

Abstract
The evolution of many microbes and pathogens, including circulating viruses such as seasonal influenza, is driven by immune pressure from the host population. In turn, the immune systems of infected populations get updated, chasing viruses even further away. Quantitatively understanding how these dynamics result in observed patterns of rapid pathogen and immune adaptation is instrumental to epidemiological and evolutionary forecasting. Here we present a mathematical theory of co-evolution between immune systems and viruses in a finite-dimensional antigenic space, which describes the cross-reactivity of viral strains and immune systems primed by previous infections. We show the emergence of an antigenic wave that is pushed forward and canalized by cross-reactivity. We obtain analytical results for shape, speed, and angular diffusion of the wave. In particular, we show that viral-immune co-evolution generates a new emergent timescale, the persistence time of the wave’s direction in antigenic space, which can be much longer than the coalescence time of the viral population. We compare these dynamics to the observed antigenic turnover of influenza strains, and we discuss how the dimensionality of antigenic space impacts on the predictability of the evolutionary dynamics. Our results provide a rigorous and tractable framework to describe pathogen-host co-evolution.
I. INTRODUCTION
The evolution of viral pathogens under the selective pressure of its hosts’ immunity is an example of rapid co-evolution. Viruses adapt in the usual Darwinian sense by evading immunity through antigenic mutations, while immune repertoires adapt by creating memory against previously encountered strains. Some mechanisms of in-host immune evolution, such as affinity maturation process, are important for the rational design of vaccines. Examples are the seasonal human influenza virus, where vaccine strain selection can be informed by predicting viral evolution in response to collective immunity[1], as well as chronic infections such as HIV [2–5], where co-evolution occurs within each host. Because of the relatively short time scales of selection and strain turnover, these dynamics also provide a laboratory for studying evolution and its link to ecology [6].
It is useful to think of both viral strains and immune protections as living in a common antigenic space [6], corresponding to an idealized “shape space” of binding motifs between antibodies and their cognate epitopes [7]. While the space of molecular recognition is high-dimensional, projections onto a low-dimensional effective shape space have provided useful descriptions of the antigenic evolution. In the example of influenza, neutralization data from hemagglutination-inhibition assays can be projected onto a two-dimensional antigenic space [8–10]. Mapping historical antigenic evolution in this space suggests a co-evolutionary dynamics pushing the virus away from its past positions, where collective immunity has developed. Importantly, the evolution of influenza involves competitive interactions of antigenically distinct clades in the viral population, generating a “Red Queen” dynamics of pathogen evolution [11, 12]. Genomic analysis of influenza data has revealed evolution by clonal interference [13]; this mode of evolution is well-known from laboratory microbial populations [14]. In addition, the viral population may split into subtypes. Such splitting or “speciation” events, which are marked by a decoupling of the corresponding immune interactions, happened in the evolution of influenza B [15] and of noroviruses [16].
The joint dynamics of viral strains and the immune systems of the host population may be modeled using agent-based simulations [17, 18] that track individual hosts and strains. Such approaches have been used to study the effect of competition on viral genetic diversity [19], to study geographical effects [20], and the effect of vaccination [21]. Alternatively, systems of coupled differential equations known as Susceptible-Infected-Recovered (SIR) models may be adapted to incorpo-rate evolutionary mechanisms of antigenic adaptation [6, 22, 23]. Agent-based simulations in 2 dimensions were used to recapitulate the ballistic evolution characteristic of influenza A [18], and to predict the occurence of splitting and extinction events [24]. In parallel, theory was developed to study the Red Queen effect [12, 25], based on the well established theory of the traveling fitness wave [26–28]. While effectively set in one dimension, this class of models can nonetheless predict extinction and splitting events assuming an infinite antigenic genome [12].
In this work, we propose a co-evolutionary theory in an antigenic interaction space of arbitrary dimension d, which is described by joint non-linear stochastic differential equations coupling the population densities of viruses and of protected hosts. We show that these equations admit a d-dimensional antigenic wave solution, and we study its motion, shape, and stability, using simulations and analytical approximations. Based on these results, we discuss how canalization and predictability of antigenic evolution depend on the dimensionality d.
II. RESULTS
A. Coarse-grained model of viral-immune co-evolution
Our model describes the joint temporal evolution of populations of viruses and immune protections in some effective antigenic space of dimension d. Both viral strains and immune protections are labeled by their position x = (x1,…, xd) in that common antigenic space, called “phenotype” (Fig. 1A). In that space, viruses randomly move as a result of antigenic mutations, as well as proliferate through infections of new hosts. Immune memories are added at the past positions of viruses. Immune memories distributed across the host population provide protection that reduce the effective fitness of the virus. We coarse-grain that description by summarizing the viral population by a density n(x, t) of hosts infected by a particular viral strain x, and immunity by a density h(x, t) of immune memories specific to strain x in the host population.
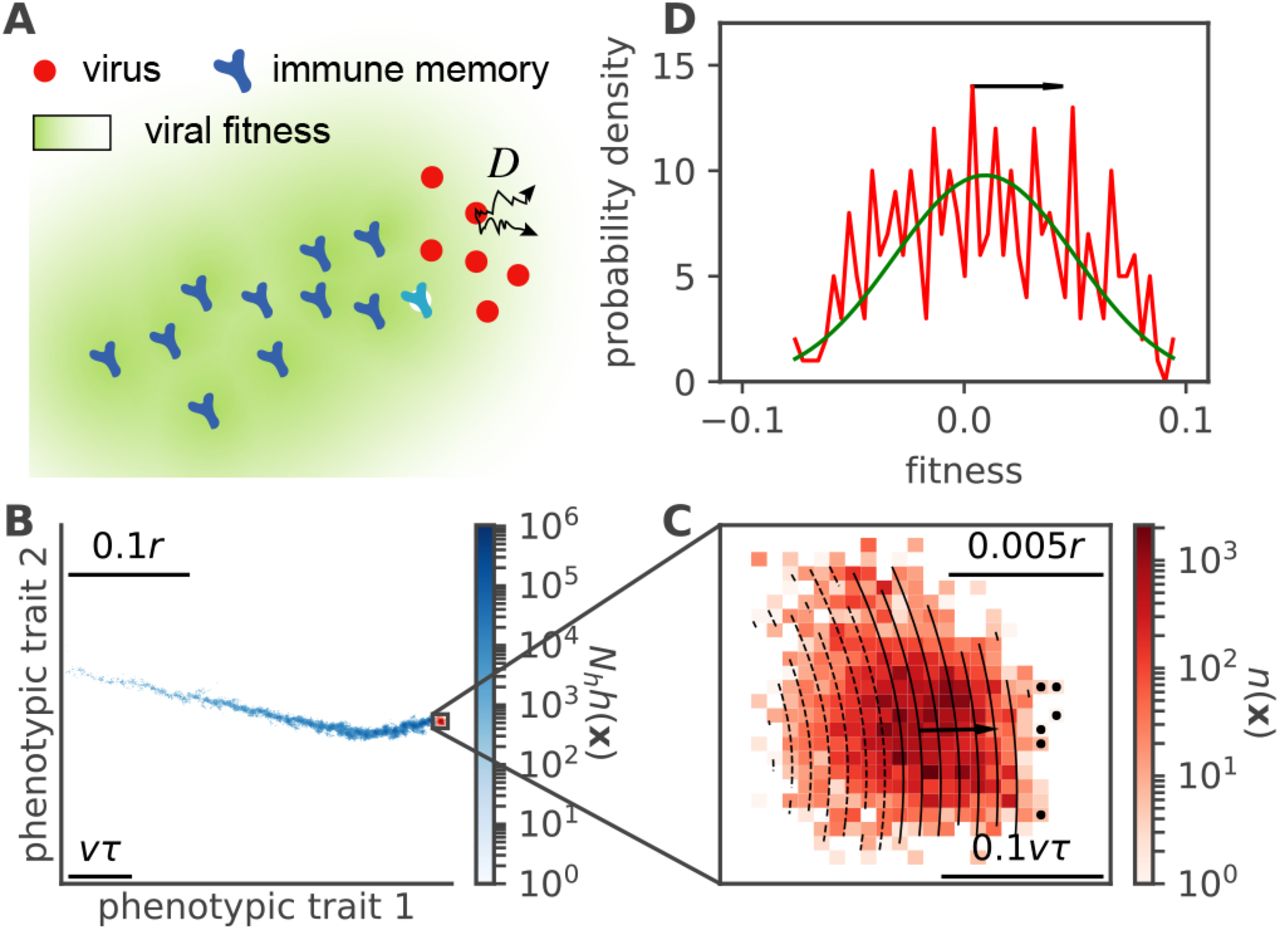
A. Schematic of the co-evolution model. Viruses proliferate while effectively diffusing in antigenic space (here in 2 dimensions) through mutations, with coefficient D. Past virus positions are replaced by immune protections (light blue). Immune protections create a fitness gradient for the viruses (green gradient) favoring strains at the front. Both populations of viruses and immune populations are coarse-grained into densities in antigenic space. B. Snapshot of a numerical simulation of Eq. 2-3 showing the existence of a wave solution. The blue colormap represents the density of immune protections h(x, t) left behind by past viral strains. The current virus density n(x) is shown in red. C. Close-up onto the viral population, showing fitness isolines. The wave moves in the direction of the fitness gradient (arrow) through the enhanced growth of stains at the edge of the wave (black dots). D. Distribution of fitness across the viral population (corresponding to the projection of B. along the fitness gradient). Parameters for B-D: D/r2 = 3 · 10−9, Nh = 108, ln R0 = 3, M = 1.
At each infection cycle, each host may infect R0 unprotected hosts, where R0 is called the basic reproduction number. However, a randomly picked host is susceptible to strain x with probability (1−c(x, t))M, where c(x, t) is the coverage of strain x by immune memories of the population, and the number M of immune memories carried by each host. Because of cross-reactivity, which allows immune memories to confer protection against closeby strains, immune coverage is given as a function of the density of immune memories:

