

abstract
Collagen fibrils are the principal supporting elements in vertebrate tissues. They account for 25% of total protein mass, exhibit a broad range of size and organisation depending on tissue and stage of development, and can be under circadian clock control. Here we show that the remarkable dynamic pleomorphism of collagen fibrils is underpinned by a mechanism that distinguishes between collagen secretion and initiation of fibril assembly, at the plasma membrane. Collagen fibrillogenesis occurring at the plasma membrane requires vacuolar protein sorting (VPS) 33b (which is under circadian clock control), collagen-binding integrin-α11 subunit, and is reduced when endocytosis is inhibited. Fibroblasts lacking VPS33b secrete soluble collagen without assembling fibrils, whereas constitutive over-expression of VPS33b increases fibril number with loss of fibril rhythmicity. In conclusion, our study has identified the mechanism that switches secretion of collagen (without forming new fibrils) to new collagen fibril assembly, at the plasma membrane.
A primary function of vertebrate cells is to synthesise the matrisome, which is an ensemble of 1000+ genes encoding extracellular matrix (ECM) and ECM-associated proteins 1. The ECM, which can account for up to 70% of the mass of vertebrates, is essential for metazoan development because it provides attachment sites for cell migration and provides biomechanical support and protection against crushing and tensile forces. However, synthesis of the matrisome is, in itself, insufficient to generate a functional ECM; scaffolding proteins, such as collagens, are assembled into defined numbers of elongated fibrils that are organised into precise three-dimensional architectures 2–4 to give tissues and organs their physical form, polarity, mechanical properties, and adaptability to environmental stimuli. Collagen fibrils are the largest (up to centimetres in length5) protein polymers in vertebrates and account for ∼25% of total body protein mass 6. They exhibit a characteristic D-periodic axial banding pattern (where D = 67 nm) 7, are roughly circular in cross section, range in diameter from ∼12 nm to ∼500 nm 8, and grow in length from their two pointed tips 9, 10. Studies in tendon and cartilage have shown that collagen fibrils are synthesised during development 4 and persist throughout life without turnover (in humans 11 and mice 12). Therefore, having the right number, length and organisation of collagen fibrils is critical for normal cell behaviour and tissue health.
Collagen can be purified from tissues and reconstituted in neutral buffers to generate fibrils with the same D-periodicity as those that occur in vivo 7. However, the fibrils lack a preferred orientation and the control of fibril number and diameter distribution is lost. Collagen fibril length is difficult to ascertain, either in vivo or in vitro; therefore, it is unknown if the centimetre lengths that are attained in vivo are reproduced in vitro. Considering that collagen can self-assemble into fibrils, but that higher-order assemblies are lost in vitro, implies that cells exert control over the fibril assembly process to produce tissue-specific matrices. We hypothesised that the endosomal system, with its complex collection of vesicles trafficking between the Golgi and the plasma membrane, could contribute to such a mechanism.
Support for this hypothesis comes from a previous report that the assembly of collagen fibrils is under the control of the circadian clock 13 and that vacuolar protein sorting (VPS) 33b (a regulator of SNARE-dependent membrane fusion in the endocytic pathway) is circadian clock regulated. VPS33b forms a distinct complex with VIPAS39 (also known as VIPAR) 14, and mutations in the VPS33B gene cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome 15, with death usually occurring within the first year of birth with renal insufficiency, jaundice, multiple congenital anomalies, predisposition to infection, and failure to thrive 16. One proposed disease-causing mechanism is abnormal post-Golgi trafficking of lysyl hydroxylase 3 (LH3, PLOD3), which is essential for collagen homeostasis during development 17. Additional support for tight cellular control of collagen fibril formation comes from electron microscope observations of collagen fibrils at the plasma membrane of embryonic avian and rodent tendon fibroblasts 18, 19. Here, one of the two tips of newly-formed collagen fibrils is enclosed within a plasma membrane invagination termed a fibripositor 20. The second tip protrudes into the extracellular matrix. One way in which cells could exert control over collagen fibrillogenesis is by separating initiation of fibril formation (nucleation at the plasma membrane) from longitudinal growth. Nucleation at the plasma membrane would give precise control of fibril number whereas propagation (involving either the proximal or distal tip) would contribute to tissue expansion. However, despite ultrastructural observations, direct mechanistic support for these observations was lacking.
Here, we showed that cells lacking VPS33b did not exhibit fibripositors and could not assemble collagen fibrils, which was in contrast to control cells that exhibited fibripositors and assembled fibrils at their plasma membrane. Instead, VPS33B knockout cells secreted soluble collagen and accumulated collagen in intracellular puncta. Using a split-GFP approach, we showed that collagen-I and VPS33b were contained in vesicles below the plasma membrane. We showed using biotin-surface labelling that col5a1 (a critical chain of collagen-V) and integrin-α11 subunit (a collagen-binding integrin involved in collagen reorganisation 21) are absent from the surface of VPS33b knockout cells. Together, these results provide insights into the importance of the endosomal system in orchestrating and regulating the assembly of collagen fibrils at the plasma membrane.
Results
Collagen-I is endocytosed and reassembled into fibrils
As a first experiment, we made Cy3 labelled collagen-I (Cy3-colI) using previously described methods22 and incubated it with freshly isolated tail tendons for 3 days, then imaged the mid-section of the tendon (Figure 1A). Cells in tendon (nuclei marked with Hoechst stain) show a clear uptake of Cy3-colI within the cytoplasm (Figure 1A, yellow box and indicated by yellow arrowheads). Additionally, fibrillar Cy3 fluorescence signals that transverse between cells were also observed (Figure 1A, grey box and indicated by white arrows). Pulse-chase experiments were performed, where Cy3-colI was added to the tendons for 3 days, followed by 5FAM-colI for a further 2 days (Supplementary Figure 1A). The results confirmed that collagen-I can be taken up by cells regardless of the fluorophore used; additionally, there are distinct areas where only Cy3-positivity was observed (Supplementary 1A, yellow box). The persistence of Cy3-colI indicated that not all collagen endocytosed by cells is directed to degradation. 5FAM-positive fibril-like structures were also observed, suggesting that collagen-I taken up by cells is recycled to the matrix (Supplementary 1A, grey box). Next, we added Cy3-colI to cultured tendon fibroblasts. Immunofluorescence staining indicated co-localisation of endogenous collagen-I with Cy3-labelled collagen-I (Figure 1B). Therefore, endocytosed collagen can be repurposed into fibrils without being targeted for degradation. Taken together, these results demonstrated that exogenous collagen-I can be taken up by cells both in vitro and in tendon tissues ex vivo.
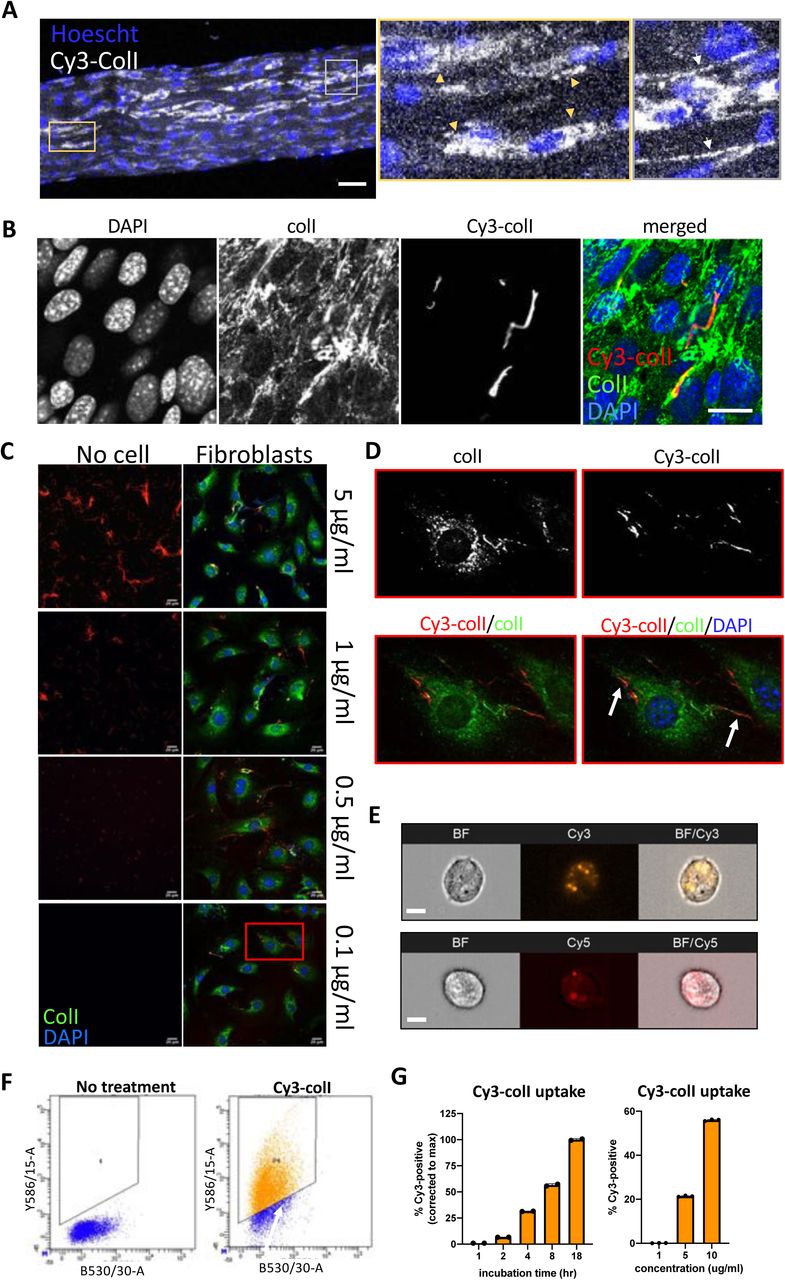
A. Fluorescent images of tail tendon incubated with Cy3-colI for 5 days, showing presence of collagen-I within the cells, and fibril-like fluorescence signals outside of cells. Hoescht stain was used to locate cells within the tendon. Area surrounded by yellow box expanded on the right, and cells with Cy3-colI present intracellularly pointed out by yellow triangles. Area surrounded by grey box expanded on the right, and fibril-like fluorescence signals indicated with white arrows. Scale bar = 50 μm. Representative of N=3.
