

Abstract
Endolysins are peptidoglycan (PG) hydrolases that function as part of the bacteriophage (phage) lytic system to release progeny phage at the end of a replication cycle. Notably, endolysins alone can produce lysis without phage infection, which offers an attractive alternative to traditional antibiotics. Endolysins from phage that infect Gram-positive bacterial hosts contain at least one enzymatically active domain (EAD) responsible for hydrolysis of PG bonds and a cell wall binding domain (CBD) that binds a cell wall epitope, such as a surface carbohydrate, providing some degree of specificity for the endolysin. Whilst the EADs typically cluster into conserved mechanistic classes with well-defined active sites, relatively little is known about the nature of the CBDs and only a few binding epitopes for CBDs have been elucidated. The major cell wall components of many streptococci are the polysaccharides that contain the polyrhamnose (pRha) backbone modified with species-specific and serotype-specific glycosyl side chains. In this report, using molecular genetics, microscopy, flow cytometry and lytic activity assays, we demonstrate the interaction of PlyCB, the CBD subunit of the streptococcal PlyC endolysin, with the pRha backbone of the cell wall polysaccharides, Group A Carbohydrate (GAC) and serotype c-specific carbohydrate (SCC) expressed by the Group A Streptococcus and Streptococcus mutans, respectively. Molecular dynamics simulations reveal a previously unidentified binding pocket that is regulated by a gatekeeper residue and uncover that a previously reported inactive PlyC mutant is locked into a ‘closed’ conformation. Docking studies with the short GAC backbone oligosaccharides expose potential protein-carbohydrate interactions and are consistent with PlyCB binding to the unmodified pRha or pRha decorated with the GAC side chains.
Introduction
Endolysins are bacteriophage-encoded PG hydrolases that normally function from within the cell to lyse the bacterial host, releasing progeny phage and completing the phage lifecycle [1]. However, the lytic activity of endolysins can be harnessed for antimicrobial use due to their ability to equally lyse bacteria when applied exogenously, without infection by a parental phage. Due to their direct lytic action on target PG, endolysins are not affected by efflux pumps, alterations in metabolism, or other mechanisms of antibiotic resistance, making them ideal candidates for development against multi-drug resistant organisms [2-4]. Notably, at least three endolysins, some of which are active against methicillin-resistant Staphylococcus aureus, are currently being evaluated in human clinical trials for their antimicrobial activity (reviewed in [5]).
Most endolysins, and in particular those from phage that infect Gram-positive bacterial hosts, are comprised of modular domains. An enzymatically active domain (EAD) is generally found in the N-terminal region, while a cell wall-binding domain (CBD) is located in the C-terminal region [6]. As the name implies, the EAD is a catalytic domain that is responsible for cleaving specific bonds in the PG, the nature of which is dependent on the mechanistic class of the EAD. Occasionally, endolysins contain two EADs, although both are not necessarily active. The CBD binds at high affinity [7] to a cell wall-specific epitope and was suggested to dictate genus, species and serovar-specificity of the endolysin. The CBD targets may be surface carbohydrates, wall teichoic acids linked to the Gram-positive bacterial cell wall, or the PG itself [8].
The endolysin now known as PlyC is one of the first described endolysins and remains one of the most studied. In 1934, Alice Evans noted a “nascent lysis” activity derived from streptococcal phage lysates on streptococcal strains that were not sensitive to the phage itself [9]. By 1957, Krause had determined that the phage used by Evans was specific for Group C Streptococci (GCS), but an “enzyme” produced by the phage could lyse Groups A, A-variant, and C Streptococci (GAS, GAVS, and GCS, respectively) [10]. These findings were confirmed by Maxted later the same year and extended to include Group E Streptococci (GES) [11]. In 2001, PlyC, then referred to as the streptococcal C1 lysin, became the first endolysin to be used therapeutically, protecting mice from GAS challenge in a nasopharyngeal model [2]. Subsequent studies revealed that PlyC is a structurally unconventional endolysin, which is not encoded by a single gene as found for all other endolysins described to date. Rather, PlyC is a nine-subunit holoenzyme encoded by two distinct genes, plyCB and plyCA, within a polycistronic operon [12]. Eight PlyCB subunits self-assemble into a ring structure and form the basis of the CBD that binds the streptococcal surface. A single PlyCA subunit contains two distinct EADs separated by an extended α-helical linker region, which interfaces with the N-terminal residues of the PlyCB octamer [13]. The PlyCA EADs consist of a glycosyl hydrolase (GH) domain and a cysteine, histidine-dependent aminohydrolase/peptidase (CHAP) domain. The very high lytic activity of PlyC relative to other endolysins is attributed to synergy/cooperativity between the two EADs.
Although the EADs of PlyCA have been extensively characterized with respect to specificity of PG bonds they cleave, active-site residues, and their synergistic activity, it is unclear how PlyCB recognizes PG of GAS, GCS and GES. Similar to other Gram-positive bacteria, the plasma membrane of streptococci is surrounded by thick cell wall that consists of a complex network of PG with covalently attached polysaccharides. Rebecca Lancefield utilized the unique immunogenicity of the surface carbohydrates in β-hemolytic streptococci to subsequently separate them into serogroups [14]. S. pyogenes is categorized as Group A Streptococcus, whilst S. dysgalactiae subsp equisimilis (SDSE) produce at least two types of carbohydrates and are annotated as Group C and G Streptococci, respectively [15]. Pioneering work by many researchers have revealed that the cell wall polysaccharides of GAS, GCS and S. mutans, consist of a pRha backbone modified with species-specific and serotype-specific glycosyl side chains (Fig. 1A) [16, 17]. In GAS, GCS and S. mutans serotype c, the polysaccharides termed the Lancefield group A carbohydrate (GAC), group C carbohydrate (GCC), and SCC, respectively, have a conserved repeating →3)α-Rha(1→2)α-Rha(1→ di-saccharide backbone [16, 17]. The β -N-acetylglucosamine (GlcNAc) side chains are attached to the 3-position of the α-1,2 linked rhamnose (Rha) in GAC [16, 18, 19]. The GCC side chains have two N-acetyl-galactosamine (GalNAc) residues attached to the same hydroxyl of Rha [16, 18, 20]. SCC carries the α-glucose (Glc) side chains attached to the 2-position of the α-1,3 linked Rha in SCC [17]. Additionally, the side chains of GAC and SCC are decorated in parts with the glycerol phosphate (GroP) moiety [21]. The GAC and SCC biosynthetic pathways are encoded by the 12-gene loci gacABCDEFGHIJKL and sccABCDEFGHMNPQ, respectively. The molecular mechanisms of GAC and SCC biosynthesis have been the focus in a number of recent studies [21-25].