Where H(x−x′) = exp(−| x−x′|/r) is a cross-reactivity Kernel describing how well memory x protects against strain x, and r is the range of the coverage provided by cross-reactivity. In summary, the effective growth rate, or “fitness”, of the virus is given by f (x, t) ≡ ln[R0(1 − c(x, t))M] in units of infection cycles.
The coupled dynamics of viruses and immune memories is then described by the stochastic differential equations:

 and η is a Gaussian white noise in time and space ⟨η(x, t)η(x′, t′)⟩ = δ(x x′)δ(t t′) accounting for demographic noise [29]. This stochastic term is crucial, as it will drive the evolution of the wave. D describes the effect of infinitesimal mutations on the phenotype,
and η is a Gaussian white noise in time and space ⟨η(x, t)η(x′, t′)⟩ = δ(x x′)δ(t t′) accounting for demographic noise [29]. This stochastic term is crucial, as it will drive the evolution of the wave. D describes the effect of infinitesimal mutations on the phenotype,  , where µ is the mean number of mutations per cycle, and
, where µ is the mean number of mutations per cycle, and  the mean squared effect of each mutation along each antigenic dimension (assuming that mutations do not have a systematic bias, ⟨δx1⟩ = 0). The continuous diffusion assumption implied by Eq. 2 is only valid when there are many small mutation effects, µ » 1 and δx « r. The total vifral population size, or number of infected hosts, N (t) = dx n(x, t) may fluctuate, but not the host population size Nh, which is constant: new added memories (first term of right-hand side of Eq. 3) overwrites existing ones picked uniformly at random (second term of r.h.s. of Eq. 3). Since each host carries M immune receptors, we have dxh(x, t) = M.
the mean squared effect of each mutation along each antigenic dimension (assuming that mutations do not have a systematic bias, ⟨δx1⟩ = 0). The continuous diffusion assumption implied by Eq. 2 is only valid when there are many small mutation effects, µ » 1 and δx « r. The total vifral population size, or number of infected hosts, N (t) = dx n(x, t) may fluctuate, but not the host population size Nh, which is constant: new added memories (first term of right-hand side of Eq. 3) overwrites existing ones picked uniformly at random (second term of r.h.s. of Eq. 3). Since each host carries M immune receptors, we have dxh(x, t) = M.
If we assume that the system reaches an evolutionary steady state, with stable viral population size N (t) = N, then Eq. 3 can be integrated explicitly:
 with τ = MNh/N. This equation shows how the density of protections reflects the past of the virus evolution.
with τ = MNh/N. This equation shows how the density of protections reflects the past of the virus evolution.
B. Antigenic waves
We simulated (2)-(3) on a square lattice (Methods) and found a stable wave solution (Fig. 1B-D). The wave has a stable population size N, and moves approximately ballistically through antigenic space, pushed from behind by the immune memories left in the trail of past viral strains (Fig. 1B). These memories exert an immune pressure on the viruses, forming a fitness gradient across the width of the wave (Fig. 1C), favoring the few strains that are furthest from immune memories, at the edge of the wave. We assume that the solution of the coupled evolution equations (2)-(3) takes the form of a moving quasispecies in a d-dimensional antigenic space,

