

Abstract
Genome editing requires insertion of DNA sequences into specific locations. Protocols involving clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated (Cas) proteins rely on homology-directed repair, require laborious vector construction, and have low efficiency. DNA oligonucleotides can be used as donors for targeted insertion via nonhomologous end joining. Our simple protocol eliminates the need for expensive equipment and vector construction by using polyethylene glycol to deliver non-modified single-stranded DNA oligonucleotides and CRISPR-Cas9 ribonucleoprotein into protoplasts. We achieved targeted insertion frequencies of up to 50.0% in Nicotiana benthamiana and 13.6% in rapid cycling Brassica oleracea without antibiotic selection. Using a 60-nt donor containing 27 nt in each homologous arm, 6 of 22 regenerated plants showed targeted insertions, and 1 contained a precise insertion of a 6-bp EcoRI site. Whole-genome sequencing showed that the DNA inserted only in the targeted positions, and genetic analysis showed that the inserted sequences transmitted to the next generation.
Introduction
To insert DNA into a specific location in the plant genome, a DNA double-strand break (DSB) must be induced at the target position and the donor DNA (DD) must be inserted into this position via homology-directed repair (HDR)1–12 or nonhomologous end joining (NHEJ).13–14 Many tools for creating DSBs are currently available.15–17 Due to its convenience and efficiency, the combination of clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR associated (Cas) proteins has become a favorite tool among genetic engineers.4 In addition to CRISPR-Cas reagents, DD must be delivered into the target cells; increasing the amount of delivered DD enhances targeted insertion (TI) efficiency. For example, in maize (Zea mays), targeted mutagenesis using CRISPR has been achieved via different methods, although TI plants were obtained only via biolistic methods rather than Agrobacterium-mediated transformation because the number of DD copies delivered by the latter method is low.2
Protoplasts offer an alternative substrate for genetic transformation and transfection, since they enable the delivery of high numbers of DNA copies18,19, and the DNA can be inserted into the genome.18 Like the transcription activator-like effector nuclease genome editing system20, CRISPR reagents [ribonucleoprotein (RNP) or plasmid DNA] can be delivered into protoplasts via polyethylene glycol (PEG)-mediated transfection, and targeted mutations can be achieved.21–28 The mutated protoplasts can then be regenerated into plants and, in contrast to other tissue culture-based transformation protocols in dicot plants where transformants are chimeric and some of the mutated alleles are not heritable, the mutated alleles are passed onto the progeny.21, 23, 24 Here, we describe a simple, high-TI-efficiency protoplast-based strategy for genome editing in plants using CRISPR that does not require expensive equipment.
Methods
Protoplast isolation, transfection, and regeneration
Nicotiana benthamiana and rapid cycling Brassica oleracea (RCBO) plants were propagated in ½-strength Murashige and Skoog (½ MS) medium supplemented with 30 g/L sucrose under a 12-h/12-h light/dark cycle at 25°C. Protoplast isolation was performed according to Lin et al.23 and Hsu et al. 28 except for the digestion solution [½ MS medium supplemented with 30 g/L sucrose, 0.4 M mannitol, 1% cellulose, 0.5% Macerozyme, 1 mg/L 1-naphthaleneacetic acid (NAA) and 0.3 mg/L kinetin] and digestion time (3 days) in N. benthamiana. The protoplasts were co-transfected with RNP and 50 μg synthetic single-stranded oligonucleotide DNA (ssODN; Genomics, Taipei, Taiwan) according to Woo et al.21 Transfected protoplasts were incubated in a 5-cm-diameter Petri dish containing liquid callus medium (½ MS, 0.4 M mannitol, 1 mg/L NAA, 0.3 mg/L kinetin) for 3 weeks. RCBO calli were additionally incubated in 1 mg/L NAA, 1 mg/L 6-benzyladenine (BA), and 0.25 mg/L 2,4-dichlorophenoxyacetic acid for 3 days in the dark. The calli were transferred to liquid shooting medium (containing 2 mg/L BA for N. benthamiana, 0.1 mg/L thidiazuron for RCBO) in a 9-cm-diameter Petri dish and incubated at 25°C for 3–4 weeks in the light (16-h/8-h light/dark, 3000 lux). Green explants larger than 5 mm were incubated in solid shooting medium and subcultured every 4 weeks. Shoot clusters with leaves were then transferred to solidified rooting medium (HB1: 3 g/L Hyponex, 2 g/L tryptone, 20 g/L sucrose, 1 g/L activated charcoal, 2.2 g/L Gelrite, pH 5.2).