B. Fluorescent image series of fibroblasts incubated with 5 µg/mL Cy3-colI for 1 hr, trypsinised and replated in fresh media, and incubated for 72 hrs. Labels on top denotes the fluorescence channel corresponding to proteins detected. Representative of N>3. Scale bar = 20 μm.
C. Fluorescent image series of Cy3-colI incubated at different concentrations for 72 hrs, either cell-free (left panel), or with fibroblasts (right panel).
D. Zoom-in (red box in Figure 1C) of Cy3-positive fibrils assembled by fibroblasts when incubated with 0.1 µg/mL Cy3-colI.
E. Flow cytometry imaging of fibroblasts incubated with 5 µg/mL Cy3-labelled (left) or Cy5-labelled (right) collagen-I for 1 hr, showing that collagen-I is taken up by cells and held in vesicular structures. Images acquired using ImageStream at 40x magnification. Scale bar = 10μm BF – brightfield channel, Cy3 – Cy3 channel, Cy5 – Cy5 channel, BF/Cy3 or BF/Cy5 – merged image of BF and Cy3 or BF and Cy5 respectively. Representative of >500 cells images collected per condition.
F. Representative dot plots from flow cytometry analysis representing Cy3 gates used in control and fibroblasts incubated with 1 µg/mL Cy3-labelled collagen (Cy3-colI) for 18 hrs. Representative of N>3.
G. Bar chart showing a progressive increase of percentage of fluorescent cells incubated with 1.5µg/mL Cy3-colI over time (left), and an increase of percentage fluorescent cells incubated with increasing concentration of Cy3-colIfor 1 hr (right), suggesting a non-linear time-dependent and dose-dependent uptake pattern. N=3.
We considered the possibility that the fluorescent fibril-like structures were the result of Cy3-colI attachment to pre-existing fibres in the extracellular matrix, or to spontaneous cell-free fibrillogenesis. Therefore, we incubated Cy3-colI at concentrations that spanned the 0.4 µg/mL critical concentration for cell-free in vitro fibril formation 23, in the presence and absence of cells (Figure 1C). In the absence of cells, Cy3-positive fibrils were observed at 1 µg/mL and 5 µg/mL with no discernible Cy3-positive fibrils at 0.1 µg/mL Cy3-colI (Figure 1C, left panel). However, in the presence of cells, Cy3-positive fibrillar structures were observed at the surfaces of cells at all concentrations of Cy3-colI examined, including 0.1 µg/mL (Figure 1C, right panel and Figure 1D). These results indicated that the cells actively recycled Cy3-colI into fibrils. As a next experiment, we incubated cells with Cy3-colI or Cy5-colI for 1 hr and released the cells from the cell culture plastic. Flow cytometry imaging revealed that tagged collagen-I is endocytosed by the cells into distinct vesicles (Figure 1E). After overnight incubation, flow cytometry analysis revealed ∼ 80% of the fibroblasts had taken up Cy3-coll (Figure 1F). Further time course analyses revealed a time-dependent, as well as concentration-dependent, increase of collagen-I uptake (Figure 1G). Similar results were obtained using Cy5-colI, indicating that soluble monomeric collagen-I can be taken up by cells regardless of the fluorophore attached (Supplementary Figure 1B, C).
The endocytic pathway controls collagen-I secretion and fibril assembly
Having shown that cells can take up and recycle collagen-I to extracellular fibrils, we asked if the endocytic process is required for collagen deposition or fibril formation. To address this, we first used Dyngo4a (Dyng) to inhibit clathrin-mediated endocytosis. We confirmed that Dyng treatment at 20 µM leads to ∼ 60% reduction in Cy3-colI uptake (Figure 2A) and does not affect cell viability even after long periods in culture (Supplementary Figure 2A).

A. Scatter plot showing Dyngo4a (Dyng), an endocytosis inhibitor, treatment for 1 hr inhibits over 50% of Cy3-colI uptake in fibroblasts. Bars show mean ± s.e.m of N=6. ****p<0.0001.
B. Western blot analysis of conditioned media taken from fibroblasts treated with DMSO (ctrl) or Dyng (Dyng) for 72 h, showing a decrease in collagen-I secretion. Top: probed with collagen-I antibody (Col-I), bottom: counterstained with Ponceau (Pon) as control. Representative of N=3.
C. Fluorescent images of collagen-I (red) counterstained with DAPI (blue) in fibroblasts treated with DMSO (top) or Dyng (bottom) for 72 hrs. Scale bar = 20 μm.
D. qPCR analysis comparing col1a1 mRNA levels in control fibroblasts (DMSO) and Dyng-treated fibroblasts (Dyng), showing a decrease in collagen-I transcripts. Bars showing mean± s.e.m of N=4. ***p=0.003.
E. Percentage Cy3-colI taken up by synchronised fibroblasts over 48 hrs. Left: raw values of % Cy3-positive over time. Meta2d analysis indicates a circadian rhythm of periodicity of 23.8 hrs. Right: fluctuation of Cy3-positive cells corrected to running average (average of 12 hrs). Meta2d analysis indicates a circadian rhythm of periodicity of 23.8 hrs. Bars show mean ± s.e.m of N=3 per time point.
We then investigated the effects of Dyng treatment on the ability of fibroblasts to assemble collagen fibrils. Murine tendon fibroblasts (which are prolific at secreting collagen-I and assembling fibrils) were treated with Dyng for 48 hrs and conditioned media (CM) collected to assess levels of secreted collagen-I. The results showed that the amount of soluble collagen-I secreted was greatly reduced in Dyng-treated cells (Figure 2B). Moreover, this was coupled with a reduction in the number of collagen-I fibres in the cell-derived matrix, as indicated by immunofluorescence (Figure 2C). qPCR analyses revealed that col1a1 transcripts were significantly reduced in cells treated with Dyng (Figure 2D), suggesting the possible existence of a feedback mechanism between endocytosis and collagen-I secretion. Taken together, these results suggest that inhibiting endocytosis in fibroblasts does not lead to accumulation of soluble collagen-I monomers in the extracellular space. Rather, endocytosis is directly involved in collagen fibrillogenesis, which in turn regulates collagen transcript levels through an unidentified negative feedback loop.
Collagen-I endocytosis is circadian clock regulated
Previously, we have demonstrated that circadian clock-synchronised fibroblasts synthesise collagen fibrils in a circadian clock rhythmic manner 13; the results here thus far indicate an involvement of endocytosis in collagen fibrillogenesis. Therefore, we tested the hypothesis that collagen-I endocytosis may also be circadian clock regulated. Time-series flow cytometry analyses on fibroblasts incubated with Cy3-colI revealed that the level of Cy3-colI endocytosed by the cells is rhythmic, with a periodicity of 23.8 hrs as determined by MetaCycle analysis (Figure 2E, left). When corrected to the running average, the rhythmic nature of Cy3-colI uptake is accentuated (Figure 2E, right). We noted that, when compared to the number of fibrils produced over time, the peak time of uptake has an inverse correlation with fibril numbers (Supplementary Figure 2B). This suggested to us that the cells may be endocytosing exogenous collagen and holding it in the endosomal compartment under circadian clock control, then trafficking the collagen to the plasma membrane for fibril formation. A possible function of this delay could be to increase the concentration of collagen-I in readiness for fibril initiation, which is supported by our findings above where fibroblasts can take a starting concentration of Cy3-colI of 0.1 µg/mL (which is lower than the 0.4 µg/mL critical concentration for fibril formation, Figure 1C, D) and concentrate it sufficiently to assemble fibrils.