Here, we have written the solution in a co-moving frame, in which a motion with constant speed v takes place in the direction of the coordinate x1, and fluctuations in the other dimensions, ρ(x2,…, xd, t), centered around xi = 0 for i > 1, are assumed to be independent. In the next sections, we will analyse solutions of this form. First, we will project the d-dimensional antigenic wave onto the one-dimensional fitness space; this projection produces a travelling fitness wave [26–28, 30, 31] that determines the antigenic speed v and the mean pair coalescence time T2 of the viral genealogy. Second, we will study the shape of the d-dimensional quasispecies and determine the fluctuations in the transverse directions. These fluctuations produce a key result of this paper: immune interactions canalize the evolution of the antigenic wave; this constraint can be quantified by a persistence time governing the transverse antigenic fluctuations. Canalization is most pronounced spaces of low dimensionality d and, as we discuss below, affects the predictability of antigenic evolution.
C. Speed of antigenic evolution
Projected onto the fitness axis f = f (x, t), the solution is approximately Gaussian (Fig. 1D). This representation suggests a strong similarity to the fitness wave solution found in models of rapidly adapting populations with an infinite reservoir of beneficial mutations [26–28, 30, 31]. To make the analogy rigorous, we must assume that the fitness gradient in antigenic space is approximately constant, meaning that fitness isolines are straight and equidistant. Mutations along the gradient direction have a fitness effect that is linear in the displacement, while mutations along perpendicular directions are neutral and can be treated independently. Note that while we will use this projection onto fitness to compute the speed of the antigenic wave, our description remains in d dimension, and we will come back to transverse fluctuations in the next sections.
There exist different theories for the fitness wave, depending on the statistics of mutational effects. Our assumption of diffusive motion makes our projected dynamics equivalent to that studied in Ref. [31], which itself builds on earlier work [27]. In the limit where the wave is small compared to the adaptation time scale, vτ » σ, the wave may be replaced by a Dirac delta function at x = (vt, 0,…, 0) in Eq. 4. One can then calculate explicitly the immune density (upstream of the wave) and coverage (downstream of the wave, using Eq. 1):

 where Θ(x) = 1 for x ≥ 0 and 0 otherwise. This idealized exponential trail of immune protections h(x, t) corresponds to the blue trace of Fig. 1B, and the coverage or fitness gradient to the isolines of Fig. 1C.
where Θ(x) = 1 for x ≥ 0 and 0 otherwise. This idealized exponential trail of immune protections h(x, t) corresponds to the blue trace of Fig. 1B, and the coverage or fitness gradient to the isolines of Fig. 1C.
In the moving frame of the wave, (u, x2,…, xd), with u = x1 − vt, the local immune protection and viral fitness can be expanded locally for u, xi « vτ (see [25] for a similar treatment in a one-dimensional antigenic space):
 where f0 = ln R0 − M ln[1 + r/(vτ)] is the average population fitness, and
where f0 = ln R0 − M ln[1 + r/(vτ)] is the average population fitness, and

is the fitness gradient. Rescaling the antigenic variable x1 as sx1, this process is equivalent to the evolution of a population where mutation effects are described by diffusion in fitness space with coefficient Ds2. This is precisely the model from which the fitness wave solution of Ref. [27, 31] was described (see Appendix). In the following we will use results from these works to describe the antigenic wave, with however the following difference concerning the regulation of the population size. In the usual fitness wave theory, population is kept constant by construction, meaning that fitness is only relevant when compared to the mean of the population. By contrast, in our model population size is left free, and fitness is defined as an absolute growth rate. However the fitness of the whole viral population undergoes continuous negative drift due to the constant adaptation of immune systems, encoded in the svt term in Eq. 8. This negative fitness drift has an analogous effect to subtracting the mean fitness in models with constant population size, making the equivalence possible.
The fitness wave theory allows us to make analytical prediction about the properties of the antigenic wave. Let us start with its population size N, which is regulated by how fast the immune system catches up with the wave. The immune turnover time τ in Eq. 4 is inversely proportional to N: the larger the population size, the faster immune memories are updated, increasing the immune pressure on current viral strains (lower f0), and thus decreasing N. As the moving wave reaches a stable moving state, its size N becomes stable over time, giving the condition (1/N)dN/dt = f0 = 0, which in turn constraints the ratio between the wave’s size and speed:

But the fitness wave theory predicts that the speed of the wave itself depends on the population size. The larger N, the more outliers at the nose the fitness wave, and the further out they may jump in antigenic space, establishing fitter ancestors of the future population. This results in a fitness wave whose speed depends only weakly on population size and mutation rate (see [31] and Appendix),

where DF = s2D and vF = sv are the diffusivity and wave speed in fitness space, which are related to their counterparts in antigenic space through the scaling factor s. Replacing this scaling into Eq. 11 yields a relation between antigenic speed and population size,
 which closes the system of equations: using the definition of s (Eq. 9), Eqs. 10 and 12 completely determine N and v as a function of the model’s parameters (through a transcendental equation, see Appendix). We validated these theoretical predictions for N and v by comparing them to numerical simulations, which show good agreement over a wide range of parameters (Fig. 2A-B).
which closes the system of equations: using the definition of s (Eq. 9), Eqs. 10 and 12 completely determine N and v as a function of the model’s parameters (through a transcendental equation, see Appendix). We validated these theoretical predictions for N and v by comparing them to numerical simulations, which show good agreement over a wide range of parameters (Fig. 2A-B).