Cas9 protein purification, single-guide RNA synthesis, and Cas9 RNP nucleofection
Preparation of Cas9 protein and single-guide RNA (sgRNA) and Cas9 RNP nucleofection were performed according to Huang et al.29 Cas9 recombinant protein was overexpressed in Escherichia coli BL21 harboring the plasmid pMJ915 (Addgene # 69090). Cas9 protein was purified and stored at −80°C in Cas9 RNP buffer (20 mM HEPES at pH 7.5, 150 mM KCl, 10% glycerol, and 1 mM β-mercaptoethanol). The sgRNAs were synthesized by in vitro transcription (IVT) using T7 RNA polymerase (New England Biolabs; M0251L). The DNA oligonucleotides used for IVT template assembly are listed in Table S10. The final sgRNA products were dissolved in Cas9 RNP buffer, quantified using a NanoDrop Lite (Thermo Fischer Scientific), and stored as aliquots at −80°C. Cas9 RNP complexes were assembled immediately before nucleofection by mixing equal volumes of 40 μM Cas9 protein and 88.3 μM sgRNA at a molar ratio of 1:2.2 and incubating at 37°C for 60 min.
Validation of targeted insertions in protoplasts and regenerants
Genomic DNA was extracted from pooled protoplasts and regenerants using a Mini GenoPlus Genomic DNA Extraction Kit (GG2002, Viogene, New Taipei City, Taiwan). To amplify the genomic region targeted by the sgRNA, the corresponding pairs of primers were designed. Primer sequences are shown in Table S10. The PCR conditions were 94°C for 5 min, 35 cycles of 94°C for 30 s, annealing at 55–63°C for 30 s, polymerization at 72°C for 30 s, followed by 72°C for 3 min. The polymerase chain reaction (PCR) products were digested using the appropriate restriction enzyme or RNP and subjected to electrophoresis. The PCR products were cloned into the T&A vector (FYC002-20P; Yeastern Biotech Co. Ltd., New Taipei City, Taiwan). Putative colonies containing the edited DNA were confirmed by sequencing.
Whole-genome sequencing for off-target DD insertion analysis
Leaves of N. benthamiana plants regenerated from protoplasts were collected for genomic DNA purification. Genomic DNA used for genome sequencing was extracted from the samples using a Plant DNA Purification Kit (DP320, Tiangen, http://www.tiangen.com/en/). Paired-end libraries were constructed from the DNA using a NEBNext Ultra DNA Library Prep Kit for Illumina with 2 × 150 bp with an average insert size of ~900 bp and sequenced on the NovaSeq 6000 platform. Three technical replicates were performed for each sample. A total of 120 Gbp reads were obtained per regenerant, with a sequencing depth >30×. High-quality Illumina reads were converted to fasta format files and used as a BLAST database for target sequence searches. The DD sequence (Exp. 1, TTTGCGATGCCTAACAAGCTTCAGGGGGAGTTCAGCCGCTT) was used as the query in a highly sensitive BLASTN search strategy (-dust no -soft_masking false -word_size 4 -gapopen 1 -gapextend 2 -penalty -1 -reward 1 -evalue 10 - perc_identity 90 -num_alignments 10000). The sequences were used as queries to perform BLASTN searches of N. benthamiana wild-type (WT) genome sequences. If the query sequence was identical to the published sequence in the genome, it was considered that the same sequences as the DD already existed and were not caused by TI. If there was a difference from the published WT genome sequence, and the difference was the same as the DD, it was regarded as an off-target TI. The raw reads were deposited in the NCBI SRA database (BioProject: PRJNA667297).
