

Abstract
The outer membrane (OM) of gram-negative bacteria is crucial for maintenance of cell viability as it functions as a selective permeability barrier. Escherichia coli periplasmic Zn-metallopeptidase BepA contributes to the maintenance of OM integrity through its involvement in the biogenesis and degradation of an essential OM protein, LptD, a β-barrel component of the lipopolysaccharide translocon. We have previously shown that BepA either promotes the maturation of LptD when it is on the normal assembly pathway (on-pathway) or degrades it when its assembly is compromised (off-pathway). BepA performs these functions possibly on the β-barrel assembly machinery (BAM) complex. However, the mechanistic details of how BepA recognizes and directs the LptD assembly intermediates to different pathways remains unclear. Here, we performed site-directed mutagenesis and crosslinking experiments to explore the interactions among BepA, LptD, and the BAM complex. We found that the interaction of the BepA edge strand located adjacent to the active site with LptD was crucial not only for proteolysis but also for assembly promotion of LptD. Site-directed crosslinking analysis indicated that the unstructured N-terminal half of the β-barrel-forming domain of an LptD assembly intermediate directly contacts with the BepA edge strand. Furthermore, the C-terminal region of the β-barrel-forming domain of the BepA-bound LptD intermediate interacted with a “seam” strand of BamA, suggesting that BepA recognized LptD assembling on the BAM complex. Our findings provide important insights into the involvement of BepA in the maintenance of OM structure and function, which can be helpful in developing OM-targeted novel drugs.
Introduction
The cell envelope of diderm bacteria is composed of two membranes, namely the inner (cytoplasmic) membrane (IM) and the outer membrane (OM). The intermembrane space, known as periplasmic space, contains a peptidoglycan layer. The OM is the outermost layer of a cell directly facing the external milieu and acts as a selective permeability barrier that prevents the penetration of toxic compounds including antibiotics (1). The cell surface localization as well as the functional impotence of the OM make its components suitable drug targets.
Outer membrane proteins (OMPs), generally exhibiting a β-barrel fold formed by more than 8 ß-strands, play important roles in maintaining the structural and functional integrity of the OM (2). Therefore, irregularities in OMP biogenesis result in elevated cellular sensitivity to toxic compounds (3, 4). After synthesis in the cytoplasm and following translocation across the IM to the periplasm through the SecYEG translocon, OMPs are delivered to the OM by periplasmic chaperones such as DegP, Skp, and SurA, and are finally integrated into the OM (2, 5, 6). The OM assembly of OMPs is mediated by the β-barrel assembly machinery (BAM) complex consisting of a β-barrel OMP, BamA, and four lipoproteins, namely BamB, BamC, BamD, and BamE (5–7). Among the BAM complex subunits, BamA and BamD are the only essential components, although recent studies have shown that certain BamA mutations render all other BAM subunits dispensable (8). The BAM complex has a silk-hat-like structure (9–13); the OM-embedded C-terminal β-barrel domain of BamA forms the “crown” and the N-terminal periplasmic polypeptide transport-associated (POTRA) domains of BamA form the “brim” together with the BamB/C/D/E lipoproteins.
Lipopolysaccharide (LPS), another major OM constituent localized in the outer leaflet of the OM, is also important for the maintenance of the structure and function of the OM (14). LPS is synthesized on the cytoplasmic side of the IM and flipped across the IM to the periplasm. After maturation, it is transported to the OM by the LPS transport (Lpt) proteins (14). A heterodimer of LptD, a β-barrel OMP, and LptE, a lipoprotein, plays roles in the final step to insert LPS into the OM (15–17). The OM assembly process of LptD is unique in that it is accompanied by rearrangement of intramolecular disulfide bonds. The mature form of LptD (LptDNC) possesses two disulfide bonds formed by non-consecutive pairs of Cys residues (C31-C724 and C173-C725) (18). It has been shown, however, that an assembly intermediate of LptD (LptDC) having disulfide bonds formed by consecutive pairs of the Cys residues (C31-C173 and C724-C725) is first generated and isomerized to LptDNC during its assembly/maturation, which is triggered by the association of LptD with LptE (19, 20). The LptDC to LptDNC conversion should occur at a later step in the OM assembly because LptD presumably associates with LptE on the BAM complex (19–22).