A) Symbolic drawings of the carbohydrate structures of GAS (GAC), polyrhamnose backbone (pRha) in GAVS and M1_dgacI, GCS (GCC), GGS (GGC) and S. mutans (SCC). For simplicity, the reported glycerol phosphate of occasionally present on the GAC and SCC side chains have been omitted. Repeat units are marked with brackets. The pRha backbone with alternating (α 1->2) and (α 1->3) linkages, whilst the α 1->2 Rha being decorated with a β1->3 side chain is a commonality among all PlyC susceptible strains. B) PlyC lysis of streptococcal pathogens. Group A and Group C Streptococci serotypes are susceptible to PlyC-mediated lysis. Group G Streptococci show limited susceptibility and S. mutans is resistant to PlyC lysis. Three SDSE isolates that produce the GAC instead of GGC (SDSE_gac) are susceptible to PlyC treatment.
Remarkably, extracted GAC is known to partially inhibit the lytic actions of PlyC [26]. Furthermore, a GAS mutant carrying the unmodified GAC (lacking the GlcNAc side chains) displayed enhanced susceptibility to PlyC [25], implicating PlyCB in recognition of the pRha backbone. Here, we identify the pRha backbone of GAC as the definite minimal binding epitope for PlyCB. Molecular dynamics simulations and docking studies based on our findings further establish a model of interaction between PlyCB and selective surface carbohydrates in streptococci.
Materials and Methods
Bacterial strains, growth conditions and media
Streptococcus pyogenes strain D471 was propagated on solid media in plates containing Todd-Hewitt broth supplemented with yeast extract (0.2%) (THY) and agar (1.4%) or in liquid THY as described by Gera et al [27]. S. mutans wild type (Xc) and mutants were grown in Todd-Hewitt broth with 1% yeast extract. All cultures were grown without antibiotics and without aeration at 37°C. E. coli genotypes DH5α (NEB, cat. No. C2988J), DH10α (Thermo Fisher, cat No. 18297010), BL21 (NEB, cat No. C2530H) and Origami 2 (Millipore Sigma, cat. No. 71346) were used for routine plasmid propagation or protein expression and grown in Lysogeny Broth (LB) medium supplemented with either 50 µg/ml kanamycin, 100 µg/ml ampicillin or 35 µg/ml chloramphenicol as needed.
Bacterial strains E. coli CS2775 were transformed with pRGP1 plasmid [28], gacABCDEFG [23] to produce pRha or empty plasmid control (pHD0131). The bacterial cells were grown overnight in LB containing erythromycin (150 µg/ml) at 37°C and used next day for whole cell Western blots and FACS and microscopy analysis.
Recombinant expression and purification of PlyCBWT and PlyCBR66E
PlyCBWT (GenBank ID: NC_004814.1:7517-7735) was expressed from a pBAD24 vector and purified from BL21 cells as previously described [12]. In brief, the culture was grown in LB and induced with 0.25% L-arabinose at OD600 ∼1.2-1.4 (Alfa Aesar, cat no. A11921). Cultures were grown at 37°C with shaking at 180 RPM for 3-4 hours, centrifuged at 4500 x g and resuspended in phosphate-buffered saline (PBS). Lysis was performed by a French press (1800 psi). Benzonase (Millipore Sigma, cat. No. 70746-3) was added and the lysate incubated at room temperature with rotation for 20-30 min. The lysate was centrifuged at 20,000 x g for 20 min and the cleared lysate was passed through a 0.45-µm filter and loaded onto a XK-26/20 column (Cytiva) with 30-35 ml ceramic hydroxyapatite (Bio-Rad, cat no. 1582000). PlyCBWT was eluted from the column with three column volumes of 1 M sodium phosphate buffer (pH 7.2). Protein was subsequently dialyzed in PBS, 10% glycerol and stored at −80°C until use. Protein purification of PlyCBR66E was performed as PlyCBWT [13]. The fluorescent labeling of PlyCBWT and PlyCBR66E was performed using the manufacturer’s recommended guidelines (Thermo Fisher, cat. No. A20174).
Purification of GAC
GAS were grown overnight in THY media at 37°C. Cultures were centrifuged at 4,500 x g. Pellets were washed and resuspended in 40 ml distilled water per each original liter of media used and combined in an 800 ml beaker. 22.5 ml 4N sodium nitrite (5 ml per liter of culture) was added to the beaker in addition to 22.5 ml glacial acetic acid (5 ml per liter of culture). An orbital shaker was used to gently mix the beaker for 15 min in a hood. The mixture was centrifuged in 500 ml bottles at 8000 x g for 15 min. The supernatant was decanted to a new beaker and neutralized with 1M sodium hydroxide. The total solution, about 300 ml, was filtered with a 0.45-micron filter assembly. 50-50 ml aliquots were deposited in a 3.5 kDa membrane and dialyzed in a 4-liter beaker overnight with water. The following day, the solution was concentrated using an Amicon 400 ml stirred cell (model 8400) filter assembly with a 76 mm diameter Ultracel® 5 kDa ultrafiltration disc (Millipore Sigma, cat. No. PLCC07610) for 2 hours with 60 psi. The ∼10 ml concentrate was loaded onto an S-100 column for final purification. Fractions were assayed for Rha, lyophilized and stored at 4°C until use.
Calculation of pRha concentration
A modified protocol by Edgar et al. [21] based on a protocol from DuBois et al. [29] was used to determine Rha concentration in purified GAC. Briefly, anthrone reagent was prepared by dissolving 0.2% w/w anthrone in H2SO4. Eighty microliters of aqueous samples or standards containing either GAC or L-Rha at known concentrations were added to a 1.5 ml microfuge tube. To this same tube, 320 µL of the anthrone reagent was added. Samples were boiled at 98°C for 10 min in a heat block. Samples were cooled to room temperature, transferred to a quartz plate, and the absorbance at 580 nm was recorded using a spectrophotometer. Rha concentration was interpolated using an L-Rha standard curve.