Results and Discussion
The protoplast regeneration protocol (Figure 1) used in the present study was modified from previously published protocols.23, 24, 28 A key to our approach is the observation that the phase of the cell cycle largely governs the choice of pathway used for DNA repair: NHEJ is the major DNA repair pathway during the G1, S, and G2 phases, whereas HDR occurs only during the late S and G2 phases.30, 31 Cell cycle synchronization is an effective strategy for enhancing TI efficiency in human embryonic kidney 293T cells.32 Here, ethynyl deoxyuridine (EdU) staining was used for detection of S-phase cell-cycle progression.33 To increase the number of cells in the late S and G2 phases, N. benthamiana leaves were incubated in solid medium containing ½ MS, 0.4 M mannitol, 1 mg/L NAA, and 0.3 mg/L kinetin (1N0.3K) for 3 days before protoplast isolation (Figure S1A). Comparison with 1N0.3K treatments, no EdU signal was identified in protoplasts incubated in ½ MS, 0.4 M mannitol medium (Figure S1B). Based on single cell analysis23, the TI efficiency increased after incubation in 1N0.3K (Figure S1C). For N. benthamiana, to simplify the procedure, we added 1 mg/L NAA and 0.3 mg/L kinetin to the digestion solution and incubated the material for 3 days. However, this method was not suitable for RCBO because most of the cells were broken after digestion.
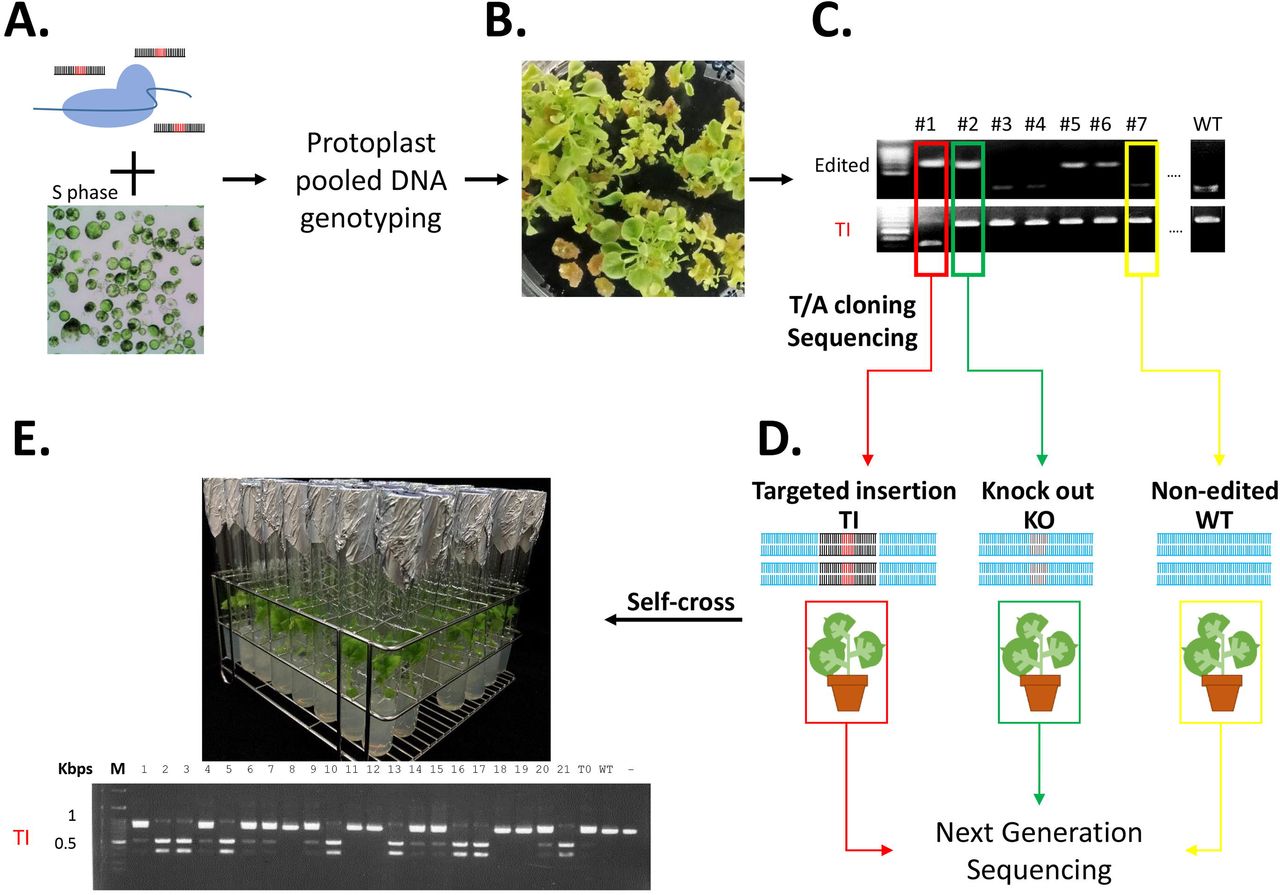
(A) Ribonucleoprotein (RNP) and single-stranded oligonucleotide DNA (ssODN) are delivered to S-phase protoplasts by polyethylene glycol–mediated transfection (Figure S1). (B) Transfected protoplasts are regenerated. (C) DNA from the regenerants is amplified by the polymerase chain reaction and genotyped using restriction enzymes or RNP. (D) DNA is purified from targeted insertion (red), knockout (green), and non-edited (yellow) regenerants and sequenced to determine whether ssODN was inserted into other positions. (E) Offspring are genotyped and sequenced to test whether the inserted sequence is heritable.