VPS33b controls collagen fibril homeostasis
VPS33b is situated in the post-Golgi space where it is involved in endosomal trafficking with multiple functions including extracellular vesicle formation 24, modulation of P53 signalling 25, and maintenance of cell polarity 26, dependent on cell type. We previously identified VPS33b to be a circadian-controlled component of the collagen-I secretory pathway in fibroblasts 13. As a result, we decided to interrogate further its role in endosomal recycling of collagen-I.
We first confirmed our previous finding that CRISPR-knockout of VPS33b (VPSko, as verified by Western blot analysis and qPCR analysis, Supplementary Figure 3A and B) in tendon fibroblasts led to fewer collagen fibrils, as evidenced by both electron microscopy (Figure 3A) and immunofluorescence microscopy (Figure 3B). The impact on matrix deposition was verified by analysis of the decellularized matrices. The results showed that VPSko fibroblasts produced less matrix (by mass) than control cells (Figure 3C, left), which was mirrored by a reduction in hydroxyproline content (Figure 3C, right). This decrease in matrix deposition was not due to changes in cell proliferation (Supplementary Figure 3C).

A. Electron microcopy images of fibroblasts plated on Aclar and grown for a week before fixation and imaging. Ctrl culture has numerous collagen-I fibrils, as pointed out by arrows. Yellow arrow points to a fibripositor, and green box is expanded to the left bottom corner, showing the distinct D-banding pattern of collagen-I fibril when observed with electron microscopy. VPSko clones all have fewer and thinner fibrils present in the culture (pointed out by red arrows). Representative of N=3. Scale bar = 0.5 μm.
B. Fluorescence images of collagen-I (red) and DAPI counterstain in ctrl and VPSko fibroblasts. Yellow arrows indicating collagen fibres, and white arrows pointing to collagen-I presence in intracellular vesicles. Representative of N>6. Scale bar = 25 μm.
C. Matrix deposition by ctrl or VPSko cells, after one week of culture. Left: decellularized matrix mass. N>5, * p=0.03. Right: hydroxyproline content presented as a ratio between ctrl and VPSko cells. N=5, ***p=0.0004.
D. Matrix deposition by ctrl or VPSoe cells, after one week of culture. Left: decellularized matrix mass. N=5. Right: hydroxyproline content presented as a ratio between ctrl and VPSoe cells. N=4.
E. Fluorescence images of collagen-I (red) and DAPI counterstain in ctrl and VPSoe fibroblasts. Representative of N>6. Scale bar = 20 μm.
F. Relative collagen fibre count in synchronised ctrl (black) and VPSoe (pink) fibroblasts, corrected to the number of fibres in ctrl cultures at start of time course. Fibres scored by two independent investigators. Bars show mean± s.e.m. of N=2 with 6 technical repeats at each time point.
G. Western blot analysis of conditioned media taken from ctrl and VPSoe after 72 hrs in culture. Top: probed with collage-I antibody, bottom: counterstained with Ponceau as control. Representative of N=3.
To further investigate the role of VPS33b in collagen-I homeostasis, we stably overexpressed VPS33b in fibroblasts (VPSoe). Overexpression was confirmed by western blot, qPCR analyses, and flow cytometry of transfected cells (Supplementary Figure 3D, E, F). The mean total matrix and mean total hydroxyproline were higher in VPSoe cultures but not significant (Figure 3D). Immunofluorescence staining of cells after 3 days in culture indicated a greater number of collagen-I fibrils in VPSoe cells (Figure 3E). Vps33b overexpressing cell lines showed equivalent proliferation to controls over the time course of these experiments (Supplementary Figure 3G), suggesting VPSoe enhanced the assembly of collagen fibrils. We then synchronised control and VPSoe fibroblasts and performed time-series immunofluorescence to quantify the number of collagen fibres formed. Similar to our previous finding in primary lung fibroblasts 13, murine tendon fibroblasts exhibited a ∼ 24 hrs rhythmic increase and decrease in collagen-I fibre numbers; however, VPSoe cells continuously deposited collagen-I fibres over the 55-hr period (Figure 3F). MetaCycle analyses indicated that the increase and decrease in fibre numbers in control and VPSoe cultures occurred at a periodicity of 22.7 hrs and 28.0 hrs, respectively (Supplementary Figure 3H). This indicates that continuous expression of VPS33b leads to loss of collagen-I fibre circadian homeostasis. Interestingly, when assessing the levels of soluble collagen-I monomers present in the culture medium from control and VPSoe cells, VPSoe cells have lower levels of soluble collagen-I (Figure 3G). In addition, when VPS33b is knocked-down using siRNA, there is an elevation of soluble collagen-I secreted into the CM; siVPS33b on VPSoe cells also increased the levels of collagen-I in CM similar to the level seen in control cells (Supplementary Figure 3I). These findings strongly indicate that VPS33b is not involved in collagen-I secretion per se, but specifically collagen-I fibre-assembly. The reduction of secreted soluble collagen-I in VPSoe cells suggests that in fibroblasts, VPS33b is directing collagen-I towards fibril assembly.
VPS33b containing vesicles carry collagen-I
To understand the mechanism of VPS33b-directed fibril-assembly, we utilised the split-GFP system 27, 28, where the 214-residue N-terminal fragment (GFP1-10) was cloned onto VPS33b, and the 17-residue C-terminal peptide (GFP11) was cloned onto the proα1(I) chain of collagen-I (colI-GFP11, Figure 4A). Using this system, a GFP signal will only be present when VPS33b interacts with collagen-I. We, and others, have previously demonstrated that insertion of tags at the N-terminal of proα2(I) or proα1(I) chain does not interfere with collagen-I folding or secretion 29, 30. To determine which terminus of the VPS33b protein GFP1-10 should be added, we inserted BFP on either the N-terminal (VPSnBFP) and C-terminal (VPScBFP) ends of VPS33b and imaged the cells. In cells with VPSnBFP, the fluorescence signal is diffuse throughout the cell, and in most instances completely excluded from vesicular structures (Supplementary Figure 4A, left). In contrast, VPScBFP-expressing cells have punctate, vesicular-like blue fluorescence signals (Supplementary Figure 4A, right). Further, previous studies investigating VPS33b interactions have also tagged VPS33b at its C-terminal end 14. Thus, we cloned GFP1-10 into the C-terminus of VPS33b (VPSbarrel). Notably, when VPScBFP was overexpressed in Dendra2-collagen expressing 3T3 cells 31, the number of Dendra2-positive fibrils synthesised by VPScBFP cells significantly increased, again verifying the effects of VPS33b overexpression in fibroblast collagen-I deposition (Supplementary Figure 4B). We noted that the average length of the fibril was not significantly different between control and VPScBFP fibroblasts, suggesting that VPS33b is important in initiating fibrillogenesis and not fibril elongation (Supplementary Figure 4C).

A. Schematic of the split-GFP detection system. GFP1-10 barrel is introduced into VPS33b (VPS-barrel), and GFP11 to Col1a1 (Col1a1-GFP11). If the two tagged proteins co-localize (e.g. in a vesicle), a GFP signal will be emitted.
B. Brightfield (top) and fluorescence (middle) images of fibroblasts expressing both VPS-barrel and Col1a1-GFP11 constructs. Green box is expanded to the bottom, to highlight the punctate fluorescence signals indicating co-localization within intracellular vesicles, as well as fibril-like structures suggestive of fibril assembly sites. Scale bar = 20 μm
C. Fluorescence images of VIPAS (green), collagen-I (red) and DAPI counterstain in fibroblasts. Representative of N=3. Green box is expanded to the right (flipped 90°) to show VIPAS co-localization with collagen-I. Scale bar = 25 μm
D. Fluorescence (left) and merged (right) images of fibroblasts expressing VPS-barrel incubated with conditioned media containing Col1a1-GFP11 for 24hr. Scale bar = 25 μm.
E. Bar chart comparing the percentage of cells that have taken up 5 µg/mL Cy5-colI after 1 hr incubation between control (ctrl), VPS33b-knockout (VPSko) and VPS33b-overexpressing (VPSoe) cells, corrected to control. N>10.