Precipitation of PlyCB with GAC
PlyCBWT and PlyCBR66E samples were defrosted from storage at −80°C. Lyophilized GAC was resuspended in PBS. Both proteins and GAC were added to a 3.5 kDa dialysis membrane and dialyzed overnight in PBS. Protein concentrations were determined using a NanoDrop spectrophotometer (Thermo Fisher ND-2000) at 280 nm and were diluted to 5 mg/ml. The GAC was also assayed and diluted with PBS to 1.6 mg/ml. One-hundred microliters of proteins and 100 µl of GAC or PBS were mixed in a 250 µl quartz plate and allowed to incubate without shaking at room temperature. Visible precipitate formed in samples in 5-8 minutes. After recording the precipitate at 340 nm using a Spectramax® M5 (Molecular Devices) spectrophotometer, the total sample volume was transferred to a 1.5 ml microfuge tube. Samples were centrifuged at 14,000 x g to pellet the precipitate and supernatants were transferred to new 1.5 ml microfuge tubes. Two-hundred microliters of 8M urea was added to the pellet. Pellets were resuspended and 5 µl of either pellet or supernatant were added to 40 µl water with 8 µl 6x Laemmli buffer with DTT. Samples were boiled at 98°C for 8 min, and then 12.5 µl were loaded onto a 7.5% SDS-PAGE and run for 32 min at 200V. Proteins were visualized using Coomassie stain.
Lysis assay
A turbidity reduction assay was used to ascertain strain sensitivity to PlyC. This assay was performed as previously described [30], except PlyC was used at 2 µM. Eight technical replicates were performed.
Sensitivity of streptococcal species to PlyC-mediated lysis was analyzed using a wide range of clinical isolates: 1) GAS isolates: M1UK, WT 5448 strain [M1], deltagacI 5448 strain [dgacI]; 2) GCS isolate: stC74A.0; 3) SDSE_gac isolates: stG245.0, stG652.0. stG485.0; GGS: stG6.0, stG485.0 and 4) S. mutans serotype c were used as negative controls. Briefly, all streptococcal strains were grown in THY at 37°C overnight in 5% CO2, except for S. mutans, which was grown in THB media. Next day, the bacterial cells were inoculated in 1:100 fresh media and grown until mid-logarithmic phase (OD600 1.0). The cells were washed in PBS and resuspended to an OD600 of 2.0. In a 96-well plate, to a 100 µl of bacterial cells, 100 µl of PlyC [1 µg/ml] was added and immediately read at an absorbance of OD600. The obtained values were standardized by subtracting from the background values. The data is plotted using GraphPad Prism version 9.
SDS-PAGE and blotting analysis
PlyCBWT binding to recombinant E. coli expressing pRha was conducted using blot analysis. Briefly, the lysate from the overnight cultures was analyzed in 20% tricine gels. SDS-PAGE and protein transfers were performed according to manufactures instructions, Atto Ae-6050 Mini Gel chamber and Novex protein separation from Thermo Fisher, respectively. The PVDF membranes were blocked with 5% non-fat dry milk with Tris-Buffered Saline, 0.1% Tween® 20 detergent prior to incubation with PlyCBWT labelled with Alexa Fluor® 647 (1:1000) for one hour at room temperature. Goat anti-rabbit GAC antibodies conjugated with IRDye® 800CW were used as a positive control (abcam ab216773). The resulting blots were imaged using the Licor Odyssey FC Imaging System. All the blots were processed in parallel under the same conditions.
Microscopy
Microscopic analysis of E. coli bacteria was performed using cells that were grown overnight in LB supplemented with antibiotics at 37°C and diluted 1:100 the next day and regrown until OD600 reached 0.5. The cells were washed twice with PBS for 5 min at 10,000 rpm and stained with 1:1000 dilution of PlyCBAF647 and left for 20 minutes on ice in the darkness. Prior to fixing the cells with 4% paraformaldehyde, the cells were washed again with PBS. The fixed cells were mounted on 1% agarose coated microscopic slides and viewed under the CY5 channel on a fluorescent Deltavision widefield microscope.
FACS analysis
E. coli cells were grown overnight as described above and diluted 1:100 the next day, grown at 37°C and used for the assay at OD600 = 0.5. The cells were washed with PBS and probed with PlyCBAF647 or PlyCBR66EAF645 at 1:1000 dilution at 1:1000 dilution. Anti-GAC antibodies conjugated with FITC (ABIN238144, antibodies-online, titer 1:50) were used as a positive control. The samples were incubated for 20 minutes on ice at dark conditions. The cells were washed twice with PBS at 14,000 rpm for 5 minutes and fixed with 4% paraformaldehyde. BD LSRFortessa Flow Cytometry software was used to analyse the samples and the data interpretation was conducted with FlowJo™ software v10.6.2.
PlyC hydrolysis of sacculi
S. mutans wild-type (WT) and the mutant strain sacculi were obtained by the SDS-boiling procedure [25] followed by four washes each with 1 M NaCl and distilled water. The sacculi were resuspended in PBS to OD600 of 1.0 and incubated with PlyC (5 µg/ml) in a 96-well plate. The lysis was monitored after 10, 20, 30, 40, 50 and 60 min as a decrease in OD600. Results were reported as fold change in OD600 of the sacculi incubated with PlyC vs the sacculi incubated without PlyC.
Molecular Dynamics
Monomers of PlyCBWT (PDB 4F87) and the PlyCBR66E mutant (PDB 4ZRZ) were simulated by Molecular Dynamics (MD). The systems for MD simulations were prepared with the utility LEaP, which is integrated in the suite of programs AMBER 16 [31]. The ff14SB force field [32] was used. The N- and C-termini of the proteins were capped with an acetyl (ACE) and methylated amino group (NME), respectively. Each simulated system was immersed in a water box (TIP3P water model) and neutralized by adding the appropriate number of counterions. This was followed by steepest-descent energy minimization to remove steric clashes. MD simulations were performed using the pmemd.cuda module of AMBER 16. The cut-off distance for the non-bonded interactions was set to 10Å. The periodic boundary conditions were used. Electrostatic interactions were treated using the smooth particle mesh Ewald method [33]. The SHAKE algorithm was applied to all bonds involving hydrogen atoms, and a time step of 2 fs was used throughout [34]. Each energy minimized system was heated to 300K, equilibrated for 10 ns, and further simulated for 2 µs without any restraints. Protein structures and MD trajectories were visually inspected and analyzed using the molecular visualization programs PyMOL [35] and VMD [36].