To avoid the need for vector construction and to reduce the instability of sgRNA and Cas gene expression, we used an RNP as the CRISPR-Cas reagent. For the DD, we used short non-modified synthetic ssODN, which is relatively inexpensive and easy to obtain. The RNP and ssODN were delivered into protoplasts using PEG-mediated transfection (Figure 1A). After 3 days of incubation in 1N0.3K liquid callus medium, genomic DNA was isolated from the protoplasts and the target gene was amplified by PCR and cloned into the T/A vector for Sanger sequencing to assess the TI efficiency. These protoplasts were cultured and regenerated (Figure 1B).23, 24, 28 The rooted plants were incubated in the growth chamber and genotyped (Figure 1C). These regenerated plants grew normally and produced seeds. DNA from two types of edited regenerants (TI) and knockout (KO) lines and one non-edited (WT) regenerant was purified for genome-wide sequencing to assess the presence or absence of off-target DD insertion (Figure 1D). DNA from the TI T1 progeny was extracted for genotyping to test whether the inserted fragment was heritable (Figure 1E).
To evaluate the effect of the length of the homologous arms and the total length of ssODN on TI efficiency, we conducted experiments using a target site in N. benthamiana PHYTOENE DESATURASE1 (NbPDS1) (Figure S2), which produces a visible phenotype when mutated.34 At the expected sgRNA target position, we inserted a HindIII site, and on the left and right sides, we added 7, 17, or 27 homologous arms to make the DD 20 nt, 40 nt, or 60 nt long (Figure S2A). We genotyped NbPDS1 PCR products from the regenerants. No TI regenerants with the 20-nt ssODN were identified, whereas the 40-nt and 60-nt ssODNs produced TI regeneration efficiencies of 27.3–31.8% without antibiotic or phenotypic selection (Figure S2B). One of the 60-nt TI regenerants (+27#6, Figure S2C, D) contained a precise insertion of a 6-bp HindIII recognition site at the target position. These results indicate that HDR also occurred during protoplast regeneration. We hypothesize that the length of the ssODN donor is an important factor in TI efficiency. In a previous study, a 59-nt ssODN (ssADHE) failed to give rise to successful insertions in 23 T0 rice (Oryza sativa) plants,13 although there was no homologous arm, and a lower DD concentration was used compared to the current study. Based on these results, we selected 40 nt as the length of the ssODN in our subsequent experiments, except for Experiment (Exp.) 2 (44 nt).
Next, we examined the effect of the insertion length in a 40-nt DD on TI efficiency (Figure 2). In Exp. 1, the HindIII site was generated by introducing a 2-nt insertion in the DD (Figure 2A), and TI efficiency in the regenerated plants was 18.2% (Figure 2B, C). In Exp. 2, the protospacer adjacent motif (PAM) position in DD was replaced, 6 nt were added, and the TI efficiency increased to 50.0%. When we increased the insertion length to 15 nt (Exp. 3), we included sites for NheI and BamHI endonucleases in the insertion, which enabled us to confirm the integrity of TI genotyping by restriction enzyme digestion. As the insertion length increased and the length of the homologous arms decreased (11 nt and 14 nt, respectively), the TI efficiency decreased slightly (40.9%). Three regenerants contained only the NheI site, indicating partial DD insertion (Figure 2B). Perhaps the ssODN DD was unstable in protoplasts and had been partially degraded before insertion, which would cause the inserted sequence to be incomplete. The presence of a phosphorothioate-linkage modification in DD stabilizes this double-stranded DNA.13 There was no phosphorothioate-linkage modification in the DD we used in this study, which may have allowed the DD to degrade during TI.

