

Abstract
DNA-based data storage platforms traditionally encode information only in the nucleotide sequence of the molecule. Here, we report on a two-dimensional molecular data storage system that records information in both the sequence and the backbone structure of DNA. Our “2DDNA” method efficiently stores high-density images in synthetic DNA and embeds metadata as nicks in the DNA backbone. To avoid costly redundancy used to combat sequencing errors and missing information content that typically requires additional synthesis, specialized machine learning methods are developed for automatic discoloration detection and image inpainting. The 2DDNA platform is experimentally tested on a library of images that show undetectable visual degradation after processing, while the image metadata is erased and rewritten to modify copyright information. Our results show that DNA can serve both as a write-once and rewritable memory for heterogenous data. Moreover, the storage density of the molecules can be increased by using different encoding dimensions and avoiding error-correction redundancy.
DNA-based data storage systems are viable alternatives to classical magnetic, optical, and flash recorders1. Macromolecular data storage platforms are nonvolatile, readout-compatible, extremely durable and offer unprecedented data densities unmatched by modern storage systems2–7. Traditional DNA-based data recording architectures store user information in the sequence content of synthetic DNA oligos within large pools that lack an inherent ordering, and user information is retrieved via next-generation or nanopore sequencing6. Despite recent progress, several challenges continue to hinder the practical implementation of molecular information storage, including the high cost of synthetic DNA, the lack of straightforward rewriting mechanisms, large write-read latencies, and introduction of errors during synthesis and sequencing which can lead to data loss in excess of 30%8.
In this work, we develop and experimentally test a hybrid DNA-based data storage system known as “2DDNA” that combines desirable features of both synthetic and nick-based9 recording by superimposing metadata stored via nicks onto images encoded in the sequence. In traditional DNA storage systems, the sequence content can carry a large amount of data, but rewriting is difficult. On the other hand, information stored in nicks generally has smaller density but can be easily rewritten. Our two-dimensional write-read system offers increased information density while simultaneously enabling rewriting of data recorded in the backbone via ligation followed by enzymatic nicking. To fully exploit the advantages of the two information dimensions, we use sequence-based encoding for large-volume image data and topological encoding for metadata in the same molecular storage construct. Other applications of the 2DDNA storage paradigm include using native DNA for the sequence content9 and nicked metadata describing the properties of the native substrate.
Image data is typically compressed before being recorded, and even a single error can cause catastrophic problems during decoding leading to unrecognizable reproductions6,10,11. Moreover, the rate of synthesis and sequencing errors may vary orders of magnitude from one platform to another. Therefore, to ensure accurate reconstruction, one needs to account for the worst-case scenario or perform write-read experiments to estimate the error rates before adding redundancies. Here, we completely eliminate the need for coding redundancy by developing specialized compression algorithms for images that can be combined with machine learning (ML) and computer vision (CV) techniques for image reconstruction and enhancement and result in high-quality replicas of the original data. For some images with highly granular details, we propose unequal error protection methods12 based on low-density parity-check (LDPC) codes13 that only introduce redundancy for facial features.
The encoding framework of 2DDNA is shown in Fig. 1. In the sequence dimension, we perform aggressive quantization and specialized lossless compression that leads to two-fold file size reduction. Compression is known to cause significant losses in image quality when errors are present, so it is common practice to include up to 30% error-correction redundancy4,7,8 which ultimately increases the cost of the storage system. Hence, we avoid error-correction redundancy and instead tailor our compression algorithm to include image processing techniques from ML and CV to restore the image to its original quality. The specialized encoding procedure involves two steps, depicted in Fig. 1a. First, RGB channel separation is followed by 3-bit quantization and separate lossless compression of the three-color channels. The latter process is performed using the Hilbert space filling curve14 which preserves local 2D image similarity and smoothness, thereby resulting in linear strings with small differences between adjacent string entries. Moreover, we further use differential encoding15 that involves taking differences of adjacent string values to create new strings with a high probability of small symbols. Differential encoding is followed by Huffman encoding16 which exploits the bias towards small symbol values. Together, these operations are performed separately on strings partitioned into eight subsets according to their intensity (brightness) levels.