Binding cavity identification and druggability assessment
The site recognition software SiteMap, implemented in the Schrodinger suite of programs [37], was used to investigate the cavities on the PlyCB wild type (PDB 4F87) and R66E mutant (PDB 4ZRZ) proteins surface, in terms of physicochemical properties (hydrophobic/hydrophilic nature), volume, exposure, and enclosure. Based on those properties, an overall “SiteScore” was generated providing an estimate value of the druggability of the pocket. Using the default settings, scores of >= 0.8 define the limit between drug-binding and non-drug-binding cavities.
Molecular docking
A low energy 3D conformation of Rha di- and tri-saccharides was generated using LigPrep in the Schrödinger platform [37]. The binding of the ligands in the Y28 gated cavity identified by MD in the wild-type structure was evaluated by molecular docking. The program Glide [37], part of the Schrodinger platform, was used in extra-precision mode with post-docking minimization. No distance or hydrogen-bond constraints were applied. A 20Å cubic box centered on the centroid of the residues Leu9, Phe10, His11, Thr12, Ser17, Tyr20 and Ile34 was used to generate the docking grid. The default settings were modified to increase the conformational sampling of rings by increasing the energy window to 6.0 kcal/mol.
Results & Discussion
Pathogenic streptococci producing the GAC are susceptible to PlyC
A major component of the GAS cell wall is the GAC, building approximately 50% of the cell wall by weight [38]. The GAC is universally conserved amongst all isolated GAS strains on the basis of the gene cluster sequence conservation [39]. A number of S. dysgalactiae subsp. equisimilis (SDSE) isolates, naturally belonging to Group G Streptococci (GGS), have been reported to have undergone homologous recombination and replaced their Group G Carbohydrate (GGC) gene cluster in parts with the GAC gene cluster [40-42]. We therefore expanded the previously reported PlyC streptococci cell lysis assay used by Nelson et al. [2] to investigate those new isolates named SDSE_gac. We also tested if PlyC was able to lyse a selection of GAS serotypes including a newly emerged isolate M1UK [43], and included negative controls GGS isolates and S. mutans serotype C (Fig. 1B). In agreement with the published literature, all tested GAS serotypes are susceptible to PlyC lysis and the two GGS isolates are resistant to PlyC. The GGC does not contain the GAC, GCC and SCC pRha backbone with →3)α-Rha(1→2)α-Rha(1→ di-saccharide repeats, but an alternating Rha-GalNAc carbohydrate [44] (Fig. 1A). All three SDSE_gac isolates tested have inherited parts of the gac gene cluster and produce the GAC instead of the GGC [41]. Strikingly, the SDSE_gac strains are all sensitive to PlyC treatment. The fact that PlyC is able to lyse SDSE_gac strains that express the GAC, but PlyC does not lyse the isogenic GGS strains producing the GGC strongly suggests that the GAC is a critical component of PlyC recognition and subsequent activity.
Importantly, all strains tested in this study that are susceptible to PlyC lysis produce a cell wall polysaccharide that contains the pRha backbone and a β-linked sugar substituent on the α1,2-linked Rha (Fig 1A). We therefore suggest that the pRha backbone with and without a side chain are both vital ligands to assist PlyC activity and the new SDSE_gac isolates will also be susceptible to PlyC treatment due to production of the GAC.
Purified GAC precipitates PlyCB -but not PlyCBR66E
The lysis assay of GAS cells, and in particular of the SDSE_gac variants, suggests that either the ubiquitous pRha or GAC in GAS cells is the ligand for PlyCB. We propose that the PlyCB octameric CBD binds GAC and/or the GAC pRha backbone. We tested this hypothesis by investigating the binding of PlyC to partially purified GAC. We hypothesized if the GAC was able to precipitate PlyCB, an interaction of the two systems must have occurred [45]. As a negative control, we employed the previously published inactive mutant PlyCBR66E, which lost the ability to bind to GAS cells [13]. The purified proteins were incubated with the extracted GAC, and precipitation was monitored at 340 nm, a standard wavelength for measuring protein aggregation [46, 47] (Fig. 2A, B). Whilst keeping the PlyCB concentration constant, we varied the concentration of GAC. Within five minutes at room temperature the solution became turbid, suggesting aggregation (Fig. 2A). When the PlyCB concentration was kept constant and the GAC concentration was varied, the turbidity correlated with PlyCB concentration in a dose dependent manner, suggesting that PlyCB requires GAC to aggregate. Importantly, PlyCB did not self-aggregate when no GAC was added in the assay. Furthermore, no aggregation was detected when PlyCBR66E was incubated with purified GAC (Fig. 2B). To demonstrate the presence of PlyCB in the precipitates, we analyzed the soluble and pellet fractions (Fig. 2C, D). A higher yield of aggregated PlyCB was found in the pelleted samples when compared to the soluble fraction (Fig. 2D). A similar precipitation effect was observed when we varied the PlyCB concentration and kept the GAC concentration constant (Fig. 2E, F), demonstrating that both species are necessary for an interaction.

Precipitation studies of purified PlyCB and GAC reveal direct interaction of PlyCB with GAC. A) The PlyCB concentration is kept constant whilst the GAC concentration is varied. Visible precipitate forms at the higher concentrations. B) The precipitate level is measured spectrophotometrically at 340 nm and compared to the mutant PlyCBR66E, which does not bind the GAC. C) Coomassie stained and D) densitometry analysis of PlyCB protein from the supernatant fraction and aggregates (pellets). E, F) The same dose dependency is observed when the PlyCB concentration is varied. Arrowhead depicts PlyCB protein at 8 kDa.
When the L-Rha monosaccharide was mixed with PlyCB, no precipitation was observed, suggesting that the GAC, or a moiety within the GAC, is a specific ligand required for PlyCB precipitation.
PlyCB binds to recombinantly produced pRha backbone
The purified GAC from bacteria contains a mixture of carbohydrates, including the fully decorated GAC with GroP[21] and a small proportion of the polysaccharide lacking the side chains [48]. PlyC is able to lyse a number of GAS mutants including GAVS and dgacI_M1 [2, 11, 25, 49], that decorate the cell wall with the unmodified GAC lacking the side chains (Fig. 1A, B), suggesting that the pRha backbone of the GAC is the minimal carbohydrate structure required for PlyCB binding. To test this hypothesis, we recombinantly produced the pRha backbone in E. coli cells. We and others have previously reported that the S. mutans sccABCDEFG gene cluster, when transformed into E. coli cells, functionally produces the pRha backbone attached to the lipid A [23, 28]. Additionally, to understand if PlyCB recognizes a specific pRha backbone, we engineered E. coli cells expressing the GAC gene cluster gacABCDEFG required for the GAC pRha production. E. coli cells carrying an empty plasmid were used as a negative control.