F. Fluorescence images of cells of different levels of VPS33b expression, fed with Cy5-colI and cultured for 72 hrs. Cultures were counterstained with DAPI. Bottom panel are zoomed-in images of the fibrils produced by the fibroblasts. Representative of N=2.
Fibroblasts stably expressing VPSbarrel and ColI-GFP11 were imaged to identify the location of the GFP fluorescence. The results showed puncta throughout the cytoplasm (Figure 4B). Intriguingly, GFP fluorescence was also observed at the cell periphery, sometimes aligned with fibrous elements (Figure 4B, zoom). This distribution was similar to the alignment of collagen fibrils seen in Dendra2-collagen expressing cells (Supplementary figure 4D, yellow arrows), suggesting that VPS33b delivers collagen-I to sites of fibrillogenesis. Immunofluorescence staining of endogenous VIPAS39 (a known VPS33b-interacting partner 14) also revealed co-localisation of VIPAS39 with collagen-I in vesicular structures (Figure 4C); in particular, in some of the co-localised vesicles, the signal of VIPAS39 is strongest surrounding collagen-I (Figure 4C, zoom), suggesting that VIPAS39 is encasing collagen-I.
We then asked if VPS33b is responsible for the endocytosis of collagen-I. Incubation of cells expressing only the VPSBarrel with culture medium collected from cells expressing only ColI-GFP11 revealed that the GFP signal was detected within cells and mostly in vesicle-like structures. However, no GFP signal was detected along the periphery of the cells, presumably where endocytosis would take place (Figure 4D). In addition, flow cytometry analyses of Cy5-colI endocytosed by control, VPSko, and VPSoe fibroblasts showed that there is no consistent change in uptake by VPSko or VPSoe cells (Figure 4E). This was verified in Cy3-colI uptake analysis of cells over-expressing VPScBFP (Supplementary figure 4E). Taken together, these results suggest that VPS33b is involved in the delivery of collagen-I to the extracellular space, and not endocytosis per se. VPSko cells replated after Cy3-or Cy5-colI uptake have conspicuously fewer fibrils when compared to control. In contrast, VPSoe cells have shorter and more Cy5-colI fibrils, as well as Cy3-colI fibrils (Figure 4F, Supplementary Figure 4F), highlighting the role of VPS33b in recycling endocytosed collagen-I to fibril assembly sites.
Integrin-α11 mediates VPS33b fibrillogenesis
To help identify other molecules involved in collagen-I fibril formation, we turned to biotin cell surface labelling coupled with mass spectrometry protein identification (LC-MS/MS). Control, VPSko and VPSoe cells were cultured for 3 days prior to biotin-labelling of intact cells in culture, followed by cell lysis, streptavidin-pulldown of biotinylated proteins, and mass spectrometry analysis. We used a “shot-gun” approach to create an overview of protein profiles in control and VPSko and VPSoe cells and visualised the results in a semi-quantitative manner using spectrum counting. In total, 2666 proteins were identified. Comparison between total lysates and biotin-enriched samples demonstrated an enrichment of 742 proteins in the biotin-enriched samples (Supplementary table 1). Gene Ontology (GO) Functional Annotation analysis 32, 33 identified the top 25 enriched terms (based on p-values) with the top 5 terms all associated with “extracellular” or “cell surface” (Figure 5A). We took this to indicate that the biotin-labelling procedure had successfully identified proteins at the cell-ECM interface.
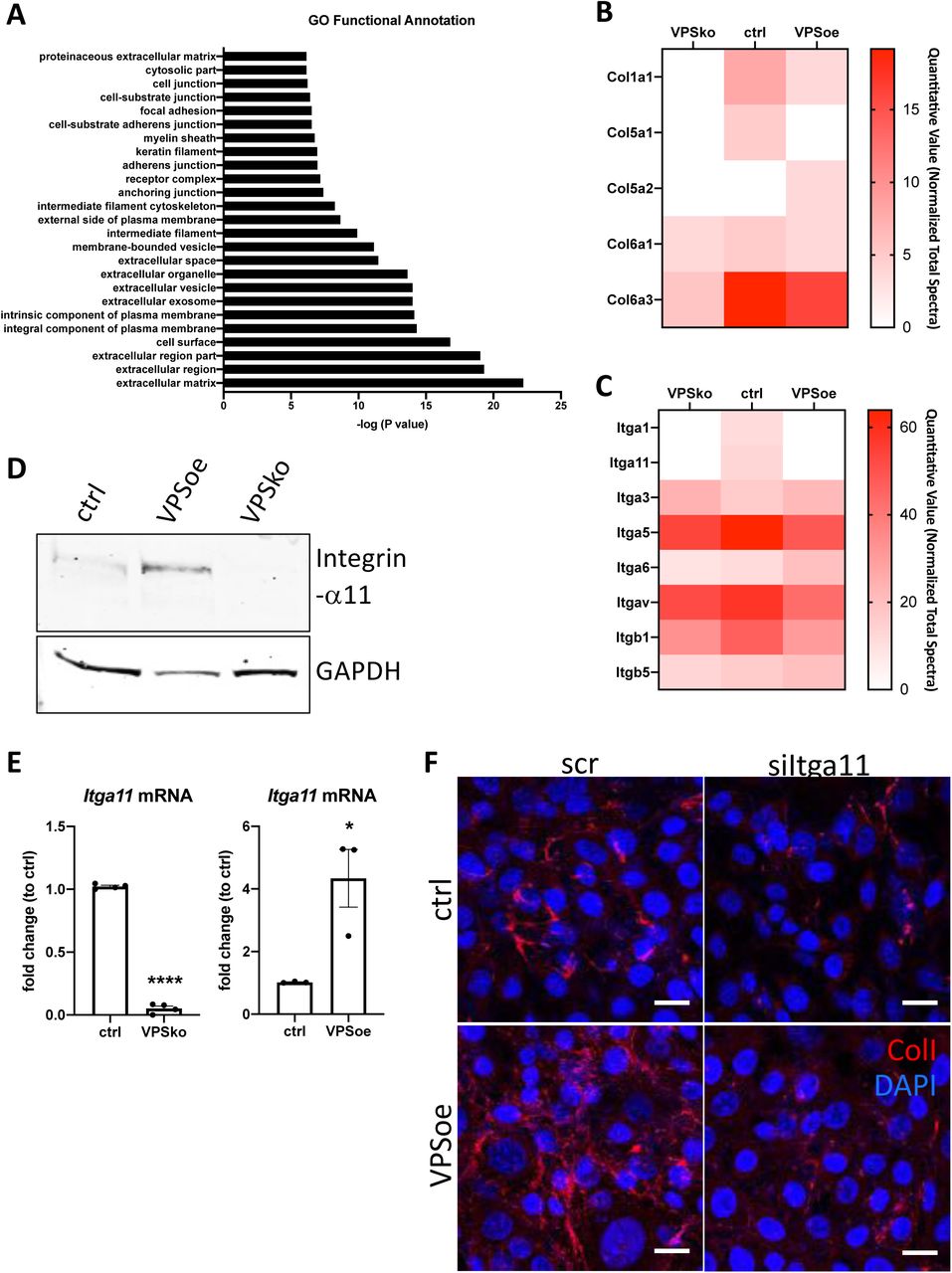
A. Top 25 Functional Annotation of proteins detected in biotin-enriched samples when compared to non-enriched samples based on p-values. Y-axis denotes the GO term, X-axis denotes –log (P value).
B. Heatmap representation of spectral counting of collagens detected in biotin-enriched surface proteins from control (ctrl), VPS33b-knockout (VPSko), and VPS33b-overexpressing (VPSoe) fibroblasts. Scale denotes quantitative value as normalized to total spectra, as determined by Scaffold analyses.
C. Heatmap representation of spectral counting of integrins detected in biotin-enriched surface proteins from control (ctrl), VPS33b-knockout (VPSko), and VPS33b-overexpressing (VPSoe) fibroblasts. Scale denotes quantitative value as normalized to total spectra, as determined by Scaffold analyses.
D. Western blot anlaysis of integrin-α11 levels in control (ctrl), VPS33b-overexpressing (VPSoe), VPS33b-knockout (VPSko) cells. Top: probed with integrin-α11 antibody, bottom: reprobed with GAPDH antibody. Representative of N=3.
E. qPCR analysis of Itga11 transcript levels in ctrl compared to VPSko fibroblasts (left), and ctrl compared to VPSoe fibroblasts (right). N>3, ****p<0.0001, *p= 0.0226
F. Immunofluorescence images of ctrl and VPSoe fibroblasts treated either with control siRNA (scr) or siRNA again Itga11 (siItga11), after 72 hrs incubation; collagen-I (red) and DAPI (blue) counterstained. Representative of N=3. Scale bar = 25 μm.