Shown are the numerical versus analytical predictions for the wave’s population size N (A), speed v (B), width σ along the wave’s direction of motion (C), and width σ⊥ in the direction perpendicular to motion (D), with d = 2 dimensions. Length are in units of the cross-reactivity range (so that r = 1, with no loss of generality). Parameters: Nh = 108 (squares), 1010 (circles), or 1012 (triangles); ln R0 = 1 (filled symbols) or 3 (empty symbols); M = 1 (small symbols) or 5 (large symbols).
D. Shape of the antigenic wave
The width σ of the wave in the direction of motion is given by Fisher’s theorem, which relates the rate of change of the average fitness to its variance in the population: ∂tf = Var(f). In our description fitness and the antigenic dimension x1 are linearly related with coefficient s, implying s2σ2 = sv. The result of that prediction for σ is validated against numerical simulations in Fig. 2C.
The wave is led by an antigenic ‘nose’ formed by few outlying strains of reduced cross-reactivity with the concurrent immune population, generating high fitness. These strains have phenotype uc = sσ4/4D = v2/(4Ds) and fitness suc. They serve as founder strains from which the bulk of the future population will derive some time ∼ uc/v = σ2/4D later (see Appendix). As a result, two strains taken at random can trace back their most recent common ancestor to some averge time ⟨T2⟩ = ασ2/2D in the past, where α ≈ 1.66 is a numerical factor estimated from simulations [31].
To explain the width σ⊥ of the wave in the other phenotypic dimensions than that of motion (xi>1), we note that in these directions evolution is neutral. Two strains taken at random in the bulk are expected to have drifted, or ‘diffused’ in physical language, by an average squared displacement  from their common ancestor, so that their mean squared distance is 4D ⟨T2⟩ = 2ασ2 along xi. If one assumes an approximately Gaussian wave of width σ⊥, the mean square distance between two random strains along xi should be equal to
from their common ancestor, so that their mean squared distance is 4D ⟨T2⟩ = 2ασ2 along xi. If one assumes an approximately Gaussian wave of width σ⊥, the mean square distance between two random strains along xi should be equal to  . Equating the two estimates yields
. Equating the two estimates yields  . Fig. 2D checks the validity of this prediction against simulations.
. Fig. 2D checks the validity of this prediction against simulations.
Both longitudinal and transversal fluctuations in antigenic space are instances of quantitative traits under interference selection generated by multiple small-effect mutations. The width of these traits is governed by the common relation  , which expresses the effective neutrality of the underlying genetic mutations [32]. This relation says that antigenic variations in all dimensions scale in the same way with the model parameters, and the wave should have an approximately spherical shape. Consistently, here we find a wave with a fixed ratio α ≈ 1.66 between transverse and longitudinal variations. This implies a slightly asymmetric shape (which may be non-universal and depend on the microscopic assumptions of our mutation model).
, which expresses the effective neutrality of the underlying genetic mutations [32]. This relation says that antigenic variations in all dimensions scale in the same way with the model parameters, and the wave should have an approximately spherical shape. Consistently, here we find a wave with a fixed ratio α ≈ 1.66 between transverse and longitudinal variations. This implies a slightly asymmetric shape (which may be non-universal and depend on the microscopic assumptions of our mutation model).
In what parameter regime is our theory valid? The fitness wave theory we built upon is meant to be valid in the large population size, N » 1. In addition, we assumed that the fitness landscape was locally linear across the wave. This approximation should be valid all the way up to the tip of wave, given by uc, since this is where the selection of future founder strains happen. This condition translates into uc « r, implying D » r2/ ln(N)2, where D is in antigenic unit squared per infection cycle. This result means that one infection cycle will not produce enough mutations for the virus to leave the cross-reactivity range. In that limit, another assumption is automatically fulfilled, namely that the width of the wave be small compared to the span of immune memory: σ « vτ. Our simulations, which run in the regime of very slow effective diffusion (D/r2≲ 10−6) and have relatively large population sizes (N ≳104), satisfy these conditions. This explains the good agreement between analytics and numerics.
E. Equations of motion of the wave’s position
The wave solution allows for a simplified picture. The wave travels in the direction of the fitness gradient (or equivalent the gradient of immune coverage) with speed v (Fig. 3A). Occasionally the population splits into two separate waves that then travel away from each other and from their common ancestor (Fig. 3B). The tip of the wave’s nose, which contains the high-fitness individual that will seed the future population, determines its future position in antigenic space. In the directions perpendicular to the fitness gradient, this position diffuses neutrally with coefficient D. This motivates us to write effective equations of motion for the mean position of the wave:

 where ξ‖ and ξ⊥ are Gaussian white noises in the directions along, and perpendicular to, the fitness gradient ∂xf/ |∂xf| = −∂xc/ |∂xc|. D‖ is an effective diffusivity in the direction of motion resulting from the fluctuations at the nose tip. These fluctuations are different than suggested by D, as they involve feedback mechanisms between the wave’s speed v, size N, and advancement of the fitness nose uc. In the following we do not consider these fluctuations, and focus on perpendicular fluctuations instead.
where ξ‖ and ξ⊥ are Gaussian white noises in the directions along, and perpendicular to, the fitness gradient ∂xf/ |∂xf| = −∂xc/ |∂xc|. D‖ is an effective diffusivity in the direction of motion resulting from the fluctuations at the nose tip. These fluctuations are different than suggested by D, as they involve feedback mechanisms between the wave’s speed v, size N, and advancement of the fitness nose uc. In the following we do not consider these fluctuations, and focus on perpendicular fluctuations instead.

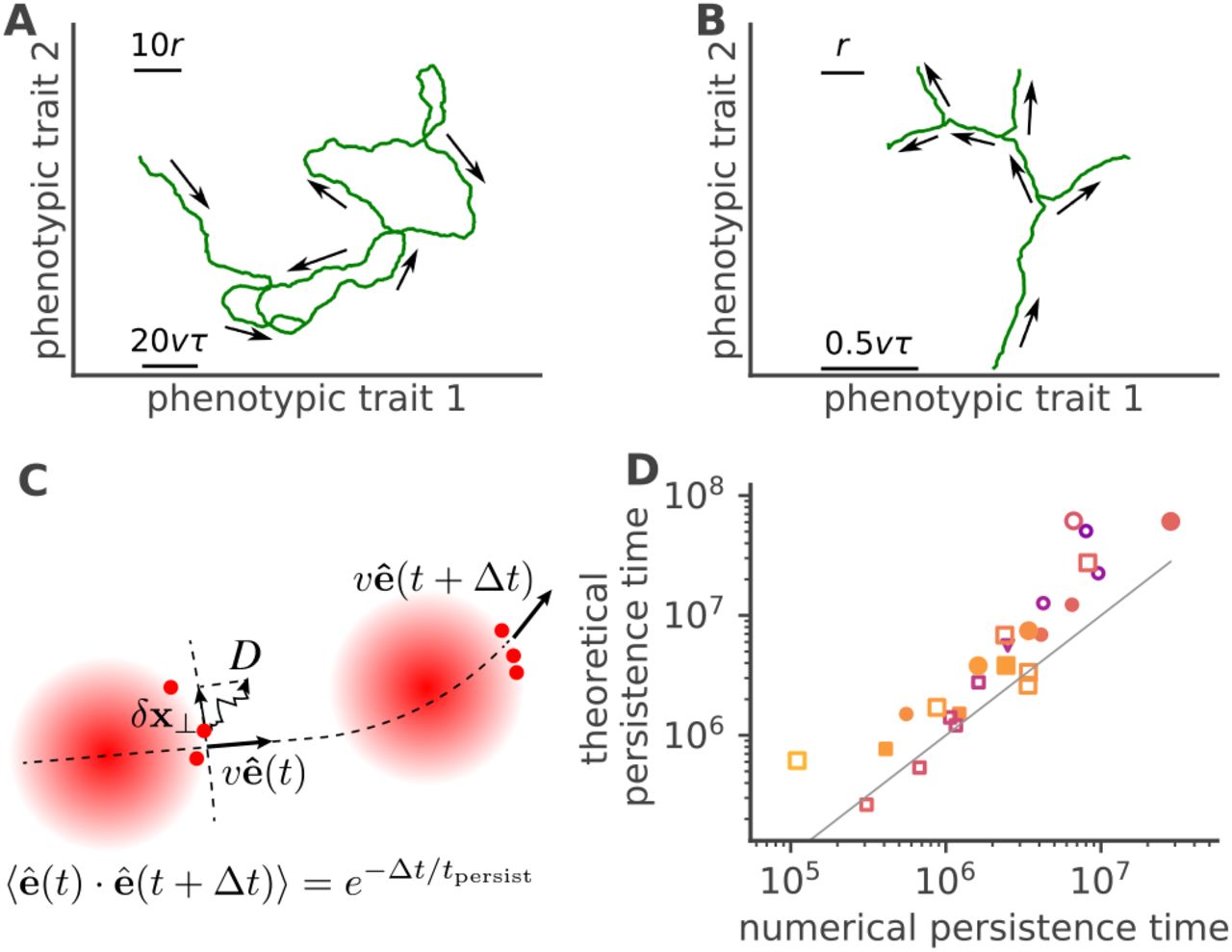
A. The wave moves forward in antigenic space but is driven by its nose tip, which undergoes antigenic drift (diffusion) in directions perpendicular to its direction of motion. These fluctuations deviate that direction, resulting in effective angular diffusion. B. When antigenic drift is large, the wave may randomly split into sub-populations, creating independent waves going in different directions. Each wave can also go extinct as size fluctuations bring it to 0. C. Cartoon illustrating the wave’s angular diffusion. Selection and drift combine to create a inertial random walk of persistence time tpersist. D. Analytical prediction (Eq. 17) for the persistence time, versus estimates from simulations. Symbols and colors are the same as in Fig. 2.
F. Angular diffusion and antigenic canalization
In the description of Eqs. 13-14, the viral wave is pushed by immune protections left in its trail. The fitness gradient, and thus the direction of motion, points in the direction that is set by the wave’s own path. This creates an inertial effect that stabilizes forward motion. On the other hand, fluctuations in perpendicular directions are expected to deviate the course of that motion, contributing to effective angular diffusion. To study this behaviour, we assume that motion is approximately straight in direction x1 = vt, and study small fluctuations in the perpendicular directions, x⊥ = (x2,…, xd), with |x⊥ | « r (as illustrated in Fig. 3C). Eqs (13)-(14) simplify to (see Appendix):
 where
where  is an effective memory timescale combining the host’s actual immune memory, and the cross-reactivity with strains encountered in the past.
is an effective memory timescale combining the host’s actual immune memory, and the cross-reactivity with strains encountered in the past.
Eq. (15) may be solved in Fourier space. Defining  , it becomes:
, it becomes:

To understand the behaviour at long times » T, we expand at small  or equivalently in the temporal domain
or equivalently in the temporal domain  . This implies that the direction of motion, e ∼∂xf/|∂xf | ∼ ∂tx/|∂tx|, undergoes effective angular diffusion in the long run:
. This implies that the direction of motion, e ∼∂xf/|∂xf | ∼ ∂tx/|∂tx|, undergoes effective angular diffusion in the long run:  . The persistence time of that inertial motion,
. The persistence time of that inertial motion,
 does not depend explicitly on the speed and size. However, a larger diffusivity implies larger N and v while reducing the persistence time. Likewise, a larger reproduction number R0 or smaller memory capacity M speeds up the wave and increases its size, but also reduces its persistence time. This implies that, for a fixed number of hosts Nh, larger epidemic waves not only move faster across antigenic space, but also change course faster.
does not depend explicitly on the speed and size. However, a larger diffusivity implies larger N and v while reducing the persistence time. Likewise, a larger reproduction number R0 or smaller memory capacity M speeds up the wave and increases its size, but also reduces its persistence time. This implies that, for a fixed number of hosts Nh, larger epidemic waves not only move faster across antigenic space, but also change course faster.
This persistence time scales as the time it would take a single virus drifting neutrally to escape the crossreactivity range, r2/D. For comparison, the much shorter timescale for a population of viruses to escape from the cross-reactivity range r,
 scales with the inverse incidence rate Nh/N. This is consistent with the whole population having been infected at least one every ∼ Nh/N infection cycles. This separation of time scales is consistent with the observation that evolution in the transverse directions is driven by neutral drift, which is much slower than adaptive evolution in the longitudinal direction. Both tpersist and tescape are much longer than the coalescence time of the viral population, uc/v ∼ σ2/4D, since they reflect long-term memory from the immune system. However, while tescape ∼ Nh/N corresponds to the re-infection period and is thus bounded by the hosts’ immune memory (itself bounded by their lifetime, which we do not consider), tpersist may be much longer than that. This is possible thanks to inertial effects, which are allowed by the high-order dynamics of Eq. 15 generated by the immune system. This very much like when, in mechanics, a massive object set in motion in a given direction will keep that direction without the need for an external force to maintain it.
scales with the inverse incidence rate Nh/N. This is consistent with the whole population having been infected at least one every ∼ Nh/N infection cycles. This separation of time scales is consistent with the observation that evolution in the transverse directions is driven by neutral drift, which is much slower than adaptive evolution in the longitudinal direction. Both tpersist and tescape are much longer than the coalescence time of the viral population, uc/v ∼ σ2/4D, since they reflect long-term memory from the immune system. However, while tescape ∼ Nh/N corresponds to the re-infection period and is thus bounded by the hosts’ immune memory (itself bounded by their lifetime, which we do not consider), tpersist may be much longer than that. This is possible thanks to inertial effects, which are allowed by the high-order dynamics of Eq. 15 generated by the immune system. This very much like when, in mechanics, a massive object set in motion in a given direction will keep that direction without the need for an external force to maintain it.
The high-frequency behaviour of (16) has a logarithmic divergence, meaning that the total power of ê is infinite unless we impose a (ultraviolet) cutoff. Such a regularization emerges from the fine structure of the wave. While the motion of the wave is driven by its nose tip, the immune pressure only extends back to the recent past of the bulk of the distribution, which stands at a distance uc away from the nose. In other words, there is a lag (and thus an gap uc in antigenic space) between the most innovative variants that drive viral evolution, and the majority of currently circulating variants which drive host immunity. Mathematically, this implies that the domain of integration of the first term in the right-hand side of (15) should start at tc = uc/v, which regularizes the divergence. A more careful analysis provided in the Appendix shows that this regularization does not affect the long-term diffusive behaviour of the wave.
G. Deflection, speciations, and predictability of antigenic evolution
We now examine how deflections of the wave in the transverse direction determines the predictability and stability of the viral quasi-species. Assuming t » T, angular diffusion causes motion to be deflected as (see Appendix)  . Crucially, this deflection depends on the dimension of the antigenic space. Higher dimension means more deviation from the predictable course of the wave, and thus less predictability. We can define a predictability time scale
. Crucially, this deflection depends on the dimension of the antigenic space. Higher dimension means more deviation from the predictable course of the wave, and thus less predictability. We can define a predictability time scale
 which is the time it takes for prediction errors to become of the order of the cross-reactivity range. In low dimensions, this time scales as a weighted geometric mean between tescape ∼ T and tpersist ∼ r2/D. However, at high dimensions tpredict may be significantly reduced, causing loss of predictability even below tescape.
which is the time it takes for prediction errors to become of the order of the cross-reactivity range. In low dimensions, this time scales as a weighted geometric mean between tescape ∼ T and tpersist ∼ r2/D. However, at high dimensions tpredict may be significantly reduced, causing loss of predictability even below tescape.
To get a sense of numbers, we can compare our results with epidemiological data, taking the evolution of influenza as an example, with an infection cycle time of 3 days. It is assumed that individuals lose immunity to the circulating strain of the flu within ∼ 5 years ∼ 500 cycles, meaning that the wave would travel a distance r in t = 500, i.e. v/r ∼ 2 ·10−3. For instance, with Nh = 109-1010, R0 = 2, D/r2 = 3 · 10−6, and M = 1, we get v/r ∼ 1.3 · 10−3 and tpersist ∼ 2 · 104 200 years. By contrast, the predictability timescale tpredict is much shorter and depends on dimension, albeit slowly, ranging from 20 years for d = 2 to about 2 years for d = 1000. We stress that these numbers are obtained by scaling laws, and should not be taken as precise quantitative predictions.
Large deflections may also cause speciations, or splits, which occur when two substrains co-exist long enough to become independent from the immune standpoint. This happens when two sub-lineages see the difference of their transverse positions Δx⊥ become larger than Δx0 ∼ r, within some limited period given by the coalescence time. We estimated the rate of such splitting events using a saddle-point approximation (see Appendix):
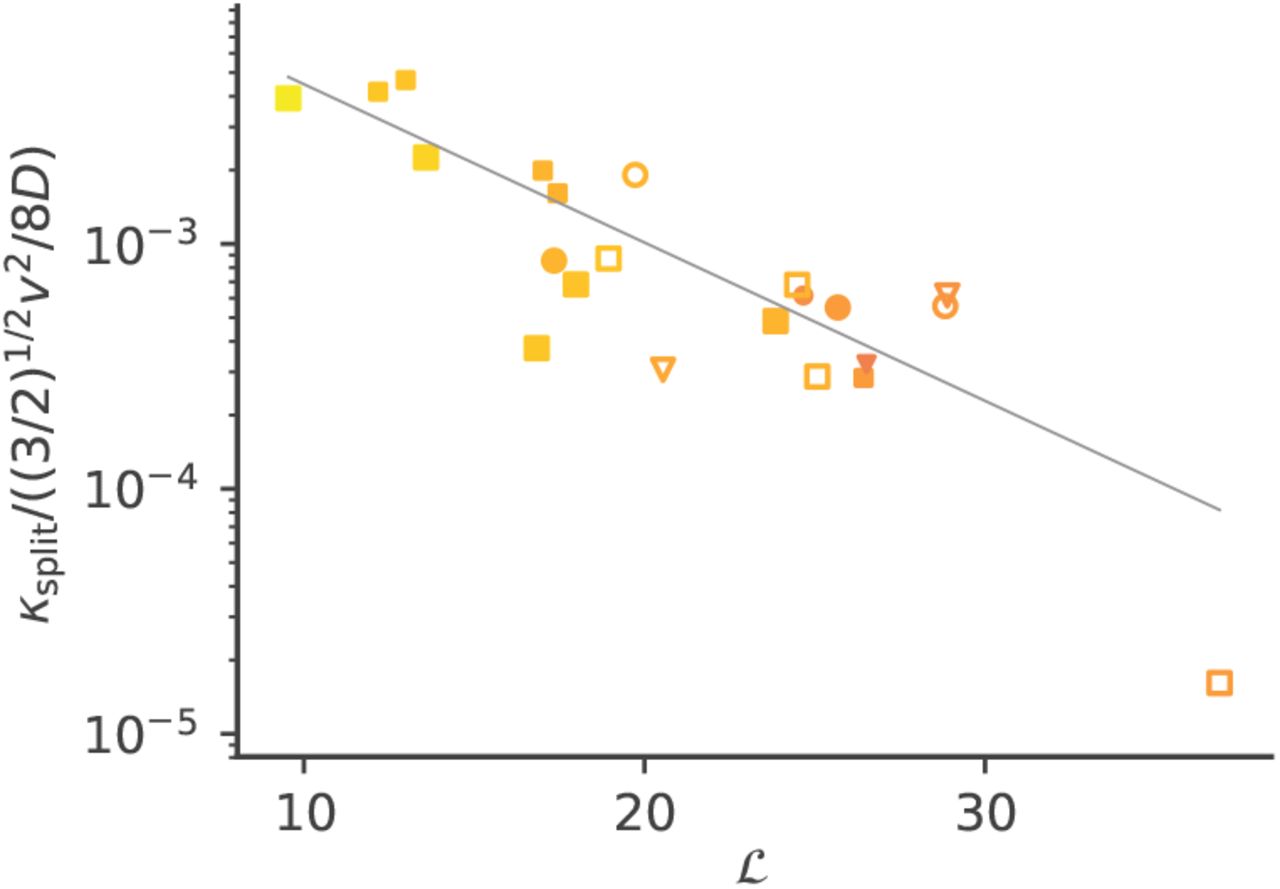
 with α some numerical factor. Simulations confirmed the validity of this scaling (Fig. 4).
with α some numerical factor. Simulations confirmed the validity of this scaling (Fig. 4).

