The presence of transposons and transposon-derived repeats strongly suggest that these elements have mediated the chromosomal rearrangement. We concluded that retrotransposons are major drivers of genetic S. stipitis genetic plasticity. Changes in transposons organisation are responsible for the bulk of genomic changes identified in two different natural isolates.

Chromosome rearrengment between S. stipitis natural isolates.
A) Circos plot displaying macrosynteny between Y-11545 (Left) and Y-7124 (Right). Chromosome (Chr) number and size is indicated. Recriprocal translocation between the two genomes is highlighted in purple and pink. B)Left: Schematics of chromosome 5 (Chr 5) and chromosome 7 (Chr 7) in Y-11545 (Left) and Y-7124 (Right). Reciprocal translocation is highlighted in purple and pink. Southern Probe is indicated. Right: Southern Blot of Y-11545 and Y-7124 chromosomes separated by CHEF gel electrophoresis. Full chromosome profiling (EtBr) and Southern Blot results (Southern) are indicated. C) Schematics of repetitive sequences associated with the translocation junction in the Y-11545 (Left) and Y-7124 (Right) genome
Genetic variability of adhesin-coding genes
Our analysis demonstrated that S. stipitis transposable elements are drivers of genetic diversity. However, it is unlikely that transposon-derived genomic changes are responsible for the different sedimentation phenotypes of Y-11545 and Y-7124 (Fig 5A) as protein-coding genes are not perturbated by a TE insertion or by the chromosomal rearrangement at the TE-rich region.
Comparison of the Y-7124 and Y-11545 nucleotide sequences reveal that the two natural isolates overall share a similar coding DNA sequence. The total number of SNPs between the two natural isolates is 50,495 SNPs, equating to one variant every 306 bases. Despite the high number of SNPs, 16,294 (74.25%) of them are synonymous, 5,622 (25.62%) are missense and only 28 (0.13%) are nonsense (Table 1).

Adhesin coding genes are diverse in S. stipites
A) Sedimentation profile (% compared to t= 0) of Y-11545 and Y-7124 natural isolates. Violin plots: average of 3 biological replicate; error bars: Standard Deviation. B) Schematics of putative adhesin proteins encoded by the Y-11545 and Y-7124 natural isolate. Predicted PFAM domains are dispayed. Green star: Proteins with a different organisation in the two isolates
Summary of the SNPs observed between the natural isolates of S. stipites NRRL Y-11545 and NRRL Y-7124.