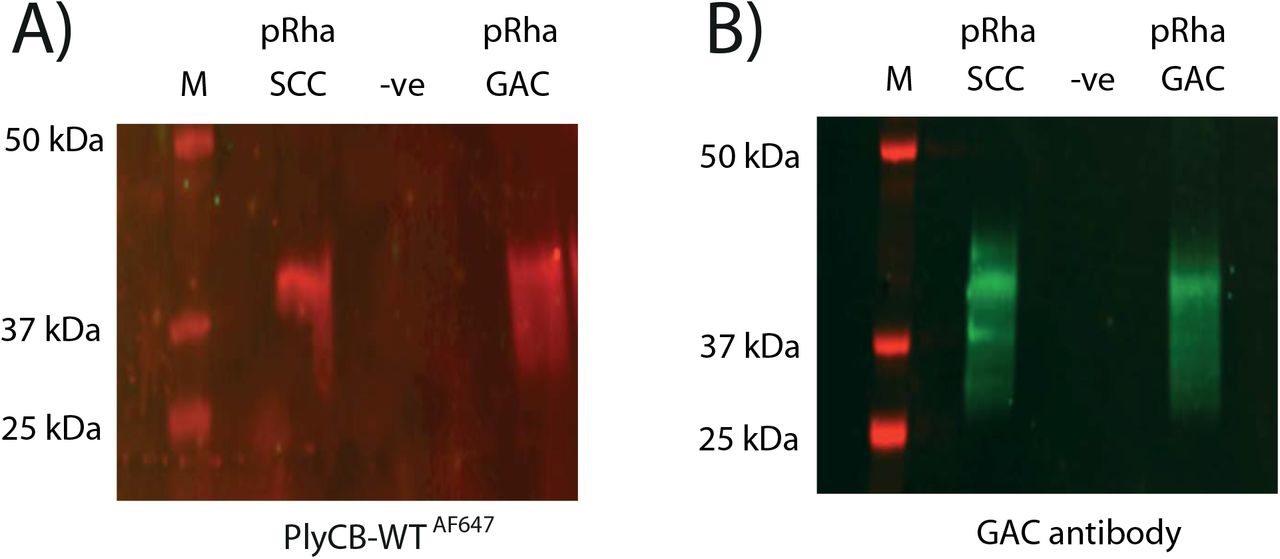
Next, we investigated the binding of PlyCB conjugated with Alexa Fluor® 647 (PlyCBAF647) to an E. coli total cell lysate expressing the pRha backbone of the SCC or GAC, respectively (Fig. 3A). The blotted membranes were incubated with PlyCBAF647, and a positive interaction between PlyCBAF647 and the E. coli produced pRha is visualized as a prominent band around 40 kDa. The size of the band agrees with the band detected by anti-GAC antibodies that were previously reported to recognize pRha [23] (Fig. 3B). Importantly, PlyCBAF647 and GAC antibodies do not interact with the cell lysate of E. coli expressing an empty plasmid (Fig. 3 A, B). We further confirmed the ability of PlyCB to bind to E. coli cells decorated with the pRha by fluorescent microscopy (S. F1). Only cells that produce the pRha are detected by the PlyCBAF647, in agreement with the results of the blot analysis.

Representation of immunoblot analysis of the cell lysate of E. coli expressing the SCC and GAC pRha and carrying an empty control plasmid (-ve). A) Blot was incubated with PlyCB-Alexa Fluor® 647 (PlyCBWTAF647). B) Probing the same samples with the GAC antibodies confirms the presence of GAC in the bands. Molecular mass markers are given in kDa.
To gather additional evidence that PlyCB interacts with the pRha backbone, we established a flow cytometry assay to analyse the binding of PlyCBAF647 to pRha-producing E. coli. Unstained E. coli cells that express pRha or carry an empty plasmid sort in the identical range (Fig. 4A). The GAC antibodies label exclusively the cells producing pRha (Fig. 4A). A similar pattern of the GAC antibodies binding is observed when the cells were incubated with PlyCBAF647 (Fig. 4B). Contrary, the PlyCBR66EAF645 mutant protein does not bind to E. coli cells, and PlyCBAF647 does not interact with the cells expressing an empty vector (Fig. 4B). Taken together, these data provide the first definitive evidence that the pRha backbone of GAC and SCC is a binding receptor of the PlyCB octameric subunit.

PlyCB binding to E. coli cells were investigated by flow cytometry after labelling with PlyCBWT AF647 and PlyCBR66EAF645 mutant proteins. Blue: -ve control cells without pRha. Red: pRha producing E. coli cells. Representative histograms are shown. A) Left panel: unstained cells. Right panel: The anti-GAC antibodies (GAC-FITC) were used as a positive control to label the E. coli cells producing pRha. The antibodies do not bind to the E. coli cells carrying an empty plasmid (-ve). B) Left panel: PlyCBWTAF647 binds to the E. coli cells producing pRha, but not to the E. coli cells carrying an empty plasmid (-ve). Right panel: PlyCBR66EAF645 does not binds to the E. coli cells producing pRha.