Schematic of the encoding and decoding procedure of our 2DDNA system. a) The encoding procedure in the first dimension (sequence content) entails splitting the color image into the Red (R), Green (G) and Blue (B) channel; aggressively quantizing the RGB channels from 256 to 8 intensity levels; performing lossless compression of individual channels through a combination of 2D to 1D conversion of the image data via space-filling curves followed by differential and Huffman encoding. Note that the encoding procedure is separately applied to each intensity level, and the generated binary vector is further augmented by channel information and addresses used to access the oligos. The scheme does not include error-correction redundancy. b) For images with granular and highly relevant image features, one can optionally use unequal error-correction coding based on low-density parity-check (LDPC) codes with only 3.3% redundancy compared to the scheme without redundancy. c) The encoding procedure in the second dimension (topological content) entails representing letters of the English alphabet in ASCII format and designating one nicking endonuclease to each of the seven bits in the format. Information is encoded using mixtures of endonucleases for which the ASCII bit is equal to 1. Rewriting is performed by sealing the nicks using the T4 DNA ligase and repeating the previously outlined procedure with different data. d) 2D data readout through the use of two subpools, one for each storage dimension. e) Image decoding is performed by reversing the steps of the encoding process in the first dimension.
Our compression algorithm involves a second step that translates the binary strings into DNA oligo sequences. Here, DNA oligos of length 196 nts are parsed into the following three subsequences (Fig. 1a): (1) a pair of primer sequences, each of length 20 nts, used as prefix and suffix, (2) an address sequence of length 10 nts, and (3) 11 information-bearing sequences of length 13 nts. Primer and address sequences are used for PCR amplification and random access5. In addition, a block of three nucleotides is prepended to the address sequence to represent the RGB color information. When converting binary data into DNA sequence content, we use two additional constrained mappings to ensure that the maximum run length of G symbols is limited to three (to avoid G quadruplexes), and that the GC content is in the range of 40 - 60%. Overall, the mapping scheme converts blocks of 16 bits into blocks of 10 nts for the address sequences, and blocks of 22 information bits into blocks of 13 nts. A detailed description of each step, including the addition of synchronizing markers, is provided in the Supporting Information (SI section 1).
In the topological dimension, we record the metadata in nicks created on the backbone of the synthetic DNA molecules by modifying our Punch-Cards system9 that was also used for specialized in-memory molecular computing17. Short binary strings are converted into positional encodings that determine the nicked/unnicked identity of a set of pre-selected nicking sites. A set of complementary nicking endonucleases is used as the writing tool and selected based on two main criteria: (1) endonucleases must be highly site-specific to prevent non-specific cleavage of the DNA template and hence preserve DNA integrity; and (2) recognition sequences should be selected with sufficiently large Hamming distances between them to prevent undesired cross-nicking (i.e., an enzyme nicking an undesired target site). The mixture composition determines which letter is stored based on the corresponding ASCII code, with the caveat that a ‘1’ is encoded through the presence of the enzyme in the mixture, whereas a ‘0’ is encoded through the absence of the enzyme. This method enables superimposing information on top of data stored in the DNA sequence content, with no need to change the synthetic platform, as shown in Fig. 1c.
To demonstrate proof-of-concept, we experimentally tested the storage platform on eight Marlon Brando movie posters, shown in Fig. 2a. The original files were of total size 8,654,400 bits, but after the two-step encoding procedure (Fig. 2b), reduced to 2,317,896 nts. The corresponding 11,826 DNA oligos were synthesized by Integrated DNA Technologies (IDT). One pool was reserved for each of the eight levels. The oPools were sequenced on an Illumina ISeq device following standard protocols described in the Methods section. Individual sequence reads may contain errors, so we first constructed a consensus sequence by aligning reads with error-free addresses, following the approach described in a previous work6. This process led to 11,739 perfectly recovered sequences and 5 sequences that contain errors but do not significantly compromise the image quality; the remaining 82 oligos were either highly corrupted or completely missing from the pool.