We then interrogated the differences between VPSko and control cells. Conspicuously, collagen α1(I) and α1(V) chains were not detected at the surface of VPSko cells (Figure 5B). The absence of α1(V) and α2(V) chains (which constitute type V collagen) from the cell surface supports the long-standing view that collagen-V nucleates collagen-I containing fibrils. Gene ontology pointed to integral components of the plasma membrane, these included multiple integrins. Whilst many integrins were detected in all samples there was an absence of integrin-α1 chain and integrin-α11 chain in VPSko cultures. It is well established that integrins are cell-surface molecules that interact extensively with the ECM 34, with integrin-α11 (partnered with integrin-β1) being a major collagen receptor 35. Interestingly, whilst the level of integrin-β1 chain detected was also reduced in VPSko cultures, it was not as drastic as the reduction observed for integrin-α11 chain. This is likely due to the promiscuous nature of integrin-β1 subunit, being able to partner with other α subunits for functions other than collagen-I interaction. The absence of integrin-α11 from VPSko cells suggested that VPS33b-mediated collagen fibrillogenesis is mediated through integrin-α11β1.
VPS33b was conspicuous by its absence from control cell samples (Figure 5B and Supplementary Figure 5A). We have previously struggled to detect VPS33b protein using mass spectrometry 13; therefore, its absence could be due to low abundance. Noteworthy, we detected VPS33b using split-GFP approaches (Figure 4A and B). VPS33b was detected at the cell surface in VPSoe samples (which over-express VPS33b) along with PLOD3 (Supplementary figure 5A), which was previously identified to be delivered by VPS33b 17. Using a complementary approach, we showed by western blot analysis of total cell lysate that integrin-α11 expression is significantly reduced in VPSko cells and is maintained, and possibly elevated, in VPSoe cells (Figure 5D and Supplementary Figure 5B). This correlation between VPS33b and integrin-α11 expression levels was confirmed at mRNA level (Figure 5E). These findings suggest that the over-abundance of collagen fibrils in VPSoe fibroblasts requires integrin-α11. To confirm this, we performed siRNA against Itga11 on control and VPSoe fibroblasts, and knock-down efficiency was confirmed with qPCR (Supplementary Figure 5C, D). siItga11 in both control and VPSoe cells reduced the level of collagen-I fibrils in culture after 3 days (Figure 5F), confirming that integrin-α11 is required for collagen-I fibrillogenesis, even in the presence of elevated levels of VPS33b. Interestingly, knocking-down integrin-α11 increases exogenous collagen-I uptake as demonstrated by flow cytometry (Supplementary Figure 5E), suggesting that integrin-α11, like VPS33b, is not involved in collagen-I endocytosis; instead, we propose that both VPS33b and integrin-α11 chain are involved in directing collagen-I monomers (endogenous or exogenous) to the formation of collagen-I fibrils.
Discussion
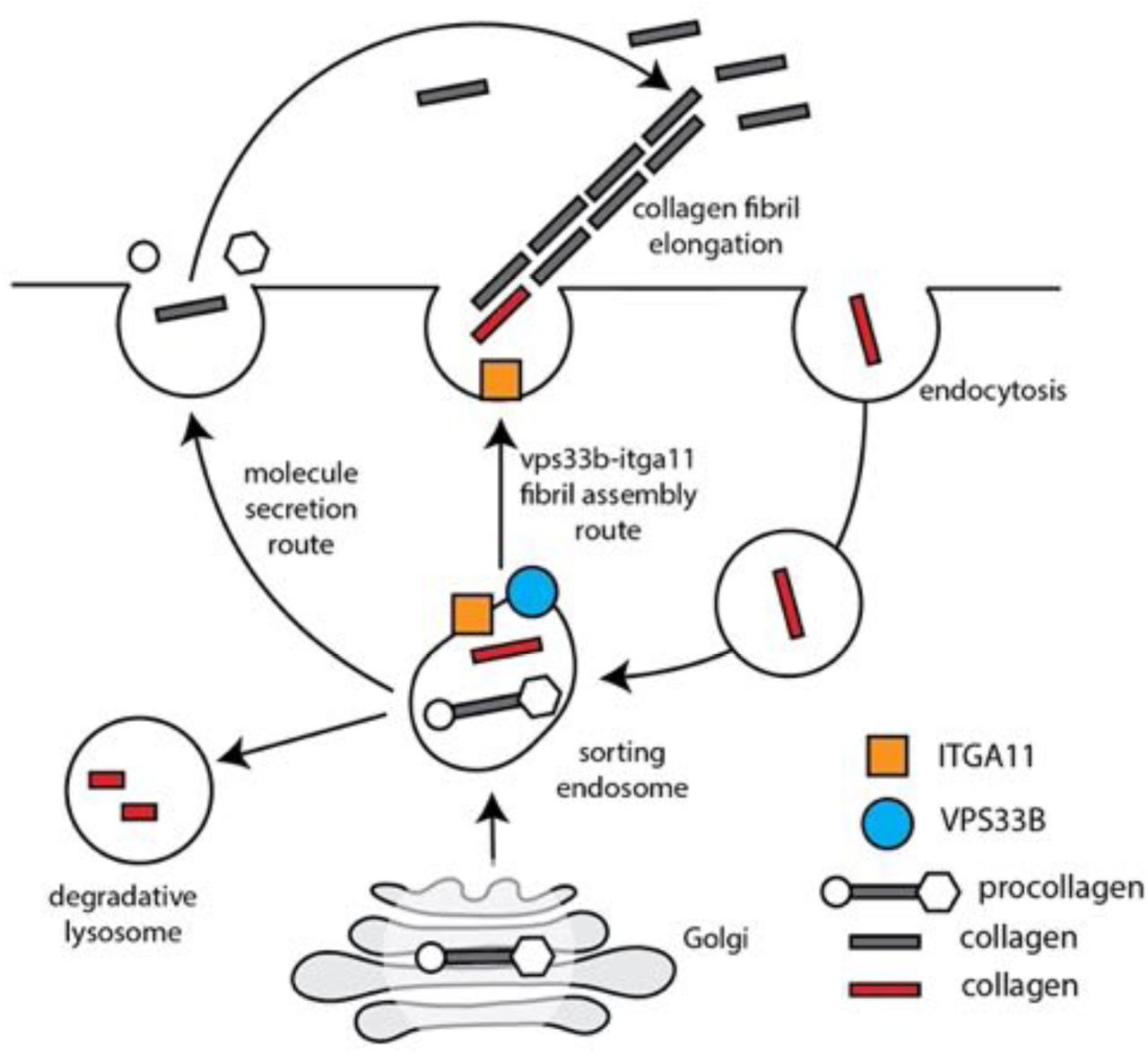
In this study we have identified a molecular mechanism for collagen fibril assembly at the plasma membrane. The major protagonists are collagen-I (the major component of the fibrils), VPS33b (an endosomal tethering molecule), and integrin-α11 subunit (a collagen-binding transmembrane receptor when partnered with integrin-β1 subunit). VPS33b is under circadian clock regulation and interacts directly with collagen-I in a VIPAS-dependent endosomal compartment (shown schematically in Figure 6). The requirement of VPS33b for fibril assembly explains the previously-observed rhythmicity of collagen fibril formation by fibroblasts, in vivo and in vitro. A detail of the mechanism is that VPS33b and integrin-α11 subunit are essential for targeting collagen-I to sites of fibril assembly at the plasma membrane but are not needed for collagen uptake.

Schematic showing how the endosomal system might direct collagen from different sources (endogenous and exogenous) to a site on the plasma membrane for fibril assembly. Grey, de novo synthesised collagen. Red, collagen that has been captured at the plasma membrane. Procollagen is shown with N- and C-propeptides (circle and hexagon, respectively).
Whilst collagen fibrils have been located within fibripositors at the plasma membrane by electron microscopy, and cell culture studies have highlighted the importance of extrinsic mechanical forces (e.g. see 36–39), cell-derived forces 40, 41, combined mechanical anchoring and TGFβ1 cell activation 42, and fluid flow 43 in generating an aligned network of collagen fibrils, mechanistic insights into how cells assemble collagen fibrils at the plasma membrane were lacking. Throughout the current study, collagen fibrils and Cy3-colI labelled fibrils were only seen in close proximity to the plasma membrane; fibrils unattached to cells and at distances from cells were not observed. In cells over expressing VPS33b (e.g. see Figure 3E, 4F), in which large numbers of fibrils were generated, all the fibrils were associated with the plasma membrane. Also, in cells in which levels of integrin-α11 subunit had been reduced, the fewer fibrils generated were associated with the plasma membrane (Figure 5F). The results demonstrate that cells exert control over collagen fibril assembly by anchoring one end of the fibril to the plasma membrane, and uses VPS33b to direct collagen-I to fibrogenic sites at the plasma membrane involving integrin-α11 subunit. The critical importance of VPS33b in the fibril assembly process was demonstrated in knockdown and knockout experiments in which its absence led to soluble collagen-I being secreted without new fibril formation.