(A) Donor sequences used in Exp. 1–3. Lowercase: insertion or replacement nucleotides. The protospacer adjacent motif (PAM) is underlined. (B) Polymerase chain reaction (PCR) of the target gene in the regenerated plants was performed in each experiment, and different restriction enzymes were used for genotyping. (black: BstNI, Edited; red: HindIII, targeted insertion). M: marker. -: wild-type control. +: restriction enzyme control. Targeted insertion PCR product sequences are shown in Tables S1–S3. (C) Edited and targeted insertion efficiencies of different ssODN donors. T: total length of DD. I: insertion. H: homologous arm. L: left arm. R. right arm. R. E.: restriction enzyme.
We sequenced all of the NbPDS1 genes in TI regenerants produced in Exp. 1, 2, and 3 and found that all regenerants contained insertions generated by NHEJ (Tables S1, S2, and S3). However, in a few TI regenerants, one end of the ODN was joined by HDR whereas the other end was joined by NHEJ (Exp. 1#12 and Exp. 2#18). In these NHEJ TI regenerants, the insertion size was 29–445 bp. These differences were caused by insertion of 1–13 repeats of the ssODN, although some of the repeats were incomplete. In rice, only 20% of TI plants had repeat insertion when modified double-stranded DD was used.13 Both orientations were observed for the TI in our regenerants. Forward and reverse insertions have also been identified in rice.13
The accurate replacement of DNA fragments is vital for gene editing. Co-expressing two sgRNAs can lead to fragment deletion in protoplasts.23 Therefore, two sgRNAs can be designed at both ends of an exon for exon replacement by NHEJ.14 To aid in TI and DNA replacement, we designed two sgRNAs (L1 and L2) based on both sides of the complementary strands of the original target site (E). These sgRNAs could form a combination of RNPs, including tail-to-head (L1+L2), tail-to-tail (L1+E), or head-to-head (E+L2) orientation (Figure 3A). The ssODN DDs were all located in the same strand of target site E (Figure 3B). In L1+L2 and E+L2, the sequence of ssODN contained 17 bp complementary to L2 sgRNA, which caused the L2 RNP to show reduced efficiency for generating DSBs (Figure S3). The notion that the strand of DD complementary to the target site can reduce RNP activity was demonstrated by experiments using L1 and L2 with complementary ssODN. These results are different from those reported for human cell lines.32 In human cells, both strands of DD can be used for gene editing. The L1+E experiment had a higher fragment deletion rate than the others (Figure 3C). Except for a decrease in E+L2, the overall TI efficiency was similar to that using a single RNP (Figure 2C, Exp. 2 and 3). Compared with the use of E RNP only, there was a decline in the TI/Edited ratio when two sgRNA RNPs were co-transfected into protoplasts (Figure 3D). Therefore, we conclude that using dual RNPs does not increase TI efficiency. Based on the insertion sequences, all of the TI regenerants contained the NHEJ insertion (Tables S4, S5, and S6).

(A) Relative positions and directions of the three ribonucleoproteins (RNPs) used. The target site is shown below. The protospacer adjacent motif is underlined. (B) Donor DNA (DD) used in each combination. Lowercase: inserted EcoRI site. (C) Restriction enzyme analysis of the target efficiency of regenerants derived from different RNP combinations and DD transfected protoplasts. TI: targeted insertion. (D) Summary of edited and TI efficiencies of RNP and single-stranded oligonucleotide DNA. R.E.: restriction enzyme.
To determine whether these insertions were heritable, we analyzed the progeny of five N. benthamiana regenerants (Figure S4A). We genotyped TI regenerant T1 seedlings and determined that all TI alleles were inherited (Figure S4B, C). Thus, these protoplast regenerants were not chimeric at the target gene. By contrast, in rice, most T0 plants appeared to be chimeric.13 In the current study, no Cas9 gene was present in the genomes of the regenerants because we used RNP as the CRISPR reagent and therefore no new edited alleles were generated.