Write-Read results for encoding information content in the sequence dimension. a) Original images with 256 RGB intensity levels, encoded by 8 bits each. b) Quantized images with 8 RGB intensity levels, encoded by 3 bits each. c) Images generated directly from the information encoded in DNA oligos without error-correction redundancy. d) Images reconstructed after applying a combination of discoloration detection and image inpainting on the results in c. e) Images refined via smoothing of the results depicted in d. f) Image enhancement results for images shown in e. g) Images reconstructed using unequal error-correcting coding for facial features. h) Images reconstructed after applying a combination of discoloration detection and image inpainting on the results in g. i) Images refined via smoothing of the results depicted in h. j) Image enhancement results for images shown in i. k) Image enhancement results for images shown in c. In summary, the best quality results – obtained using our image processing techniques – are given in i and j (boxed).
The images generated from this procedure are depicted in Fig. 2c. Upon close inspection, it is apparent that the encoded images suffer from visible degradation, and in particular, large blocks of discolorations. These artifacts can be removed by applying a carefully designed combination of ML and CV image processing techniques (see Fig. 3), tailor-made to operate on images compressed according to our method.
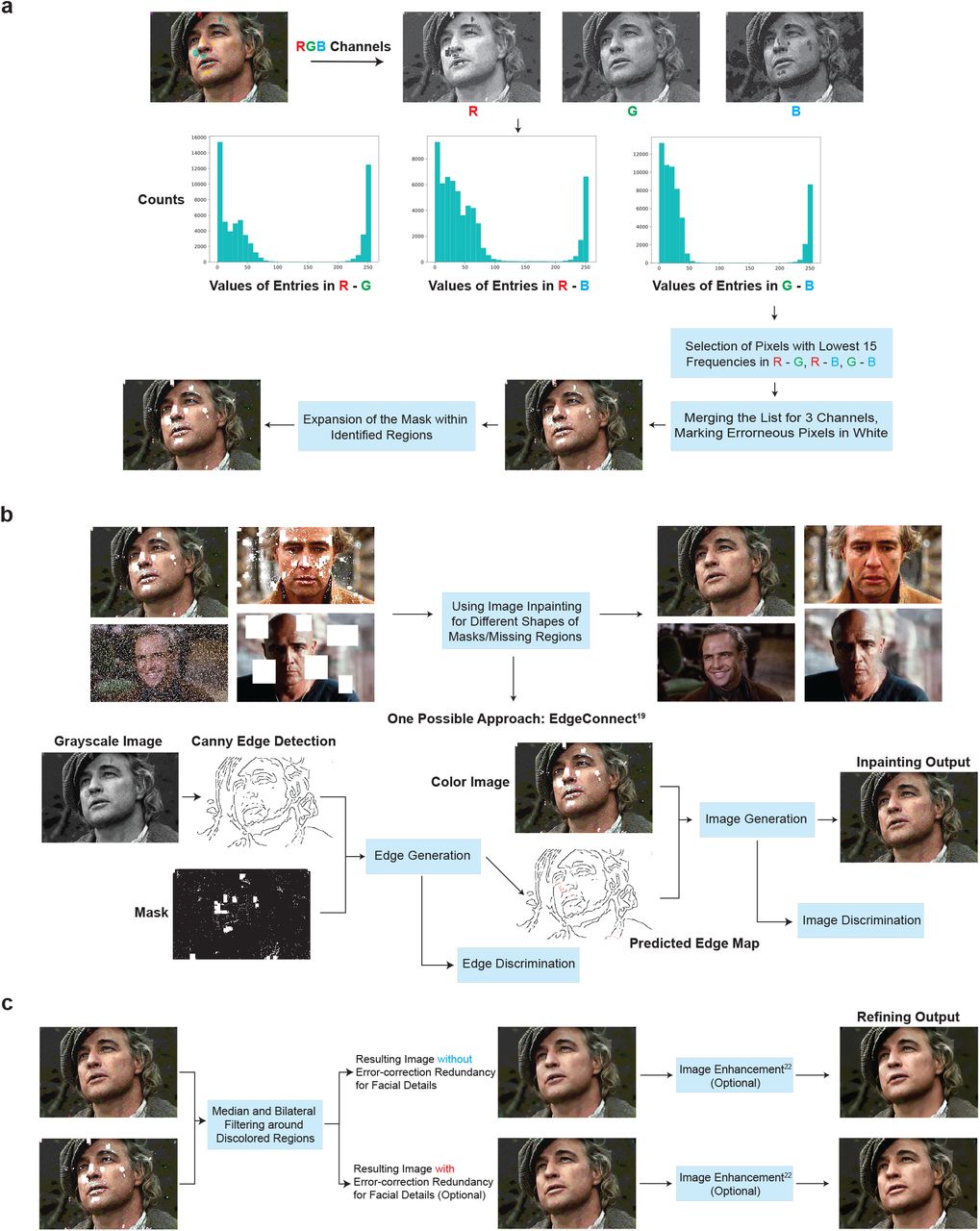
Diagram of the ML postprocessing techniques used to reconstruct images encoded in oPools. a) Automatic discoloration detection based on the natural redundancy in the three RGB color channels. The histograms reflect the frequency counts of the pairwise differences in channel intensity levels which are used to assess which color channel may contain errors (see SI section 3). b) Pixel masking and inpainting via deep-learning architectures. c) The smoothing and image enhancement procedures.