PlyC lyses engineered S. mutans producing the GAC
Despite the fact that the SCC pRha backbone is identical to the GAC, S. mutans is resistant to PlyC lysis (Fig. 1A). To get a better understanding why S. mutans is resistant to PlyC, we compared PlyC-induced lysis of the sacculi purified from S. mutans WT and a number of mutant strains producing different SCC variants (Fig. 5). First, we examined the ΔsccH mutant producing the GroP-deficient SCC [21]. Similar to S. mutans WT, ΔsccH was resistant to PlyC-mediated lysis (Fig. 5). Second, we tested the sccN deletion mutant, ΔsccN, that is deficient in the enzyme required for generation of the Glc side chains [50]. A time dependent lysis is observed for ΔsccN. Expression of the WT copy of sccN in ΔsccN (the ΔsccN:psccN strain) fully restored the resistance of the bacteria to PlyC (Fig. 5). These observations clearly suggest that PlyC is able to bind to the S. mutans cells producing the unmodified pRha backbone, and the Glc side chains in SCC hinders PlyC binding. We then investigated whether the addition of the GAC GlcNAc side chains to the pRha backbone affects sensitivity of the engineered S. mutans sacculi to PlyC-induced lysis. We expressed the GAS genes gacHIJKL required for the formation and addition of the GlcNAc side chains and GroP to GAC [21], in the ΔsccN background strain in two versions, creating the ΔsccN:pgacHI*JKL and ΔsccN:pgacHIJKL strains [50]. The plasmid pgacHI*JKL contains an inserted stop codon in the gacI gene required for generation of the GlcNAc side chain, and, therefore, the ΔsccN:pgacHI*JKL strain produces the unmodified SCC lacking any side chains (Fig. 5). As expected, the sacculi isolated from this strain remains susceptible to PlyC lysis. We previously showed that in ΔsccN:pgacHIJKL, the Glc side chains are replaced with the GlcNAc side chains [50]. Interestingly, expression of gacHIJKL in ΔsccN did not restore the resistance of the bacteria to PlyC (Fig. 5), indicating that the GlcNAc side chains do not obstruct PlyC binding. Lastly, we analyzed PlyC-mediated lysis of sacculi purified from the ΔrgpG mutant, which is deficient in SCC expression [50]. The RgpG protein catalyzes the first step in SCC biosynthesis [51]. In comparison to ΔsccN, PlyC-induced lysis of ΔrgpG was less pronounced (Fig. 5), indicating the importance of the pRha backbone of SCC in PlyC activity and supporting the findings that the pRha backbone is a ligand contributing to PlyC binding. These studies reveal that if the SCC is ‘unmasked’ i.e., stripped of the Glc and Glc-GroP side chains, it becomes a ligand for PlyCB and that S. mutans is PlyC susceptible if SCC is replaced with GAC.

The PlyC-mediated lysis of sacculi purified from S. mutans strains. The lysis was monitored after 10, 20, 30, 40, 50 and 60 min as a decrease in OD600. Results are presented as a fold change in OD600 of the sacculi incubated with PlyC vs. the sacculi incubated without PlyC. Data points and error bars represent mean values of four biological replicates and standard deviation, respectively. P-values were determined by two-way ANOVA with Dunnett’s multiple comparisons test.
Molecular Dynamics simulations identify novel binding pocket in PlyC
Our biochemical investigations revealed that PlyCB interacts directly with the pRha backbone of the GAC as shown in the cellular context of the native streptococcal cells, engineered E. coli cells and S. mutans mutant strains. In contrast, traditional biochemical techniques such as size exclusion chromatography or isothermal titration calorimetry could not detect the binding of PlyCB when either the short or long pRha fragments were used (data not shown). These results corroborate the previous studies on related proteins, showing that the binding between endolysins to their respective isolated or purified cell wall ligands is challenging to characterize [52, 53]. However, the CBD is a carbohydrate binding module, which often contains the prominent binding sites to selectively coordinate the binding of linear polysaccharides (reviewed in [54]). To identify the prominent binding sites for pRha, we investigated the previously reported crystal structures of PlyCBWT and PlyCBR66E with the ligand site identification software SiteMap [55]. The software identifies cavities on the molecular surface of a protein structure and provides a ‘druggability score’, which indicates the likelihood that an identified site can bind a small molecule. Interestingly, for both PlyCBWT and PlyCBR66E protein structures, no druggable sites were identified (SF2). In the PlyCBWT structure, a small shallow cavity in proximity of R66 was identified, but with a very poor druggability score (0.4). Shen et al., had previously reported a putative phosphate binding site in PlyCB [56], however, we concluded from our studies that no pocket or cavity is large enough to accommodate the pRha structure. Protein crystal structures represent a fixed conformation state that does not have to reflect the native fold in solution or in the context of the respective ligands. Riley et al. reported previously a PlyCB SAXS in solution structure [57] where a high degree of flexibility and movement of the PlyCB octameric ring was revealed. We therefore conducted Molecular Dynamics (MD) simulations on the PlyCBWT structure (PDB 4F87) [13] to investigate how the protein structure changes over time. The structure was simulated as a monomeric protein for 500 ns in the explicit solvent mode. Strikingly, the PlyCBWT structure changed into an ‘open’ conformation, where Y28 rotates and remains stable in a new conformation, generating a novel pocket (SF3). In the WT crystal structure, R29 establishes a salt bridge with E36 in the same β-hairpin and has a repulsive interaction with R66 on the α-helix. This repulsive effect results in a higher mobility of the terminal part of the β-strand which allows the stabilisation of an open conformation of Y28 (SF3). The stabilisation of the open conformation of Y28 opens a cavity that is clearly visible on the molecular surface of the WT structure and is identified by SiteMap as a druggable binding site (score 0.8) (SF2).
It is well reported in the literature that a PlyC holoenzyme formed by PlyCA and the PlyCBR66E mutant protein is not able to lyse GAS [56]. Our biochemical studies have revealed that the PlyCBR66E mutant does not bind pRha. We therefore conducted MD simulations also on the reported PlyCBR66E crystal structure. In agreement with the PlyCBWT protein simulations, the structure was stable. Contrary to the PlyCBWT protein, the rotation of Y28 was not observed and subsequently, no cavity/pocket is formed (SF4). We inspected the conformation of the mutated residues in PlyCBR66E. The side chain of R66E forms a stable salt bridge with R29, increasing the structural rigidity and preventing the opening of Y28 (SF4). Interestingly, the site controlled by Y28 is not at the protein-protein interface, but within the monomeric structure, suggesting that a ligand binding pocket exists within the monomeric protein and is not at the interface of two neighboring monomers (SF2 1B, Fig. 6).