Previous research on endocytosis of collagen-I in fibroblasts has focussed on degradation or signalling as being the endpoint (e.g., 44–48). However, our results show that collagen-I endocytosis can also contribute towards fibril formation (Figures 1, 2; Supplementary figure 1). This is highlighted when endocytosis is inhibited and fibroblasts can no longer efficiently assemble collagen-I fibrils (Figure 2C). The decision to flip the endocytosed collagen from degradation to recycling may depend on cell type and on the environment in which the cell finds itself. In complex environments such the site of wound healing, for example, both degradation (of denatured collagen molecules) and recycling (of structurally sound collagen molecules to generate new fibrils) could be beneficial. Nevertheless, we suggest that fibroblasts can either assemble fibrils by a canonical ‘inside-out’ route using collagen that has been synthesised in the ER and trafficked to the plasma membrane via the Golgi apparatus, or, by an ‘outside-in-out’ route involving the endocytic pathway.
These results highlight decision-making processes in the endosomal pathway, where endocytosed collagen-I can be switched between degradation to fibril assembly and involves VPS33b and integrin-α11 subunit. We have previously reported that collagen homeostasis is circadian clock controlled. Here, we confirmed that fibroblasts produce collagen-I fibrils in a rhythmic manner in vitro (Supplementary Figure 2B, Figure 3F) and went on to show that endocytosis of exogenous collagen-I also fluctuates with time (Figure 2E). Collagen can spontaneously form fibrils in the absence of cells in vitro; however, the concentration of collagen must exceed 0.4 µg/mL, which is the critical concentration of fibril formation at 37 °C 23. Here, we showed that fluorescently-tagged collagen-I can spontaneously assemble into fibril-like structures at concentrations greater than 0.4 µg/mL but the aggregates formed at lower concentrations are short. In contrast, when cells are present, fluorescently-tagged collagen-I assembles into fibrils even at concentrations where spontaneous aggregation does not occur (Figure 1C, D). Further, overlaying the endocytosis profile with fibril numbers profile indicates a peak of endocytosis prior to a spike in fibril numbers (Supplementary figure 2B); thus, we propose that endocytosis concentrates the collagen-I to facilitate fibrillogenesis.
Our results showed a direct correlation between VPS33b levels and collagen-I fibrils (Figure 3) and that VPS33b delivers collagen-I directly to cell surface fibrillogenic sites (Figure 4). Importantly, secretion of soluble collagen-I and assembly of collagen-I into fibrils are regulated differently; it is possible for cells to secrete soluble collagen without assembling fibrils. Thus, the abundance of collagen-I secretion into the media is not correlated with the level of VPS33b or the number of fibrils. Instead, VPS33b is responsible for directing collagen-I (either endogenous or captured) to fibre assembly sites. The rhythmicity of VPS33b expression in normal fibroblasts determines the rhythmicity of collagen-I fibres, as highlighted by the loss of fibril rhythmicity in VPSoe fibroblast cultures. Conversely, VPSko fibroblasts lack the capacity to recycle labelled collagen-I to assemble fibres, despite still being able to endocytose collagen-I. In turn, VPS33b requires the presence of the collagen-I binding integrin-α11β1 to produce a fibrous collagen-I matrix. Thus, the distinction between secretion of soluble collagen and fibril assembly is important in studies of disease, in which simple measurement of collagen expression or secretion does not equate to fibrogenesis. This distinction between collagen secretion and assembly was also suggested in a recent study on Pten-knockout mammary fibroblasts, where SPARC affects collagen fibrillogenesis and not secretion, acting through fibronectin 49. Of note, whilst SPARC and fibronectin were detected in our previously-reported time-series mass spectrometry analyses (in the SLDOC soluble fraction and high-salt insoluble fractions respectively), they were not circadian clock rhythmic 13.
Finally, whilst previous research has demonstrated that collagen-I can be secreted in a matter of minutes 20, other studies have demonstrated a relatively slow emergence of collagen-I fibres over days 31. This difference may be due in part to the requirement to concentrate the collagen in the endocytic pathway to a level that supports fibril formation, as discussed above. In addition, nucleation of a fibril is expected to involve collagen-V, a minor fibril-forming collagen that is necessary for the appearance of fibrils containing collagen-I in vivo 50. Another long-standing view is that fibronectin may tether collagen to a fibronectin-binding integrin and thereby function as a proteolytically cleavable anchor (see ref 51 and reviewed by 52). Whilst this study did not focus on the role of fibronectin, biotin-surface labelling mass spectrometry results indicate that the levels of the major fibronectin-binding integrins (integrin-α5β1 and αVβ3) were not altered between control, VPSko, and VPSoe fibroblasts (Supplementary table 2), suggesting that the mechanism by which VPS33b modulates collagen fibrillogenesis does not act directly via fibronectin. Interestingly, it was previously demonstrated that in fibronectin knock-out liver cells, exogenous collagen-V could be added to initiate collagen-I fibrillogenesis 53. Further, whilst collagen α1(V) could only be detected in surface biotin-labelled control and not VPSko or VPSoe cells, collagen α2(V) was only detected in VPSoe cells (Figure 5B). Of note, we have previously shown that collagen α2(V) protein was rhythmic and in phase with collagen α1(I) and collagen α2(I). Whether collagen-V co-traffics with collagen-I in VPS33b vesicles to fibrillogenic sites at the cell periphery, or collagen-V is directed to the nucleation site by another mechanism, is an important question to address in future studies.
Materials and Methods
Cell culture
Unless otherwise stated, all cell culture reagents were obtained from Gibco, and all cells were maintained at 37°C, 5% CO2 in a humidified incubator. Immortalised mouse tail tendon fibroblasts (iTTF 13), and NIH3T3 cells with Dendra2-tagged collagen-I expression (Dendra2-3T3 31) were cultured at in DMEM/F12 with sodium bicarbonate and L-glutamine (supplemented with 10% fetal calf serum (FCS), 200 μM L-ascorbate-2-phosphate (Sigma), and 10,000 U/mL penicillin/streptomycin), and DMEM with sodium bicarbonate and L-glutamine (supplemented with 10% new born calf serum, 200 μM L-ascorbate-2-phosphate, and 10,000 U/mL penicillin/streptomycin) media respectively. HEK293T cells were cultured in DMEM with sodium bicarbonate and L-glutamine, supplemented with 10% FCS. Prior to imaging, media was changed to FluoroBrite media with the appropriate supplements.
CRISPR-Cas9-Mediated Knockout
iTTFs were treated with CRISPR-Cas9 to delete VPS33b gene as previously described 13. Gene knockout was confirmed by western blotting and qPCR.
Transfection and stable infection of over-expression vectors in cells
Three different vectors were used to stably over-express VPS33b protein in iTTF and 3T3 cells – untagged VPS33b overexpression (with red fluorescent protein expression as selection), N-terminus BFP-tagged VPS33b overexpression, and C-terminus BFP-tagged VPS33b overexpression. All vectors were cloned onto a pLV vector backbone using Gibson Assembly® (NEB). For split-GFP experiments, vectors expressing GFP11-tagged collagen-I and GFP1-10-tagged VPS33b were ordered from VectorBuilder. Lentiviral particles were generated In HEK293T cells, and cells were infected in the presence of 8 μg/mL polybrene (Millipore). Cells were then sorted using Flow Cytometry to isolate RFP-positive or BFP-positive cells. Alternatively, cells were treated with puromycin (Sigma) or G418 (Sigma) depending on selection marker on the vectors.
Fluorescence labelling of collagen-I
Cy3 or Cy5 NHS-ester dyes (Sigma) were used to fluorescently label rat tail collagen-I (Corning), using a previously described method 22. Briefly, 3 mg/mL collagen-I gels were made, incubated with 50mM borate buffer (pH 9) for 15 min, followed with incubation with either Cy3-ester or Cy5-ester (Sigma) dissolved in borate buffer in dark overnight at 4°C, gently rocking. Dyes were then aspirated and 50 mM Tris (pH 7.5) were added to quench the dye reaction, and incubated in the dark rocking for 10 min. Gels were washed with PBS (with calcium and magnesium) 6x, incubating for at least 30 min each wash. The gels were then resolubilized using 500 mM acetic acid, and dialyzed in 20 mM acetic acid.