To correct for image discolorations, we implement a three-step postprocessing procedure. The first step includes detecting the locations with discolorations and masking them out, as shown in Fig. 3a. To pinpoint the discolored regions without direct visual inspection (i.e., in an automated fashion), we leverage the separate information content in the three distinct RGB color channels. Due to the random nature of errors, it is highly unlikely to have correlated errors in multiple channels for the same pixel. Hence, the three-color decomposition naturally acts like a 3-repetition code, because two of the three color channels are likely to be unperturbed. A detailed explanation of the technique can be found in the SI section 3 (with our preliminary approach described in18). The second step involves using a deep learning technique known as image inpainting19–21 to replace the masked pixels with values close to the original. Neural networks are well-suited for inpainting because they can be trained on massive datasets. For our system, we use the state-of-the-art GatedConvolution19 and EdgeConnect21 methods. The basic architecture of EdgeConnect is shown in Fig. 3b, and the results after applying discoloration detection and image inpainting are shown in Fig. 2d. Finally, the third step involves smoothing the image to reduce blocking effects caused by quantization and blending mismatched inpainted pixels, as shown in Fig. 3c. Here, we use bilateral22 and adaptive median smoothing23 on the coarsely inpainted images, and we include additional image enhancement features24 to further improve image quality.
The results of image smoothing are depicted in Fig. 2e, and the enhanced images are shown in Fig. 2f. As shown in Figs. 2e, f, some facial details in highly granular images remain blurred even after applying the learning methods. To address these issues, we further use unequal error-protection for such images, which implies adding highly limited redundancy only to oligos bearing facial features (e.g., eyes, lips), as shown in Fig. 1b and explained in SI section 1. Redundancy is added through a regular systematic LDPC code of rate 3/4, resulting in 391 additional oligos and an overall overhead of 3.3%. Images generated from this redundant pool are shown in Fig. 2g, whereas Figs. 2h, i, j parallel the results of Figs. 2d, e, f for the case of no unequal error-correction redundancy. Note that using image enhancement directly on the images generated from the information encoded in DNA oligos without error-correction redundancy results in very poor image quality because classical image enhancement methods cannot automatically correct discolorations (Fig. 2k).