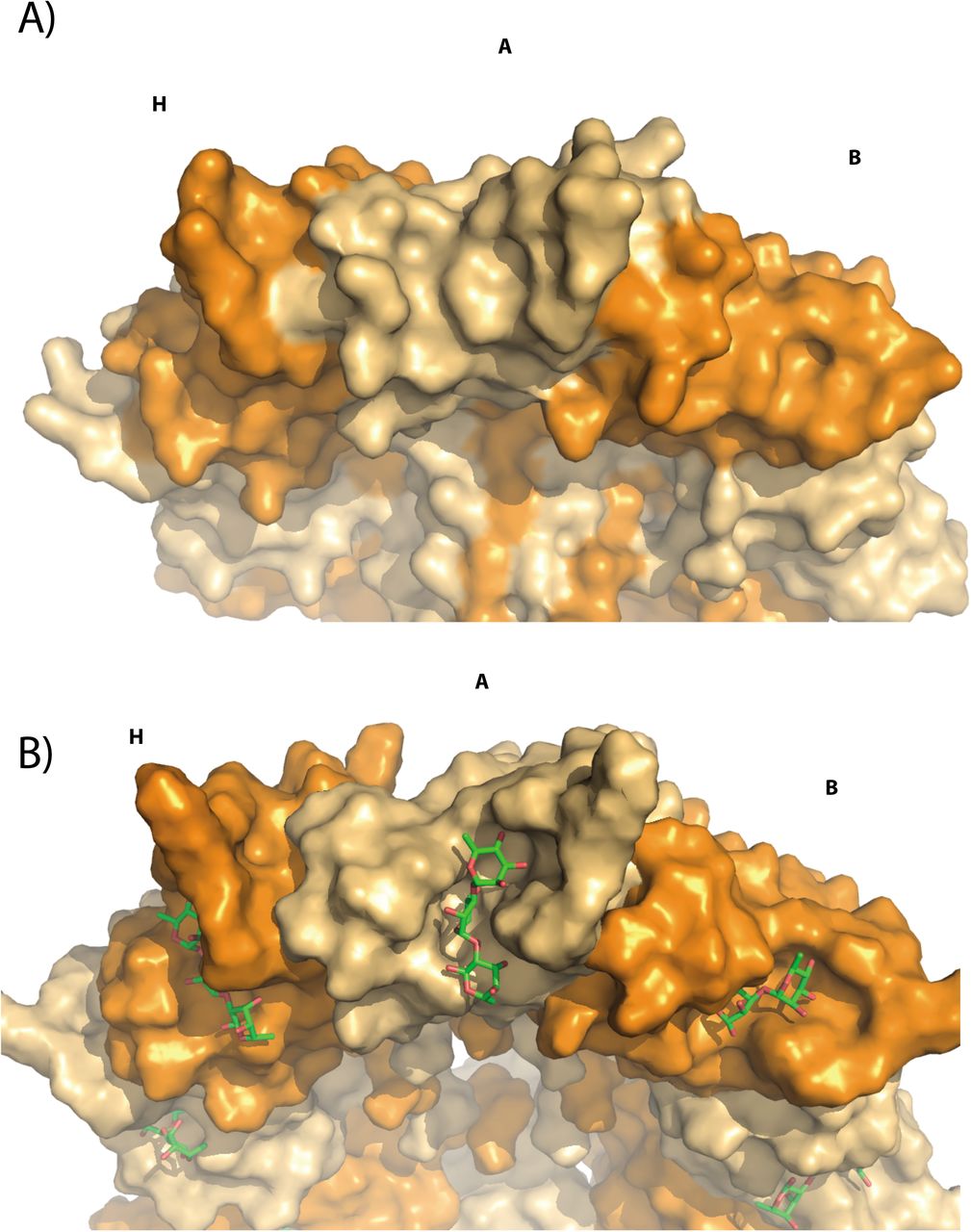
Bottom view of the PlyCB protein structures in surface representation. The octameric PlyCB structure with brown A, C, E, G and orange B, D, F, H monomers. A) WT (GH and CHAP domain omitted, 4f87.pdb) B) WT after MD with docked tri-Rha. The MD simulations have revealed a novel pocket that could accommodate the binding of short Rha-polysaccharides.
PlyCB-rhamnose di- and tri-saccharide docking reveals potential interactions
The MD simulations revealed a potential ligand binding site in PlyCBWT that is generated after the gatekeeper Y28 rotated towards the α-helix. We conducted ligand-protein docking using a Rha di- and tri-saccharide, with both possible linkage configurations. The Rha disaccharide and trisaccharide with a non-reducing end α1,2-Rha (SF5) produced the congruent docking results, which we further analyzed (Fig. 6, SF6). The Rha disaccharide with the non-reducing end (1->3) linkage appears to be stabilized by four hydrogen bond interactions (SF7 A, B). The terminal reducing end Rha is within the hydrogen bond distance to S17 and T12, whilst the α1,3-linked non-reducing end Rha is able to interact with the carbonyl backbone of F10. The identical interactions are observed for the docked trisaccharide (SF7 C, D). In addition, the non-reducing end Rha of the trisaccharide also interacts with the side chain hydroxyl groups of S54 and Q49 (SF7 C, D). When we carefully inspected the structural context of the Rha trisaccharide with the terminal α1,2-linked Rha moieties, we detected the adjacent pockets that could potentially bind the GlcNAc side chain linked to these Rha moieties (SF8 A, B). It is beyond the scope of this study to confirm that these pockets have the ability to bind the GAC β-GlcNAc sidechain or in fact the Group C Carbohydrate (GCC) β-GalNAc-GalNAc disaccharide. However, this model is consistent with PlyCB binding the unmodified pRha backbone and the backbone decorated with the GAC or GCC side chains. The model also explains why the S. mutans SCC — with the α-2-Glc substitution on the α1,3-linked Rha — is not a ligand for the PlyCB protein. The identified pocket does not allow the binding of the SCC with the side chain in this configuration (SF8 A, C).
Polyrhamnose binding pocket suggests the PlyC catalytic mechanism
We further investigated the structural context of this binding pocket, which is perpendicular to the PlyCB monomer-monomer interface. We superposed the reported PlyCA structure that also contains the GlyH and CHAP amidase domains (Fig 7). This led to the intriguing discovery that this model would not only explain the specificity of the enzyme, but also the substrate recognition by the CHAP amidase and GlyH domains. The proposed pRha binding pocket is perpendicular to the GlyH and CHAP amidase domains, that bind the PG backbone and crosslinked peptides. Similar to wall teichoic acids[58], the GAC and GCC are likely directly linked via their reducing end sugars to N-acetylmuramic acid (MurNAc) of PG. We observed that the reducing end sugar of the docked complex points towards the GlyH and CHAP amidase domains. When we compare this structural alignment with the architectural arrangement of the crosslinked PG decorated with the GAC, the model greatly assists our understanding of PlyCAactivity: the CHAP amidase domain cleaves the PG peptide bond. This allows the PG to open up and slide into the GlyH domain, which removes the PG GlcNAc-MurNAc from the reducing end. Concurrently, the GAC attached to the PG binds to the octameric PlyCB domains. We docked a PG tetrasacharide into the reported PlyCA GlyH domain (Fig. 7). This positions the PG in 5-7Å, the distance that the terminal GAC sugar, GlcNAc, could occupy [23].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A schematic model of the proposed PlyC binding to the streptococcal cell wall. A) PlyC in the complex with docked PG tetra-saccharide in the GlyH domain (red) and pRha tri-saccharides in PlyCB subunits. B) A schematic model showing PlyC binding to peptidoglycan and pRha. Sugars are drawn to scale, with green triangles (Rha), blue square (GlcNAc) and beige square (MurNAc). Proposed PlyC activity: The EAD cleaves the PG peptide to open up the PG backbones that subsequently enter the GlyH domain (red) for PG cleavage.