Rescaled rate of splitting events, defined as the emergence of two substrains at distance Δx0 = 0.1r from each other in antigenic space, meaning that they are becoming antigenically independent. The predicted scaling, ksplit ∼ (v2/D)e− ℒ, as well as the definition of the collective variable ℒ as a function of the model parameters, are given by Eq. 20. The line shows a linear fit of the logarithm of the ordinate.
The splitting rate grows with the dimension, consistent with the intuition that departure from canalized evolution is easier when more directions of escape are available. Splitting events are expected to strongly affect our ability to predict the future course of the wave. However, the rarity of such events (exponential scaling of ksplit) means that they will have a lower impact on predictability than deflections. These results provide a theoretical and quantitative basis from which to assess the effect of dimension on predictability, and possibly estimate d from antigenic time course data of real viral populations.
III. DISCUSSION
In this work we have developed an analytical theory for studying antigenic waves of viral evolution in response to immune pressure. We showed that predictabilty is limited by two features of antigenic evolution, directional diffusion and lineage speciations of the antigenic wave.
Unlike previous efforts that considered one- [25] or infinite-dimensional antigenic spaces [12], we explicitly embedded the antigenic phenotype in a d-dimensional space. This description allows for the possibility of compensatory mutations, and makes it easier to compare results with empirical studies of viral evolution projected onto low-dimensional spaces [8, 9]. Unlike these studies however, our work does not address the question how an effective dimension of antigenic space arises from the molecular architecture of immune interactions. Rather, we focused on the implications of the dimensionality of antigenic space for phenotypic evolution and its predictability.
Our results suggest a hierarchy of time scales for viral evolution. The shortest is the coalescence time ⟨T2⟩, which determines population turnover. Then comes tescape, which is the time it takes the viral population to escape immunity elicited at a previous time point. The longest timescale is the persistence time tpersist, which governs the angular diffusion of the wave’s direction. That time scale is due to inertial effects rather than rely directly on the hosts’ immune memories, and may thus exceed their individual lifetimes. Finally, the prediction timescale tpredict, beyond which prediction accuracy falls below the resolution of cross-reactivity, scales between tescape and tpersist at low dimensions. However, unlike the other timescales, it decreases with the dimension of the antigenic space, and may become arbitrarily low at very high dimensions.
Our framework should be applicable to general host-pathogens systems. For instance, co-evolution between viral phages and bacteria protected by the CRISPR-Cas system [33] is governed by the same principles of escape and adaptation as vertebrate immunity. Even more generally, our theory (Eqs. 2,3) may be relevant to the coupled dynamics of predators and preys interacting in space (geographical or phenotypic), opening potential avenues for experimental tests of these theories in synthetic microbial systems.
IV. METHODS
We simulated discrete population dynamics of infected hosts n(x, t)) and immune protections nh(x, t) ≡ Nhh(x, t) (all integers) on a 2D square lattice with lattice size Δx ranging from 10−5r to 0.1r. Each time step corresponds to a single infection cycle, Δt = 1. At each time step: (1) viral fitness f is computed at each occupied lattice site from the immune coverage Eq. 1; (2) viruses at each occupied lattice site are grown according to their fitness, n(x, t + 1) ∼ Poisson[(1 + f Δt)n(x, t)]; (3) viruses are mutated by jumping to nearby sites on the lattice; (4) the immune system is updated according a discrete version of Eq. 3, by implementing nh(x, t+1) = nh(x, t) + n(x, t) and then removing N (t) protections at random (so that Nh remains constant).
To implement (1), we used a combination of exact computation of Eq. 1 and approximate methods, including one based based non-homogeneous fast Fourier transforms [34, 35]. Details are given in the Appendix.
To implement (3), we drew the number of mutants at each occupied site from a binomial distribution Binomial(n(x, t), 1 − e−µΔt). The number of new mutations m affecting each of these mutants is drawn from a Poisson distribution of mean µΔt conditioned on having at least 1 mutation. The new location of each mutant is drawn as x + δx, with  (rounding is applied to each dimension), where Ei is a vector of random orientation and modulus drawn from a Gamma distribution of mean δ 2Δx and shape parameter 20. This distribution was chosen so as to maximize the number of non-zero jumps while maintaining isotropy. We then define
(rounding is applied to each dimension), where Ei is a vector of random orientation and modulus drawn from a Gamma distribution of mean δ 2Δx and shape parameter 20. This distribution was chosen so as to maximize the number of non-zero jumps while maintaining isotropy. We then define  .
.
To find the wave solution more rapidly, the viral population was initialized as a Gaussian distribution centered at (0, 0) with size N and width σ in all dimensions, to which 0.1% additional viruses are randomly added within the interval (0; uc) along x1 (N, σ, and uc being all given by the theory prediction). Immune protections are placed according to Eq. 6. The first 20,000 time steps serve to reach steady state and are discarded from the analysis. When a population extinction (N = 0) or explosion (N = Nh/2) occurs, the simulation is resumed at an earlier checkpoint to avoid re-equilibrating. Simulations are ended after 5 106 steps of after 20 consecutive extinctions or explosions from the same checkpoint.
In order to analyze the organization of viruses in phenotypic space, we save snapshots of the simulation at regular time intervals. For each saved snapshot we take all the coordinates with n > 0 and then cluster them into separate lineages through the python scikit-learn DB-SCAN algorithm [36] [37] with the minimal number of samples min samples = 10. The ϵ parameter defines the maximum distance between two samples that are considered to be in the neighborhood of each other. We perform the clustering for different values of ϵ and select the value that minimizes the variance of the 10th nearest neighbor distance. Clustering results are not sensitive to this choice. This preliminary clustering step is refined by merging clusters if their centroids are closer than the sum of the maximum distances of all the points in each cluster from the corresponding centroid.
From the clustered lineages we can easily obtain a series of related observables, such as its speed v obtained as the derivative of the center’s position. The width of the lineage profile in the direction of motion σ as well as in the perpendicular direction σ⊥ are obtained by taking the standard deviaton of the desired component of the distances of all the lineage viruses from the lineage centroid. Reported numbers are time averages of these observables. We can track their separate trajectories in antigenic space. A split of a lineage into two new lineages is defined when two clusters are detected where previously there was one, and their distance is larger than Δx0.
To estimate the persistence time, we first subsample the trajectory so that the distance between consecutive points is bigger than 6(⟨σ⟩ + std(σ)) so that fast fluctuations in the population size do not affect the inference. We take the resulting trajectory angles and smooth them with a sliding window of 5. Then we divide the trajectory into subsegments, and compute the angles mean squared displacement (MSD) over all lineages and all subsegments. We consider time lags only bigger than twice the typical smoothing time, and if the MSD trace is long enough we also require the time lag to be bigger than 2T. Finally we only keep time lag bins with at least 10 datapoints. We fit the resulting time series to a linear function ax + b, and get the persistence time as  . We compute the reduced χ2 as a goodness-of-fit score. Results are shown for simulations that had enough statistics to perform the fit, lasted at least 105 cycles, and had a reduced χ2 below 3.
. We compute the reduced χ2 as a goodness-of-fit score. Results are shown for simulations that had enough statistics to perform the fit, lasted at least 105 cycles, and had a reduced χ2 below 3.
Acknowledgements
The study was supported by the European Research Council COG 724208 and ANR-19-CE45-0018 “RESP-REP” from the Agence Nationale de la Recherche and DFG grant CRC 1310 “Predictability in Evolution”.
Appendix A: Fitness wave theory
We decompose the density of viral strains according to the main direction of the wave x1:

where ϕ is normalized to 1. Projecting and linearizing Eq. (2) of the main text yields:
 with s defined by (9), and η1 = dx2 · dxd η(x, t), so that
with s defined by (9), and η1 = dx2 · dxd η(x, t), so that  . The change of variable
. The change of variable  , yields the traveling wave equation of Ref. [31]:
, yields the traveling wave equation of Ref. [31]:
 with
with  and
and  .
.
Note that this continuous description differs from that used in Ref. [12], which also describes a fitness wave in antigenic space. Their approach relies on a discrete evolutionary model where each mutation confers a fixed fitness advantage, as described by the fitness wave solution of Desai and Fisher [28].
Applying the formulas of that theory yield in the limit of large populations:

Or
 and
and

The fittest in the population is ahead of the bulk by uc = sσ4/4D in phenotypic space, with

Plugging in (9) yields:



From the stationarity condition (10) we obtain a self-consistent equation for N :

The condition uc « r, implies that r scales with N faster than uc, r » ln(N). We also want σ « vτ, therefore  , which is automatically satisfied by the previous condition.
, which is automatically satisfied by the previous condition.
Appendix B: Fluctuations in the direction perpendicular to motion
The dynamics of the wave in the directions that are orthogonal to x1 is governed by the projection of (13) onto x⊥ = (x2,…, xd):