BepA (formally called YfgC) is a bi-functional periplasmic zinc metallopeptidase that plays an important role in maintaining OM integrity (20). We have previously shown that BepA is involved in the biogenesis and quality control of LptD. While BepA promotes the LptDC to LptDNC conversion (chaperone-like function) (20), it also degrades the stalled or misassembled LptDC molecules that are generated due to an lptD mutation (LptD4213) or decreased availability of or weakened interaction with LptE (protease function) (20, 23). The BepA protein consists of an N-terminal M48 metallopeptidase domain and a C-terminal tetratricopeptide repeat (TPR) domain that are associated closely to form a compact structure (Fig. 1A) (24, 25). Our previous study suggested that the TPR domain of BepA directly interacts with LptD and with the BAM complex with its TPR domain inserted into the interior of the periplasmic part (brim) of the BAM complex (20, 26). A mutational study has suggested that these interactions are important for BepA functions (24). Recent study have also shown that the H246 residue of BepA that coordinates the zinc ion at the proteolytic active site acts as an ON/OFF switch (His switch) for the proteolytic activity of BepA (25, 27). The dual functions of BepA should be appropriately regulated because the unregulated expression of the proteolytic activity of BepA caused by an H246 mutation leads to the degradation of the normally assembling LptD intermediate (27). However, information on the molecular mechanism of this regulation and the modes of the BepA-LptD interaction in each BepA function remains elusive.

(A) Crystal structure of BepA (PDB code: 6AIT). The peptidase and the TPR domains of BepA are shown in gray and orange, respectively. The edge strand, the proteolytic active site (the HExxH motif and the third zinc ligand, Glu-201) and the regulatory His-246 residue (His switch) in the peptidase domain are shown in red, blue, and green, respectively, and the coordinated zinc atom is shown in yellow. An enlarged view of the active site region is shown in right. (B) Protease activities of the BepA edge strand mutants. Cells of SN56 (ΔbepA) carrying pTWV228-lptD-his10 and either pSTD689 or pSTD689-bepA plasmids were grown at 30°C in L-medium until early log phase and induced with 1 mM IPTG for 1 h. Total cellular proteins were acid-precipitated and analyzed by 7.5 or 10% Laemmli SDS-PAGE and immunoblotting with the indicated antibodies. (C) In vivo photo-crosslinking analysis of the BepA edge strand. Cells of SN56 carrying pEVOL-pBpF and pUC18-bepA(E137Q, amb)-his10 plasmids were grown at 30°C in L medium containing 0.02% arabinose and 0.5 mM pBPA until early log phase, and induced with 1 mM IPTG for 1 h to express the indicated BepA(pBPA) variants. The cultures were divided into two portions, each of which was treated with or without UV-irradiation for 10 min at 4°C. Proteins of the total membrane fractions were subjected to pull-down with Ni-NTA agarose. Purified proteins were analyzed by 7.5 % Laemmli SDS-PAGE and immunoblotting with the indicated antibodies. Open triangles indicate unknown crosslinked products. (D) Chaperone-like activities of the BepA edge strand mutants. Cells of SN56 carrying pSTD689 or a pSTD689-bepA plasmid were grown at 30°C in M9-based medium until early log phase, induced with 1 mM IPTG for 15 min, pulse-labeled with 35S-Met for 1 min and chased for the indicated periods. At each time point, total cellular proteins were acid-precipitated, subjected to IP with an anti-LptD antibody, and analyzed by 7.5 % Laemmli SDS-PAGE followed by phosphorimaging. The ratio of the band intensities of LptDNC to that of total LptD (LptDC + LptDNC) at the each time point was quantitated and the mean values were plotted with S.D. (n = 2).
Here, we investigated the mechanism by which BepA established interaction with LptD in promotion of its assembly and degradation. Our results suggest that a conserved ²-strand (“edge strand”) located adjacent to the BepA active site directly contacts with LptD and plays important roles in not only proteolysis but also in assembly promotion of LptD. Crosslinking experiments demonstrated that BepA could interact with an LptD molecule assembling on the BAM complex. Based on these observations, we propose a model explaining the functions of BepA in LptD assembly.
Results
Interaction of the BepA edge strand with LptD is crucial not only for proteolysis but also for assembly promotion of LptD by BepA
Zinc-metallopeptidases generally possess a β-strand, called edge strand, located close to their proteolytic active sites (28, 29). The edge strand directly interacts with a substrate polypeptide and converts it into an extended conformation for its presentation to the active site and proteolysis. The solved structures of Escherichia coli BepA (24, 25) show that it has a β-strand (β2) that is conserved among the M48-peptidases and is located adjacent to the active site (Fig.1A and Fig. S1), suggesting that this strand presumably acts as an edge strand. To examine the role of the β2 strand in BepA functions, we constructed BepA mutants by introducing Pro at each position in β2 (from Asn-105 to Phe-110; Fig. 1A). Cells defective in BepA functions exhibit elevated sensitivity to high-molecular-mass antibiotics including erythromycin (EM) (20, 30). This phenotype was suppressed by the expression of wild-type BepA but not by several Pro mutants (N105P, A106P, F107P, and A108P), even though these mutants accumulated at a level comparable to that of wild-type BepA (Fig. S2A). These results indicate that the β2 strand is functionally important.