Concluding remarks
Strikingly, our PlyC molecular dynamics simulations revealed a previously unknown conformational change that exposes a potential carbohydrate binding site. Our simulations agree with the reported flexibility of the PlyCB structure in solution, as determined by SAXS studies [57]. The reported crystal structure represents a locked state that appears to be the most stable configuration in the crystallization conditions, in absence of any ligand. The side chain of Y28 acts as gatekeeper and conformational changes allow opening of a cavity that is likely capable of recognizing the compounds in the size of several sugar residues. These structural changes are possible due to an intrinsic flexibility introduced by the repulsion between the side chains of R29 (adjacent to Y28) and R66 (SF3). Moreover, we propose a structural feature of the PlyCB protein that explains why only certain streptococci are susceptible to PlyC’s lytic activity. The pRha decorated with an α–linked side chain sugar is not compatible with the PlyCB ligand binding site and therefore only those streptococci expressing pRha decorated with β-linked substituents, such as GlcNAc and GlcNAc-GroP are susceptible. This could potentially be exploited for diagnostic purposes or in the case of SCC and Group G Streptococci, opens up the potential for novel therapeutic approaches. If SCC was treated by PlyC in combination with an additional enzyme that removes the α–linked side chains in these streptococcal carbohydrates, this would expose the pRha backbone and subsequently make these strains susceptible. The identification of the pRha ligand site could also be further exploited by directed evolution approaches to generate PlyCB protein variants that are capable to bind the carbohydrates from, for example, SCC and Group G Streptococci. Since our proposed ligand binding site is on the surface of each PlyCB monomer mutagenesis would most likely not impact the multimerization interface of the PlyCB octameric ring.
Whilst much has been learned about the structure and function of PlyC in the past 20 years, many questions remain, specifically with respect to its interaction with the PG. Considering the average length of the cellular pRha is 7-10 kDa [25] and that our MD simulations shows an individual PlyCB monomer can bind its target ligand, the α1,2-1,3-pRha with or without β-configured GlcNAc/GalNAc-side chains, it is inviting to speculate that an element of avidity is responsible for tight binding of the PlyCB octamer to the streptococcal surface. We have envisioned a model in Fig. 7, although further proof is needed to substantiate this hypothesis. Another question lies in the actions of the EADs relative to the PlyCB octamer. PlyC clearly has a high turnover as demonstrated in multiple biochemical assays. However, it is unknown if PlyCB “docks” to the surface and the flexibility of the holoenzyme allows the PlyC EADs to cleave multiple bonds in a localized area weakening the overall superstructure of the PG. Alternatively, the enzymatic turnover could be dictated by a balance of on and off rates of the EADs and CBD monomers leading to widespread hydrolysis of the PG. Lastly, it is unknown whether PlyC binds, cleaves, and releases the PG at random points on the streptococcal surface or works its way down a single strand of PG in a processive manner. It is noteworthy that cellulase enzymes, which cleave the β1,4 glycosidic linkages in cellulose, possess a catalytic domain, a flexible linker, and a cellulose binding domain, analogous to the traditional endolysins. It has been demonstrated that energy is stored in the flexible linker can adopt compact and extended configurations that allows the cellulase to move in a “caterpillar-like” motion down a chain of cellulose [59, 60]. Although PlyC does not contain an equivalent flexible linker, the octameric nature of PlyCB invites the possibility that it may interact with the successive pRha strands allowing PlyC to depolymerize the PG in a zipper-like fashion.
In conclusion, the α1,2-1,3-pRha is the definite, minimal carbohydrate substrate for the PlyCB subunit. We validated this by comprehensive experiments, using pRha recombinantly produced in E. coli, and the S. mutans variants of the Rha-based polysaccharides. Furthermore, the MD simulations and subsequent docking studies revealed a potential binding site for Rha tri-saccharides. Further studies are needed to detail the dynamics of the PlyC holoenzyme on the streptococcal surface and the interdomain interactions. Nonetheless, the work described here provides valuable insight into the molecular interactions that define a PlyC’s host specificity, which can inform the future studies as well as engineering approaches.
Data Availability Statement
No mandated datasets are associated with the paper.
FUNDING
The HCD laboratory is supported by Wellcome and Royal Society Grant 109357/Z/15/Z and the University of Dundee Wellcome Trust Funds 105606/Z/14/Z and Tenovus Scotland Large Research Grant [T17/17]. HK is supported by the National Institute of Standards and Technology (NIST). VAF is supported by Rockefeller University laboratory funds. NK is supported by NIH grants R01 DE028916 from the NIDCR and R01 AI143690 from the NIAID.
ACKNOWLEDGEMENTS
We thank the CDC Streptococcus Lab and the Active Bacterial Core surveillance program for sharing isolates isolate 20170556 (stG6.0), 20171682 (stC74A.0), 20154376 (stG245.0), 20173686 (stG652.0), 20170560 and 20176966 (stG485.0). We also thank Ryan Heselpoth, Yang Shen, and Sara Linden for reagents, technical advice, and helpful discussion.
Abbreviations (alphabetical order)
- (CBD)
- Cell wall-binding domain
- (EAD)
- enzymatically active domain
- (Glc)
- Glucose
- (GroP)
- glycerol phosphate
- (GlyH)
- Glycosyl Hydrolase
- (GAC)
- Group A Carbohydrate
- (GAS)
- Group A Streptococcus
- (GAVS)
- Group A-variant Streptococcus
- (GCC)
- Group C Carbohydrate
- (GCS)
- Group C Streptococcus
- (GGC)
- Group G Carbohydrate
- (GlcNAc)
- N-acetyl-glucosamine
- (GalNAc)
- N-acetyl-galactosamine
- (MurNAc)
- N-acetyl-muramic acid
- (PG)
- Peptidoglycan
- (pRha)
- polyrhamnose
- (Rha)
- Rhamnose
- (SDSE)
- S. dysgalactiae subsp. equisimilis
- (SCC)
- S. mutans serotype c carbohydrate
References