From (14) we have

Taking the derivative along x⊥ yields:
 where we assumed |x⊥| « r, τ/v. This derivative is small compared to the gradient along the x1, so that we may approximate
where we assumed |x⊥| « r, τ/v. This derivative is small compared to the gradient along the x1, so that we may approximate  .
.
Replacing into (B1), we obtain:
 with 1/T = 1/τ + v/r. Using integration by part, this equation can be rewritten as an auto-regressive process on ∂tx⊥:
with 1/T = 1/τ + v/r. Using integration by part, this equation can be rewritten as an auto-regressive process on ∂tx⊥:
 where
where  has the property
has the property  . Computing the Fourier transform of E1(t/T)/T, which is given by
. Computing the Fourier transform of E1(t/T)/T, which is given by  and using the rule of convolution in Fourier space yields (16). Eq. (B5) can be re-written in terms of the direction of motion ê = ∂tx/|∂tx| ≈ ∂tx/v:
and using the rule of convolution in Fourier space yields (16). Eq. (B5) can be re-written in terms of the direction of motion ê = ∂tx/|∂tx| ≈ ∂tx/v:

Focusing on long-term behaviour yields the angular diffusion equation,  , for the direction of motion ê. The two-point function of ê follows the equation
, for the direction of motion ê. The two-point function of ê follows the equation
 where η(t) is a unit Gaussian white noise, leading to:
where η(t) is a unit Gaussian white noise, leading to:
 with tpersist = v2T 2/(4D) is defined as the persistence time. Going along the curviline coordinate that follows the trajectory with speed v, we obtain a persistence length of vtpersist.
with tpersist = v2T 2/(4D) is defined as the persistence time. Going along the curviline coordinate that follows the trajectory with speed v, we obtain a persistence length of vtpersist.
The logarithmic divergence at high frequencies in (16), which is also apparent in the logarithmic divergence in the temporal domain at small t in the auto-regressive Kernel E1(t/T)/T. This divergence may be regularized by realizing that there is a lag uc/v between the nose of the wave, which drives the behaviour of the wave, and its bulk. This implies that the integral over the past trajectory encoding the immune memory extends only up t − uc/v in the past:
 or after integration by parts:
or after integration by parts:
 with ϵ = tc/T « 1.
with ϵ = tc/T « 1.
In Fourier space this reads:
 where now the K is the Laplace transform of the operator:
where now the K is the Laplace transform of the operator:

Since E1(z) goes to 0 for large z, (z − K(z))−1 goes as 1/z for z → ∞ (small time scales), which means that at high frequency fluctuations of ∂tx⊥ (and thus of the direction of motion ê) track those of ξ⊥.
Since E1(x) ∼ − γ − ln(x) + x at small x, then E1(ϵ) − E1(ϵ (1 + z)) ≈ ln(1 + z) − ϵz for moderate z and small ϵ, and K(z) ≈ z − z2/2 + O (z2ϵ), so that (z − K(z))−1 ∼2/z2. We thus recover that at long time scales ∂tx⊥ diffuses with diffusivity 4D/T 2.
Appendix C: Rate of speciation
A speciation, or split, occurs when two strains starting at the tip of the nose of the fitness wave, and continuing through their progenies, co-exist long enough for them to become independent from the viewpoint of the immune pressure. This happens when their distance in the x⊥ direction become larger than some threshold scaling with the cross-reactivity range, Δx0 ∼ r.
Assuming t » T, angular diffusion causes a strain to bend from the main direction of the wave as:

After integration, the expected deviation reads:
 assuming x⊥(t = 0) = 0. Now if there are two strains a and b co-existing, their divergence in the perpendicular direction is Gaussian distributed with:
assuming x⊥(t = 0) = 0. Now if there are two strains a and b co-existing, their divergence in the perpendicular direction is Gaussian distributed with:

Two strains are expected to co-exist at the leading edge for a time t before one of them gets absorbed into the bulk and goes extinct. The expected time for that scales as ∼τsw = uc/v = σ2/4D = v/4Ds. Assuming splitting events are rare, they occur when two co-existing strains both survive for an unusually long time. The distribution of such rare events is asymptotically given by the probability density function  . The probability that the two strains have drifted by at least r before that happens is then given by:
. The probability that the two strains have drifted by at least r before that happens is then given by:

Since we assume that this even is rare, P (Δx > Δx0; coexist) « 1, we make a saddle-point approximation (Laplace method) in the t variable. We look for the maximum of
 with respect to t, ∂tℒ = 0, which gives:
with respect to t, ∂tℒ = 0, which gives:

Applying Laplace’s method with Δx = Δx0 along with a linear approximation of ℒ in the vicinity of Δx > Δx0, we obtain:

Replacing  and τsw = v/(4Ds) yields:
and τsw = v/(4Ds) yields:

We check self-consistently that our approximation of angular diffusion is correct for Δx0 ∼ r. The condition is met when t* » T, or

This condition is equivalent to:
 which is (barely) satisfied for large population sizes (Eq. 12).
which is (barely) satisfied for large population sizes (Eq. 12).
Finally, to get the rate of splitting events, we must multiply the probability of a successful splitting event, P (Δx > Δx0; coexist), by the rate with which branches sprout from the main trunk of the phylogenic tree. Since mutations are modeled by continuous diffusion in antigenic space, such a new branch occurs whenever the individual virus on the trunk of the tree (defined as the virus that will eventually seed the entire future population) reproduces, as the two offspring immediately become antigenically distinct because of diffusion, and thus make two distinct branches. This happens with rate f (uc) = ucs = v2/4D, so that the overall rate of speciation should scale as:

Replacing Δx0 = ar, with a a numerical scaling factor, gives the result of the main text.
Appendix D: Details of simulation implementation
To update the fitness at each time step, we used either an exact computation of Eq. 1, or a faster approximate method based non-homogeneous fast Fourier transforms [34, 35]. For the exact computation, c (x, t +Δt) –c (x, t) was calculated at each time step by convolving the Kernel H with nh (x, t + Δt) −nh (x, t) (exploiting the fact that this sum is sparse because not all positions get updated). Both algorithms can be preceded by an extra-approximation for positions x′ with nh (x′, t + Δt) > 0 that are far enough from the viral cloud. We approximate the Kernel H by reducing |x−x′| to its projection along the direction given by x′−⟨x⟩ n, with ⟨x⟩ n = ∫ dx xn(x)/N. This allows us to pre-compute the contributions of all these x′ to Eq. 1 with a mere 1D convolution, ⩝x where n(x) > 0, speeding up the computation considerably. We choose the desired combination of approximations based on the convolution computational complexity, driven by the number of positions with n(x, t + Δt) > 0 and nh (x, t + Δt) −nh (x, t) > 0. To limit errors accretion we compute the update to the convolution exactly as explained above depending on a proxy for the fitness errors. In addition, the full convolution was recalculated with no approximation every 10000 steps.





{kind=link}
{kind=link}
{kind=link}
{kind=link}