RNA isolation and Quantitative Real-Time PCR
RNA was isolated using TRIzol Reagent (Thermo Fisher Scientific) following manufacturer’s protocol, and concentration was measured using a NanoDrop OneC (Thermo Fisher Scientific). Complementary DNA was synthesized from 1 μg RNA using TaqManTM Reverse Transcription kit (Applied Biosystems) according to manufacturer’s instructions.
SensiFAST SYBR kit reagents were used in qPCR reactions. Primer sequences used were as followed:
Col1a1 AGAGCATGACCGATGGAT and AGGCCTCGGTGGACATTA Itga11 AGATGTCGCAGACTGGCTTT and CCCTAGGTATGCTGCATGGT, Rplp0 ACTGGTCTAGGACCCGAGAAG and CTCCCACCTTGTCTCCAGTC Gapdh CAGCCTCGTCCCGTAGACAA and CAATCTCCACTTTGCCACTGC
Vps33b GCATTCACAGACACGGCTAAG and ACACCACCAAGATGAGGCG
Protein extraction and Western blotting
For lysate experiments, proteins were extracted using urea buffer (8M urea, 50mM Tris-HCl pH7.5, supplemented with protease inhibitors and 0.1% β-mercaptoethanol). For conditioned media (CM) media, cells were plated out at 200,000 cells per 6-well plate, and left for 48-72 hrs before 250 μl was sampled. Samples were mixed with 4xSDS loading buffer with 0.1% β-mercaptoethanol and boiled at 95°C for 5 minutes. The proteins were separated on either NuPAGE Novex 10% polyacrylamide Bis– Tris gels with 1XNuPAGE MOPS SDS buffer or 6% Tris-glycine gels with 1XTris-glycine running buffer (all Thermo Fisher Scientific), and transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare). The membranes were blocked in 5% skimmed milk powder in PBS containing 0.01% Tween 20. Antibodies were diluted in 2.5% skimmed milk powder in PBS containing 0.01% Tween 20. The primary antibodies used were: rabbit polyclonal antibody (pAb) to collagen-I (1:1,000; Gentaur), mouse mAb to vinculin (1:1000; Millipore), rabbit pAb to integrin-α11 (1:1000; from Donald Gulberg) and mouse mAb to VPS33B (1:500; Proteintech). Horseradish-peroxidase-conjugated antibodies and Pierce ECL western blotting substrate (both from Thermo Fisher Scientific) were used and reactivity was detected on GelDoc imager (Biorad). Alternatively, Licor rabbit-anti-mouse 680, mouse-anti-rabbit 800 were used and reactivity detected on an Odyssey Clx imager.
Flow cytometry and imaging
Cy3- or Cy5-tagged collagen was added to cells and incubated at 37°C, 5% CO2 in a humidified incubator for predetermined lengths of times before washing in PBS, trypsinised, spun down at 2500 rpm for 3 min at 4 °C, and resuspended in PBS on ice. Cells were analysed for Cy3- or Cy5-collagen uptake using LSRFortessa (BD Biosciences). For imaging, cells were prepared as described, and analysed on Amnis® ImageStream®XMk II (Luminex).
Imaging and immunofluorescence
For fixed immunofluorescence imaging, cells plated on coverslips were fixed with 100% methanol at −20°C and then permeabilized with 0.5% Triton-X in PBS. Primary antibodies used were as follows: rabbit pAb collagen-I (1:400, Gentaur OARA02579), rabbit pAb VIPAS (1:50, Proteintech 20771-1-AP), mouse mAb FN1 (1:400, Sigma F6140 or Abcam ab6328). Secondary antibodies conjugated to Alexa-488, Cy3, and Cy5 were used (ThermoScientific), and nucleus were counterstained with DAPI (Sigma). Coverslips were mounted using Fluoromount G (Southern Biotech). Images were collected using a Leica SP8 inverted confocal microscope (Leica) using an x63/0.50 Plan Fluotar objective. The confocal settings were as follows: pinhole, 1 Airy unit; scan speed, 400 Hz bidirectional; format 1024 x 1024 or 512 x 512. Images were collected using photomultiplier tube detectors with the following detection mirror settings: DAPI, 410-483; Alexa-488, 498-545 nm; Cy3, 565-623 nm; Cy5, 638-750 nm using the 405 nm, 488 nm, 540 nm, 640 nm laser lines. Images were collected in a sequential manner to minimize bleed-through between channels. When acquiring three-dimensional optical stacks, the confocal software was used to determine the optimal number of Z sections.
For fluorescence live-imaging, cells were plated onto Ibidi μ-plates and imaged using Zeiss LSM880 NLO (Zeiss). For split-GFP experiment, cells were seeded for 24h onto Ibidi μ-plates before imaging with an Olympus IXplore SpinSR (Olympus) with 100x oil magnification.
Decellularisation of cells to obtain extracellular matrix
Cells were seeded out at 50,000 in a 6-well plate and cultured for 7 days before decellularization. Extraction buffer (20mM NH4OH, 0.5% Triton X-100 in PBS) was gently added to cells and incubated for 2 minutes. Lysates were aspirated and the matrix remaining in the dish were washed gently twice with PBS, before being scrapped off into ddH2O for further processing.
Hydroxyproline Assay
Samples were incubated overnight in 6 M HCl (diluted in ddH2O (Fluka); approximately 1 mL per 20 mg of sample) in screw-top tubes (StarLab) in a sand-filled heating block at 100 °C covered with aluminium foil. The tubes were then allowed to cool down and centrifuged at 12,000 xg for 3 min. Hydroxyproline standards were prepared (starting at 0.25 mg/mL; diluted in ddH2O) and serially diluted with 6 M HCl. Each sample and standard (50 μL) were transferred into fresh Eppendorf tubes, and 450 μL chloramine T reagent (0.127 g chloramine T in 50% N-propanol diluted with ddH2O; brought up to 10 mL with acetate citrate buffer (120 g sodium acetate trihydrate, 46 g citric acid, 12 mL glacial acetic acid, 34 g sodium hydroxide) adjusted to pH 6.5 and then made to 1 litre with dH2O; all reagents from Sigma) was added to each tube and incubated at room temperature for 25 min. Ehrlich’s reagent (500 μL; 1.5 g 4-dimethylaminobenzaldehyde diluted in 10 mL N-propanol:perchloric acid (2:1)) was added to each reaction tube and incubated at 65 °C for 10 min and then the absorbance at 558 nm was measured for 100 μL of each sample in a 96-well plate format.
Electron Microscopy
Unless otherwise stated, incubation and washes were done at room temperature. Cells were plated on top of ACLAR films and allows to deposit matrix for 7 days. The ACLAR was then fixed in 2% glutaraldehyde/100mM phosphate buffer (pH 7.2) for at least 2 hrs and washed in ddH2O 3 x 5 min. The samples were then transferred to 2% osmium (v/v)/1.5% potassium ferrocyanide (w/v) in cacodylate buffer (100 mM, pH 7.2) and further fixed for 1 hr, followed by extensive washing in ddH2O. This was followed by 40 minutes of incubation in 1% tannic acid (w/v) in 100 mM cacodylate buffer, and then extensive washes in ddH2O. and placed in 2% osmium tetroxide for 40 min. This was followed by extensive washes in ddH2O. Samples were incubated with 1% uranyl acetate (aqueous) at 4 °C for at least 16 hrs, and then washed again in ddH2O. Samples were then dehydrated in graded ethanol in the following regime: 30%, 50%, 70%, 90% (all v/v in ddH2O) for 8 min at each step. Samples were then washed 4 x 8 min each in 100% ethanol, and transferred to pure acetone for 10 min. The samples were the infiltrated in graded series of Agar100Hard in acetone (all v/v) in the following regime: 30% for 1 hr, 50% for 1 hr, 75% for overnight (16 hrs), 100% for 5 hrs. Samples were then transferred to fresh 100% Agar100Hard in labelled moulds and allowed to cure at 60 °C for 72 hrs. Sections (80 nm) were cut and examined using a Tecnai 12 BioTwin electron microscope.