As a proof of concept for storage in the topological dimension, we superimposed information on the same Marlon Brando images (see Fig. 4). In the writing experiment, we recorded the word “ILLINOIS,” comprising 56 bits in ASCII code, across eight different intensity-level DNA pools. We selected seven nicking endonucleases, each representing one bit of the 7-bit ASCII code. These enzymes have recognition sites that exist in at least one oligo of each of the eight pools, and the sites are used as recording positions. In the ASCII code, ‘1’ translates into inclusion, whereas ‘0’ translates into exclusion of the corresponding enzyme. Upon nicking, the pools are sequenced using the procedure described in Fig. 1d. In this way, the nicked oligos were denatured, resulting in ssDNA fragments of various lengths dictated by the position of the nicks. The fragments were subsequently converted into a dsDNA library and sequenced via Illumina ISeq. To verify the existence of short-length fragments capped at both ends by enzyme recognition sites, we developed a new detection algorithm (see SI section 4). To rewrite the data, we performed the process outlined in Fig. 1c, which involves treatment of the nicked DNA with the T4 DNA ligase. This erasure method completely removed the recorded metadata. Note that the ligase was perfectly effective in so far that each original oligo was accounted for in the sequenced pool. We then rewrote the word “GRAINGER” using the same topological nicking process with error-free reconstruction.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic of metadata encoding and identification using DNA nicking. The numbers in the middle column of the leftmost and rightmost tables represent the number of oligos in the sequenced pool capped by the recognition sequences of the nicking enzymes. The numbers listed in red correspond to the labels of nicking enzymes not used in the encoding of the letter to the left. As may be seen from both tables, the largest red numerical value is significantly smaller than the smallest black value for all encodings (e.g., 4<<81, 5<<40 in the leftmost table) and the second round of writing resulted in no spurious nicks whatsoever. The quality of the results in the rewriting experiment may be attributed to a more suitable choice of nicking enzymes determined upon inspection of the results of the first round. Hence it is recommendable to use the second collection of enzymes for recording purposes. Also note that we shuffled the symbol encodings for the rewriting experiment in order to test more combinations of nicking enzymes. In the erasure step, the T4 DNA ligase was used in a single step reaction to seal all the nicks. No nicks were found after the ligation reaction, showing that the ligase perfectly erased the data (middle table). Note that when recording “ILLINOIS” and “GRAINGER” only six and five enzymes were effectively used for the ASCII code, respectively, due to the choice of the letters in the words.
To read the content stored in both dimensions, two separate subpools are retrieved for each level. The sequence content is reconstructed by first sealing the nicks in one of the two subpools via ligation, as done during rewriting, followed by sequencing. Alternatively, to avoid ligation for the sequence content readouts, one may choose to only record the topological information on a subpool of oligos. The content in the nicks is retrieved using the second subpool. After sequencing, the reads are aligned to the now known full-length reads obtained from the first subpool. The results of the alignment are used in the algorithmic procedure to determine which enzymes were used for nicking and consequently, reconstructs the ownership metadata (see SI section 4).
Existing technologies for DNA synthesis, editing, and sequencing allow for writing and reading diverse information in multiple dimensions or molecular features. Our 2DDNA platform exploits these tools to enable recording data in two DNA dimensions, including sequence context and backbone structure, thereby opening the door for multidimensional macromolecular storage systems that can use multiple molecular properties (including molecular concentration). Our results show that the 2DDNA system takes advantage of powerful deep learning methods for image processing and enhancement to substantially improve the quality of the stored images without error-control redundancy. This represents a fundamental advancement in molecular storage which departs from prior methods and reduces the cost of data storage by greatly minimizing or eliminating the need for synthesizing redundant oligos. The tailor-made learning methods also overcome reliability issues that cannot be addressed by off-the-shelf JPEG compression and joint source-channel coding methods (see SI section 5). Moreover, our approach results in an exceptionally high physical storage density of the order of zettabytes/g and reconstructed images visually indistinguishable from the originals even under error-rates 0.0038/nt (before nicking) and 0.0053/nt after nicking and ligation (see SI section 9). In conclusion, 2DDNA provides the foundations for storage of heterogeneous datasets with rewriting capabilities and at the same time empowers the use of DNA media for nontraditional applications such as parallel in-memory computing.