In several yeast species, including S. cerevisiae and C. albicans, adhesins are cell-surface proteins that play a critical role in cell to cell adhesion (41). Variation in adhesin protein organisation and structure has been shown to influence cell to cell adhesion (65). Therefore, we hypothesised that the distinct sedimentation phenotype of Y-7125 and Y-11545 is due to differences in adhesin proteins organisation. To test this hypothesis, we categorised S. stipitis genes encoding for proteins with adhesin-like domains (See Material and Methods). This analysis identified 26 genes encoding for adhesin-like proteins. The number of genes found in the two strains is conserved. However, their predicted protein organisation is often different, as 17/26 adhesin-like proteins have a distinct protein organisation. The most common difference between the two isolates is variation in the number of serine/threonine-rich repeats (Fig 5B).
Therefore, the distinct sedimentation phenotype of Y-7125 and Y-11545 correlates with a different organisation of the adhesin encoding genes.
S. stipitis real-time evolution leads to extensive genomic changes
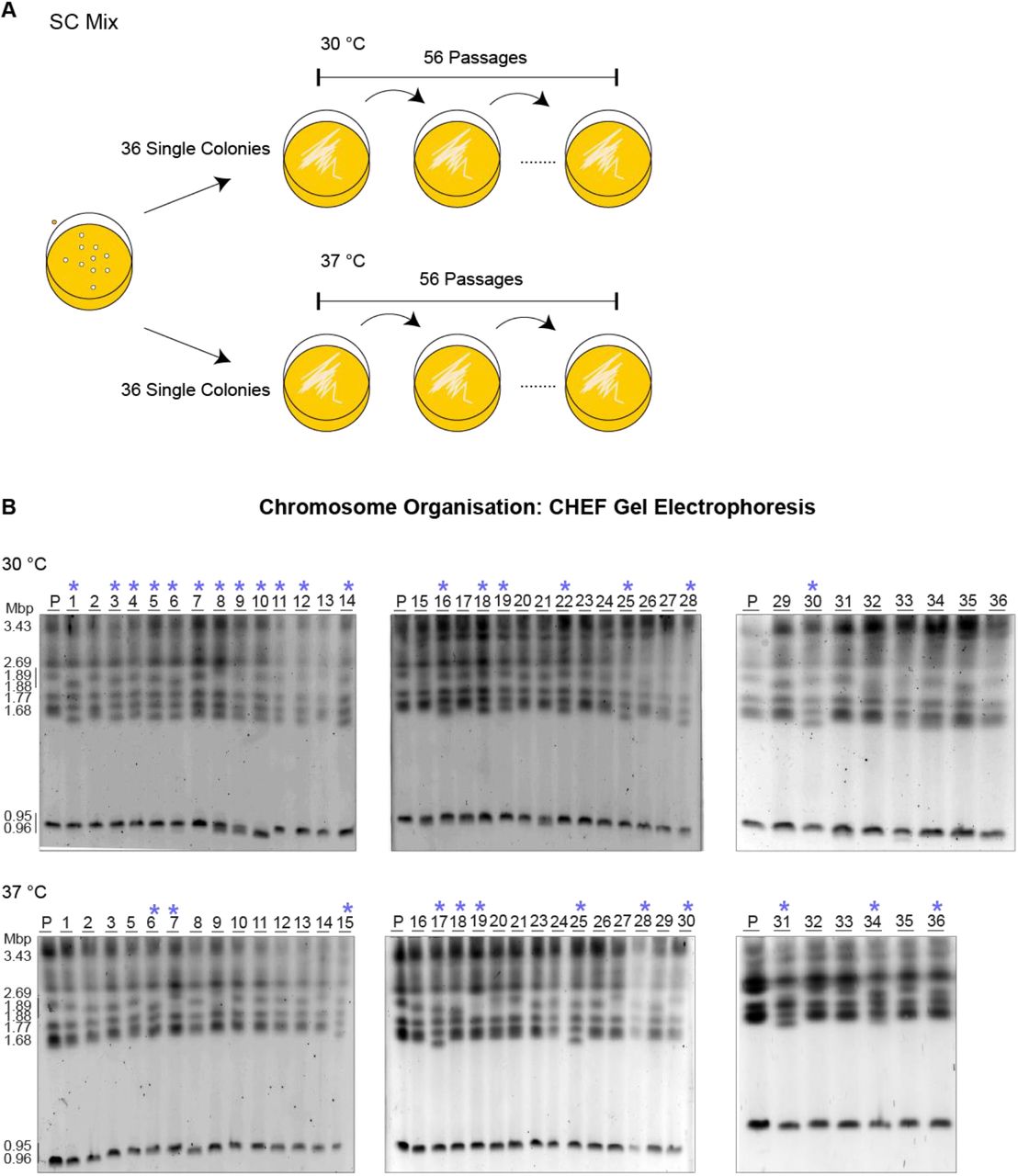
Results presented in this study demonstrates that intraspecies genetic diversity is common in S. stipitis. However, the evolutionary history of the analysed S. stipitis natural isolates is unknown. Therefore, it is difficult to predict whether the observed genomic changes are due to the selection of rare genomic rearrangements events. To determine S. stipitis genome evolution’s time scale, we investigated the genome organisation of strains following passaging for 8 weeks daily (56 passages, ∼672 divisions) (Fig 6A). To this end, 72 single colonies were passaged every day on SC-Mix media at 30 (36 colonies) or 37 (36 colonies) °C. The genome of the parental and evolved strains was then analysed by CHEF gel electrophoresis. This analysis identifies significant differences in chromosome organisation following in vitro evolution as genome rearrangements were detected in 19/36 strains evolved at 30°C and 12/36 strains evolved at 37 °C (Fig 6B). Therefore, genome plasticity is a defining feature of the S. stipitis genome and its genome can rapidly change in mitotic cells propagated in vitro.

The S. stipitis genome is plastic following real-time evolution
A) Schematics of laboratory evolution strategy B) Karyotype organisation of S. stipitis colonies following 8 weeks of laboratory evolution at 30 °C and 37 °C (*) indicates variation in the structure from the parental (P: NRRL Y-7124) at day 0.
DISCUSSION
The CTG-Ser1 clade is an incredibly diverse yeast group that includes many important human pathogens and non-pathogenic species with high biotechnological potential (66). Given the widespread focus in understanding and combating virulent organisms, we still know very little about the biology of non-pathogenic CTG(Ser1)-clade members. However, understanding how CTG (Ser1) non-pathogenic species adapt to environmental changes might be critical for identifying pathogenic adaptation strategies and/or developing important biotechnologies.