Surface biotinylation-pulldown Mass Spectrometry
Cells were grown in 6 well plates for 72 hrs prior to biotinylation protocol. Briefly, cells around 90% confluence were kept on ice and washed in ice-cold PBS, following with incubation with ice-cold biotinylation reagent (prepared fresh, 200 μg/mL in PBS, pH 7.8) for 30min at 4°C gently shaking. Cells were then washed twice in ice-cold TBS (50 mM Tris, 100 mM NaCl, pH 7.5), and incubated in TBS for 10 min at 4 °C. Cells were then lysed in ice-cold 1% TritonX (in PBS, with protease inhibitors) and lysates cleared by centrifugation at 13,000 xg for 10 min at 4 °C. Supernatant was then transferred to a fresh tube and 1/5 of the lysates were kept as a reference sample. Streptavidin-sepharose beads are then aliquoted into each sample and incubated for 30min at 4°C rotating. Beads were then washed 3x in ice cold PBS supplemented with 1% TritonX, followed by one final wash in ice cold PBS and boiled at 95°C for 10min in 2x sample loading buffer, followed by centrifugation at top speed for 5min follow by sample preparation for Mass Spectrometry analysis. The protocol used for sample preparation was as described previously 54. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.5.1). Mascot was set up to search the SwissProt_2018_01.fasta; Trembl_2018_01 database (selected for Mus musculus, 84416 entries) assuming the digestion enzyme to be non-specific. Mascot was searched with a fragment ion mass tolerance of 0.020 Da and a parent ion tolerance of 10.0 PPM. Carbamidomethyl of cysteine was specified in Mascot as a fixed modification. Oxidation of methionine was specified in Mascot as a variable modification. Scaffold (version Scaffold_4.10.0, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 50.0% probability by the Peptide Prophet algorithm 55 with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 95.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm 56. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Statistics and reproducibility
Data are presented as the mean ± s.e.m. unless otherwise indicated in the figure legends. The sample number (N) indicates the number of independent biological samples in each experiment, and are indicated in the figure legends. Data were analyzed as described in the legends. The data analysis was not blinded, and differences were considered statistically significant at P<0.05, using Student’s t-test or One-way Anova, unless otherwise stated. Analyses were performed using Graphpad Prism 8 software. Significance levels are: * P<0.05; ** P<0.01; *** P<0.005; ****P<0.0001. For periodicity, analysis was performed using the MetaCycle package57 in the R computing environment 58 with the default parameters.
Author contributions
JC and KEK conceived the project. JC performed experiments and interpreted data. AP and RG performed experiments, and YL performed electron microscopy analyses. DG provided reagents and data interpretation. All authors co-wrote the manuscript.
Competing interests
The authors declare no competing interests.

A. Fluorescent images of tail tendon incubated with fluorescently-tagged collagen-I for 5 days – Cy3-colI was added for the first 3 days, with 5FAM-labelled collange-I (FAM-colI) added in the last two days. Images show presence of collagen-I within the cells, and fibril-like fluorescence signals outside of cells. Hoescht stain was used to locate cells within the tendon. Area surrounded by yellow box expanded on the right highlighting a cell with only Cy3-colI present intracellularly. Area surrounded by grey boxes expanded on the right, highlighting fibril-like fluorescence signals that are either FAM-colI positive only, or have co-localization of both Cy3-colI and FAM-colI. Representative of N=2.
B. Representative dot plots from flow cytometry analysis representing Cy5 gates used in control and fibroblasts incubated with 1 µg/mL Cy5-labelled collagen (Cy5-colI) for 18 hrs. Representative of N>3.
C. Bar chart showing a progressive increase of % of fluorescent cells incubated with 1.5 µg/mL Cy5-colI over time (left), and an increase of % fluorescent cells incubated with increasing concentration of Cy5-colI for 1 hr (right), suggesting a non-linear time-dependent and dose-dependent uptake pattern. N=3.

A. Alamar Blue assay showing that prolonged treatment of 20 μM Dyngo4a (Dyng) does not inhibit fibroblast proliferation.
B. Percentage of Cy3-colI taken up by synchronised cells, corrected to the maximum percentage uptake of the time course (pink, bars show mean ± s.e.m of N=3 per time point), compared to the percentage collagen fibre count over time, corrected to the maximum percentage fibril count of the time course (black, fibres scored by two independent investigators. Bars show mean± s.e.m. of N=2 with 6 technical repeats at each time point). Data presented here in maximum percentage fibril count over time (black) are taken from the control as in Figure 5F, presented in a different way.

A. Western blot analysis of VPS33b knock-out (VPSko) clones compared to control (ctrl) fibroblasts. Top: probed with VPS33b antibody, bottom: probed with GAPDH antibody. Representative of N=3.
B. qPCR analysis of VPS33b expression in the 3 selected clones. *** p=0.0002, ** p=0.0039, * p=0.0163
C. Alamar blue analysis of proliferation rates of ctrl fibroblasts and VPSko fibroblasts. Representative of N=3.
D. Western blot analysis of VPS33b protein levels in control (ctrl) and VPS33b over-expressing (VPSoe) cells. Top panel probed with VPS33b antibody, bottom panel re-probed with vinculin antibody. Representative of N=4.
E. qPCR analysis of VPS33b expression in ctrl and VPSoe cells. N=4, **** p<0.0001.
F. Single parameter histograms of Flow cytometry analysis on ctrl (left) and VPSoe (right) fibroblasts, showing a shift in increase of Cy3 fluorescence and thus expression of VPSoe vector. Representative of N>4.
G. Alamar blue analysis of proliferation rates of ctrl fibroblasts and VPSoe fibroblasts. Representative of N=3.
H. MetaCycle analyses of the fibril counts showed a rhythmicity of circa 23 h in ctrl fibroblasts compared with circa 28 hrs in VPSoe fibroblasts.
I. Western blot analysis of conditioned media taken from ctrl and VPSoe treated either with siRNA scrambled control (scr) or siRNA against Vps33b (siVPS), and cultured for 72 hrs. Top: probed with collage-I antibody, bottom: counterstained with Ponceau as control. Representative of N=2.

A. Fibroblasts expressing BFP-tagged VPS33b. Left: BFP tagged on the N-terminal end of VPS33b (VPSnBFP), Right: BFP tagged on the C-terminal end (VPScBFP). Images taken in Airy mode. Representative of N>4. Scale bar = 10 μm.
B. Quantification of average number of fibrils per cell in control Dendra-colI expressing 3T3 cells (ctrl) and Dendra-colI expressing 3T3 overexpressing VPScBFP (VPScBFP). >500 cells quantified per condition. N=12. *p =0.048.
C. Measurement of average fibril length in arbitrary units (A.U.) in control Dendra-colI expressing 3T3 cells (ctrl) and Dendra-colI expressing 3T3 overexpressing VPScBFP (VPScBFP). >500 cells quantified per condition. N=12.
D. Image of Dendra-colI in 3T3 fibroblasts. Image taken in Airy mode. Yellow arrows highlighting fibril structures at cell periphery. Representative of N>5. Scale bar = 10 μm.
E. Bar chart comparing the percentage of cells that have taken up 5 µg/mL Cy3-colI after 1 hr incubation between control (ctrl), VPS33b-knockout (VPSko) and VPScBFP overexpressing (VPSoe) cells, corrected to control. N>10
F. Cy3-fed ctrl or VPSoe fibroblasts after 72 hrs incubation. Scale bar = 20 μm.

A. Spectral counts of Plod3 and VPS33b as detected by Mass Spectrometry, scale bar represents quantitative value as normalized to total spectra. Gene names of proteins detected shown here.
B. Western blot analysis of total lysates taking from control (ctrl), VPS33b-overexpressing (VPSoe), VPScBFP-overexpressing (VPScBFP), VPS33b-knockout (VPSko), probed with ITGA11 and reprobed with β-tubulin or vinculin as a loading control.
C. qPCR analysis of Itga11 mRNA expression in ctrl fibroblasts treated either with scrambled control (scr) or siRNA against ITGA11 (siITGa11), collected after 96 hrs. N=3, **P=0.0091.
D. qPCR analysis of Itga11 mRNA expression in VPSoe fibroblasts treated either with scrambled control (scr) or siRNA against ITGA11 (siITGa11), collected after 96 hrs. N=3, ****P<0.0001.
E. Bar chart comparing the percentage of cells that have taken up 5 µg/mL Cy5-colI after 1 hr incubation between fibroblasts treated with scrambled control (ctrl) or siRNA against ITGA11 (siITGa11), corrected to scr. N=3. **p=0.0062.
Supplementary Table 1 Report from Scaffold Viewer on the Quantitative Value (Normalized Total Spectra) detected of each protein in each sample.
Specific spectral counts of various proteins as detected by Mass Spectrometry, presented in quantitative value as normalized to total spectra. Gene names of proteins detected shown here.
Acknowledgements
The research was generously funded by Wellcome in the form of a Senior Investigator Award (110126/Z/15/Z) and Centre Award (203128/Z/16/Z) to KEK, and a Norwegian Centre of Excellence grant (grants 2233250) and a grant from Nasjonalföreningen for folkhelsen (Project 16216) to DG. The proteomics was performed at the Biological Mass Spectrometry Facility in the Faculty of Biology, Medicine and Health (University of Manchester) with the assistance of Stacey Warwood and Ronan O’Cualain. Imaging was performed at the Bioimaging Facility in the Faculty of Biology, Medicine and Health (University of Manchester). Some of the illustrations were created with Biorender.com.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}