We also examined the TI efficiency in RCBO, targeting BoSnRK1 and BoGA4.a35 (Figure 4). RCBO contains two BoSnRK1 genes: BoSnRK1a and BoSnRK1b (Figure 4A). DD was inserted into the target sites (Figure 4B, C, Tables S7 and S8). Sequencing indicated that DD was inserted at an efficiency of 4.5–13.6% (Figure 4C), which is lower than that demonstrated in N. benthamiana.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Target site of BoSnRK1 and the donor sequence. The protospacer adjacent motif is underlined. Lowercase: insertion sequence. (B) Genotyping of the regenerants derived from ribonucleoprotein (RNP) and single-stranded oligonucleotide DNA (ssODN) transfected protoplasts. The edited efficiency by RNP was assessed. Targeted insertion was assessed using the EcoRI site inserted by the ssODN. (C) Summary of edited and targeted insertion efficiencies. R.E.: restriction enzyme site inserted by ssODN. HA: homologous arm. TI: targeted insertion.
Although we identified an off-target DD insertion in BoGA4.b in RCBO, no off-target DD insertion was detected in N. benthamiana. N. benthamiana contains two NbPDS genes, Niben101Scf01283g02002.1 (NbPDS1) and Niben101Scf14708g00023.1 (NbPDS2), and the sgRNA matched PDS1 but not PDS2. The PDS2 genes in the regenerants were analyzed, revealing that no off-target mutagenesis or TI occurred. These results indicate that the off-target DD insertion that occurred in RCBO BoGA4.b was caused by off-target DSBs that provided an opportunity for DD insertion.
Since NHEJ is the main mechanism for TI, we reasoned that the presence of a homologous arm on the DD designed for HDR might not be necessary. To investigate whether ssODN DD with a non-homologous arm could be used for TI, we co-transfected RCBO protoplasts with BoGA4.a sgRNA RNP and NbPDS1 DD. TI was observed in BoGA4.a in only 4.2% of the 24 regenerants (1/24), and none was observed in BoGA4.b, even though the editing efficiency was 95.8% (Figure 4C, Table S9). These results indicate that the presence of a homologous arm is not absolutely necessary for TI, but affects the efficiency of this process.
Finally, to explore the off-target DD insertion that occurred in the presence of DSBs without homologous sequences, we performed whole-genome sequencing of three types of plants, in which the regenerant NbPDS1 gene was (1) the same as the WT, (2) a heterozygous mutant (KO) but without TI, or (3) a bi-allelic TI (TI; Figure 1). The ssODN insertion did not occur in the WT or KO genome. In the genome of the TI regenerant, the DD insertion occurred only in the DSB position created by the CRISPR-Cas RNP. By contrast, in rice that had been transformed using DNA CRISPR-Cas reagent via the biolistic method, quantitative PCR revealed multiple copies (2–10 per plant) of the donor inserts in T1 plants, which suggested that frequent off-target DD insertion occurred in addition to the intended target site insertions.13
Conclusion
In this study, we used protoplast regeneration, RNP, and ssODN to establish a simple and inexpensive DNA TI system that can be used in N. benthamiana and RCBO. A synthesized 40-nt ssODN can be used as DD directly without the need for plasmid construction, and the PEG-mediated protoplast transfection method can provide a large amount of DD for TI. In addition, the plant material can be treated during protoplast isolation to increase the proportion of cells in S phase. In stable transformation systems, the expression of Cas protein is important for knock in,3 but in this study, we used the RNP as the CRISPR reagent to avoid low expression, and TI occurred after CRISPR-Cas-created DSBs had formed. Thus, this insertion method should be applicable to any gene target site in any plant species that can be regenerated from protoplasts. Because the TI efficiency is high, TI regenerants can be obtained without the need for antibiotic selection or phenotypic screening. Similar to other protocols,13, 14, 36 the major mechanism of TI in this study was NHEJ. Although the HDR ratio was low, we still obtained a precise insertion regenerant.
Funding information
This research was supported by Academia Sinica, Innovative Translational Agricultural Research Administrative Office (AS-KPQ-107-ITAR-10; AS-KPQ-108-ITAR-10; AS-KPQ-109-ITAR-10), Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-108-116), and the Ministry of Science and Technology (105-2313-B-001-007-MY3; 108-2313-B-001 -011 -; 109-2313-B-001 -011 -), Taiwan.
Author Disclosure Statement
No competing financial interests exist.
Author contributions
CSL, YCL, SL, MCS, and JS conceived and designed the experiments. CTH, QWC, and YHY performed the CRISPR-Cas9 experiments. SL performed SpCas9 protein purification. CTH, QWC, YHY, and CSL conducted the protoplast regeneration. CTH, QWC, YHY, and FHW performed the molecular biology experiments and targeted mutagenesis analysis. YCL performed whole genome sequencing and bioinformatics analysis. YCL, MCS, JS, and CSL wrote the manuscript with input from all co-authors. All authors read and approved the final manuscript.