The S. stipitis genome is highly unstable
C. albicans, is the best-studied member of the CTG-Ser1 clade as it is a leading cause of death (67). C. albicans, a commensal organism that lives in the human host, display remarkable genotypic plasticity that is instrumental for host adaptation (29). C. albicans can generate genetic diversity through several mechanisms including a parasexual cycle that involves mating of diploid cells to generate tetraploid cells. C. albicans tetraploid cells do not go through a meiotic cycle but undergo random concerted chromosome loss that reduces DNA content to approximately a diploid state (68). In addition, dramatic genome changes can appear quite rapidly in C. albicans mitotic cells propagated in vitro as well as in vivo (69–71). Here, we established that genome plasticity is not exclusively associated with pathogenic CTG (Ser1)-clade species that lack meiosis. We demonstrated that S. stipitis natural isolates display distinct phenotype and have a genome with a dissimilar chromosomal organisation. Importantly, we detected extensive genomic changes following passaging of mitotic S. stipitis cells in vitro. Studies conducted in C. albicans demonstrated that stress exacerbates mitotic genome instability (72, 73). Although we cannot exclude that the media (SC-Mix) used for the evolution experiment imposes mild stress, our results suggest that S. stipitis genome instability is not regulated by stress and that the S. stipitis genome is intrinsically unstable. Indeed, we detected a similar rate of chromosomal rearrangements at two different temperature (30 °C and 37 °C). Our findings have important implications for the development of second-generation biofuels as the genome of superior biofuel-producer strains is likely to be unstable, and the genetic drivers of improved phenotypes might be lost over time.
We demonstrated that non-centromeric retrotransposons are major drivers of S. stipitis genome diversity. Indeed, we identified six novel families of non-centromeric retrotransposons: the LTR retrotransposons Ava, Bea and Caia and the LINE retrotransposons Ace, Bri and Can. The Y-11545 and Y-7124 genomes contain several full-length copies of these retrotransposons in addition to solo LTR and truncated copies. However, the number and genomic position of both LTR and LINE retrotransposons vary in the two isolates. Furthermore, we found that a transposon-rich region is a translocation site between chromosome 5 and chromosome 7. Significantly, this transposon-mediate genome diversity does not disrupt any coding regions. However, transposon might alter S. stipitis gene expression by insertion with gene regulatory regions (36). Our results strongly suggest that S. stipitis transposons are active and generate genome diversity by two principal mechanisms: transposition into different genomic locations or by triggering complex chromosomal rearrangements probably due to faulty repair of double-strand breaks generated during transposable elements excision or homologous recombination of nearly identical TE copies (36). Both mechanisms have been described in other organisms, including yeast, human and plants (35, 74, 75). It is important to note that, although transposon activity has been detected in C. albicans (76), transposons do not seem to be the major drivers of C. albicans genetic diversity. Indeed, recent sequence analyses demonstrate that the C. albicans genome contains fewer than initial predicted full-length transposable elements. Different types of repeats with no homology to transposon drive genomic diversity (47, 73, 77–79).
Distinct sedimentation phenotypes are linked to diver adhesin protein organisation
Our phenotypic analysis demonstrated that the Y-11545 reference strain and the highly used Y-7124 strain differ in their ability to sediment. Sedimentation is of considerable importance for the ethanol industry, as it provides an environmental-friendly and cost-effective strategy to remove yeast cells at the end of fermentation (62). We found that distinct sedimentation phenotypes correlate with a different organisation of putative adhesin-like proteins. We found that the S. stipitis genome contains 26 genes encoding for putative adhesins including three genes encoding for proteins with similarity to S. cerevisiae FLO11 gene and 23 genes encoding for proteins with similarity to C. albicans ALS genes. S. cerevisiae Flo 11 is responsible for adhesion to substrates (80), while C. albicans ALS proteins confer adhesion to mammalian host tissues (81). Given that S. stipitis does not colonise the human host, it is unlikely that ALS proteins are involved in mammalian host tissue interaction and are more likely to be important for cell to cell or cell to environment interactions. Similarly to our finding in S. stipitis, ALS proteins organisation is often different between C. albicans clinical isolates. The genetic diversity associated with ALS genes leads to distinct adhesin-driven phenotype such as biofilm formation (41). Therefore, we postulate that the different adhesin proteins affect the ability of S. stipitis strains to flocculate.
In summary, our study demonstrates for the first time that S. stipitis genome is highly unstable. Understanding the cause and effect of this extensive genome plasticity is of paramount importance to understand the biology of the CTG(Ser1)-clade of fungi.
DATA AVAILABILITY
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JADGGA000000000. The version described in this paper is version JADGGA010000000. Illumina and nanopore sequence data associated with this work have been deposited on the Sequence Read Archive (SRA) under BioProject PRJNA609885.
FUNDING
This work was supported by the University of Kent Vice-Chancellor’s Research Scholarship (to SV), BBSRC grants (Grant number BB/L008041/1 to AB, BB/P020364/1 to RJH) and an MRC grant (MR/M019713/1 to AB)
CONFLICT OF INTERESTS DISCLOSURE
None declared.
ACKNOWLEDGEMENTS
We thank Dr Patricia Slininger, members of the Buscaino Lab, the Kent Fungal Group and Dr Jan Soetaert, for discussion and critical reading of the manuscript.
BIBLIOGRAPHY
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.
- 79.↵
- 80.↵
- 81.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}