Author Contributions
C.P., SMH.T.Y. and O.M. conceived the ML image compression, inpainting and unequal-error protection scheme and performed detailed data analysis. SK.T., C.M.S., SMH.T.Y. and O.M. designed the nicking experiments while SK.T. performed the experiments. A.G.H. performed the sequencing experiments and helped with the data analysis. All authors contributed towards the system development and participated in the writing of the manuscript.
Competing Interests
University of Illinois at Urbana-Champaign has filed a (pending) nonprovisional patent on behalf of C.P., O.M., C.M.S., SMH.T.Y., A.G.H. and SK.T., with application number: 17/102,143.
Methods
Machine Learning and Computer Vision methods
A detailed description of our compression algorithms and the supporting automatic discoloration detection, inpainting, smoothing and enhancement methods is relegated to the SI sections 1 and 3. due to the technical nature of the methodology used. The nick-detection method based on suffix-prefix matchings is also described in the SI section 4.
Nicking Experiments
All nicking experiments were performed using the manufacturer’s protocols available at: https://www.neb.com/products/restriction-endonucleases/hf-nicking-master-mix-time-saver-other/nicking-endonucleases/nicking-endonucleases.
oPool PCR Amplification and Sequencing
The oPools and corresponding primers were ordered from Integrated DNA Technologies (IDT): https://www.idtdna.com/pages/products/custom-dna-rna/dna-oligos/custom-dna-oligos/opools-oligo-pools.
All oPools were diluted to 5ng/ul. The primers were diluted to 10uM. Each oPool was amplified in separate reactions using forward and reverse primers for each of the 8 levels. Reactions were set up with 5ng of oPool, 1ul of each forward and reverse primer diluted to 10uM, 22ul of water and 25ul of Kapa HiFi DNA Polymerase (Roche, CA) with the following PCR cycling conditions: denaturation at 98°C for 45s, 8 cycles of 98°C for 15s, annealing at 51°C for 30s and extension at 72°C for 30s, followed by a final extension at 72°C for 1 min and hold to 4°C.
After PCR, the individual reactions were cleaned up with 50ul of AMPure beads (Agilent, CA) and eluted in 20ul of 10mM Tris. The PCR products were quantitated with the Qubit 3.0 fluorometer and run on a Fragment Analyzer (Agilent, CA) to determine the presence of a band of the correct size and the absence of free primers or primer-dimers. The PCR products from each level were pooled in equimolar concentration and the pool was converted into a sequence-ready library with the Kapa Hyper Library Construction kit (Roche, CA) with no PCR amplification. The final library was quantitated with Qubit and evaluated in a Fragment analyzer and further quantitated by qPCR. The library was loaded on a MiSeq (Illumina, CA) and sequenced for 250 cycles from each end of the library fragments with a Nano V2 500 cycles kit (Illumina). The raw fastq files were generated and demultiplexed with the bcl2fastq v2.20 Conversion Software (Illumina).
ssDNA Nicking Products Preparation for iSeq Sequencing
All nicked products were purified using the Qiaquick PCR purification kit (QIAGEN) and eluted in ddH2O. They were then denatured at 98°C for 5min and immediately cooled down to 4°C. The ssDNA samples were first quantified via the Qubit 3.0 fluorometer. Next, the Accel-NGS® 1S plus DNA library kit (Swift Biosciences) was used for library preparation following the manufacturer’s recommended protocol. Prepared libraries were quantitated using Qubit and then run on a DNA Fragment Analyzer (Agilent, CA) to determine fragment sizes, pooled in equimolar concentration. The pool was further quantitated by qPCR. All steps were performed for each sample separately and no nicked DNA samples were mixed. The pooled libraries were loaded on an iSeq device and sequenced for 250 cycles from each end of the library fragments with a Nano V2 500 cycles kit (Illumina). The raw fastq files were generated and demultiplexed with the bcl2fastq v2.20 Conversion Software (Illumina).
Acknowledgements
The work was funded by the DARPA Molecular Informatics Program, the NSF+SRC SemiSynBio program under agreement number 1807526 and the NSF grant 1618366.
Footnotes
Author ORCID updated
References