Supplementary Material
Figure S1. 5-ethynyl-2’-deoxyuridine (EdU) staining of different materials. (A) N. benthamiana leaves were cut into strips, placed in 1N0.3K (1/2 MS, 0.4 M mannitol, 1 mg/L NAA, 0.3 mg/L kinetin), and incubated for 0, 24, 48, or 72 hours. The incubated leaves and the protoplasts derived from these leaves were stained with EdU. N. benthamiana protoplasts derived from BY2 cells during cell division were used as a positive control. Bar = 50 μm. (B) Protoplasts obtained using different incubation media (1/2 MS, 0.4 M mannitol 1N0.3K) were stained with EdU. Bar = 50 μm. (C) Protoplasts obtained using different incubation media were subjected to targeted insertion (TI) using RNP (target sequence: TTGCGATGCCTAACAAGCCAG) and donor DNA (TGCGATGCCTAACAAGCaagcttCAGGGGAGTTCAGCCGC). NbPDS1 was subjected to PCR and analyzed by digestion with BstNI (for Edited) and HindIII (for TI). M: DNA marker. %: the efficiency of Edited (black) and TI (red).
Figure S2. Effect of homologous arm length and total length of the single-stranded oligonucleotide DNA donor DNA on targeted insertion in NbPDS1 in the N. benthamiana regenerant. (A) Sequences of donor DNA (DD). The protospacer adjacent motif (PAM) is underlined. Lower case: restriction enzyme site. (B) Protoplasts transfected with different lengths of single-stranded oligonucleotide DNA (ssODN). Regenerant genotypes were determined. PCR of the target gene NbPDS1 was performed, and the PCR product was analyzed by digestion with BstNI (for Edited) and HindIII (for TI). (C) Editing and TI efficiency for different lengths of ssODN. T: total length. I: insertion. H: homologous arm. L: left arm. R: right arm. R. E.: restriction enzyme. (D) Results of Sanger sequencing of +27#6, the precise HindIII insertion regenerant (blue background). (E) BLASTN results of +27#6 NbPDS1 (Query) and wild-type NbPDS1 (Sbjct).
Figure S3. Editing efficiencies of regenerants from Exp. 1–3 assessed using restriction enzyme digestion and ribonucleoprotein.
Figure S4. Progeny of targeted insertion regenerants in N. benthamiana. (A) DNA was isolated from 21 progeny seedlings from Exp. 3#7 and the target gene NbPDS1 was amplified by PCR. The products were digested using two restriction enzymes (NheI and BamHI), for which sites were added in the donor single-sranded oligonucleotide DNA (ssODN). (B) Summary of the five targeted insertion (TI) regenerants in the T1 progeny. The genotype was determined as described in (A). (C) Summary of sequences of the TI alleles. Detailed sequence information for each regenerant is shown in Table S3 (Exp.3#7) and Table S5 (L1+E#2, 6, 9, and 10).
Table S1. Exp. 1 sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. L: left homologous arm; I: insertion; R: right homologous arm. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S2. Exp. 2 sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. –number: deletion length. L: left homologous arm; I: insertion; R: right homologous arm. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S3. Exp. 3 sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. –number: deletion length. L: left homologous arm; I: insertion; R: right homologous arm. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S4. L1+L2 sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. –number: deletion length. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S5. L1+E sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S6. E+L2 sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S7. BoSnRK1 sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. L: left homologous arm; I: insertion; R: right homologous arm. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S8. BoGA4.a sequences. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. L: left homologous arm; I: insertion; R: right homologous arm. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S9. BoGA4.a sequences of the RCBO TI regenerants by using N. benthamiana NbPDS1 donor DNA. (A) Summary of the TI regenerants. Underlined: PAM. +number: insertion length. –number: deletion length. L: left homologous arm; I: insertion; R: right homologous arm. + in Orientation column: forward insertion; -: reverse. (B) Insertion sequences of TI regenerants. Underlined: PAM.
Table S10. Primer information.
Acknowledgements
We thank Yu-Jung Cheng for tissue culture. We thank Miranda Loney and Plant Editors for editing. We thank the Academia Sinica Advanced Optics Microscope Core Facility for microscope imaging technical support.
References