We then investigated the effects of β2 mutations on the proteolytic activity of BepA against overproduced LptD. When LptD is overproduced from a multi-copy plasmid, it mainly accumulates in the form of LptDC possibly due to the limited availability of its partner protein, LptE (26, 27). This species probably represents a “normal” assembly intermediate as it is associated with the BAM complex (See below) and can be converted to the mature form (LptDNC) when LptE is co-expressed (31). As reported previously (27, 32), overproduced LptD was degraded by co-expressed wild-type BepA to generate discrete degradation products (Fig. 1B). We found that the expression of a few BepA mutants (N105P, A106P, F107P, and A108P) led to a significantly decreased generation of the LptD degradation products (Fig. 1B). Furthermore, some of these mutations compromised the degradation of BamA in a ΔsurA strain (26) and the self-cleavage of BepA-His10 (BepA possessing a C-terminal His10-tag) within the His10-tag (20) (Fig. S3A and B). These results strongly suggest that the β2 strand is important for the proteolytic activity of BepA.
We next analyzed whether the β2 strand directly interacts with a substrate by using a site-directed in vivo photo-crosslinking approach (33, 34). We expressed derivatives of BepA-His10 harboring a photoreactive amino acid analog (pBPA) at each position in the β2 strand in a ΔbepA strain and examined their complementation activity regarding EM sensitivity of the cell and self-cleavage of BepA-His10. The results showed that certain mutants (F107pBPA, F109pBPA, and F110pBPA) were as functional as the wild-type, but others exhibited neither significant complementation activity nor self-cleavage (Fig. S44 and B). For the photo-crosslinking experiments conducted in this study, we used BepA variants harboring the E137Q mutation in the H136ExxH motif for two reasons. First, this mutation would repress possible degradation of LptD by the BepA derivatives. Second, our previous study has shown that this mutation stabilizes the interaction of BepA with LptD (26). Following UV irradiation of cells expressing each of the BepA mutants, BepA-His10 and its crosslinked products were purified from the membrane fractions by affinity isolation using Ni-NTA agarose and subjected to immunoblotting analysis (Fig. 1C). The N105pBPA, A106 pBPA, F107pBPA, and A108pBPA derivatives of BepA generated, in a UV-dependent manner, evident crosslinked products that reacted with both anti-BepA and anti-LptD antibodies. Taken together, these results imply that the β2 strand of BepA directly interacts with LptD and plays an important role in their degradation. The structure, intramolecular disposition, as well as involvement in substrate interaction and proteolysis of the β2 strand strongly supports the hypothesis that it indeed acts as the edge strand to recognize and present a substrate to the active site. We have thus referred to the β2 strand as “the edge strand” hereafter. The differential effects of the mutations on the degradation of LptD and BamA and the self-cleavage of the C-terminal tag (Fig. 1B and Fig. S3A and B) might reflect their different interaction properties and/or affinities for the BepA edge strand. Our previous study also suggested substrate-specific interaction of an edges strand for another metallopeptidase, RseP (29).
BepA not only degrades the stalled or misassembled LptD (when LptD is on the normal (on-) pathway) but also promotes the maturation of LptD through facilitating its assembly with the partner protein LptE (chaperone-like activity) (on the off-pathway) (20). Therefore, we examined the effects of the edge strand Pro mutations on the chaperone-like activity of BepA. LptD possesses two intramolecular disulfide bonds. While the mature (fully assembled) LptD has non-consecutive disulfide bonds, an assembly intermediate form of LptD (LptDC) that accumulates in the absence of functional BepA possesses consecutive disulfide bonds. Consistent with our previous results (20), expression of the wild-type BepA in a ΔbepA strain markedly decreased the accumulation of LptDC. On the contrary, only partial decrease in accumulation was observed with the expression of the E137Q mutant. We found that the A106P mutant was defective in reducing LptDC accumulation, suggesting that a mutation in the edge strand could affect the chaperone-like function (Fig. S2A). Subsequently, we examined the effects of two edge strand mutations—A106P and F1107P—that markedly compromised LptD degradation on the chaperone-like function of BepA using pulse-chase experiments. Expression of the wild-type BepA in ΔbepA cells significantly accelerated the conversion of LptDC to LptDNC, whereas expression of the protease-dead E137Q mutant demonstrated weakened conversion (Fig. 1D). The acceleration of the LptDC to LptDNC conversion by the F107P mutant was weaker than that by the wild-type BepA (almost the same or slightly stronger as compared with the E137Q mutant), and no detectable acceleration was observed with the A106P mutant (Fig. 1D). These results unexpectedly revealed that the edge strand was also important for the chaperone-like activity of BepA.
BepA derivatives with a Cys substitution (note that BepA intrinsically does not possess a Cys residue), instead of a Pro substitution, at the positions of Asn-105, Ala-106, or Phe-107 in the β2 strand exhibited almost normal chaperone-like and proteolytic functions (Fig. S2B and C). This suggests that the secondary structure of the edge strand, but not the individual amino acid residues, is important for its function.
The LptD assembly intermediate interacts with the N-terminal half of the BepA β-barrel-forming domain
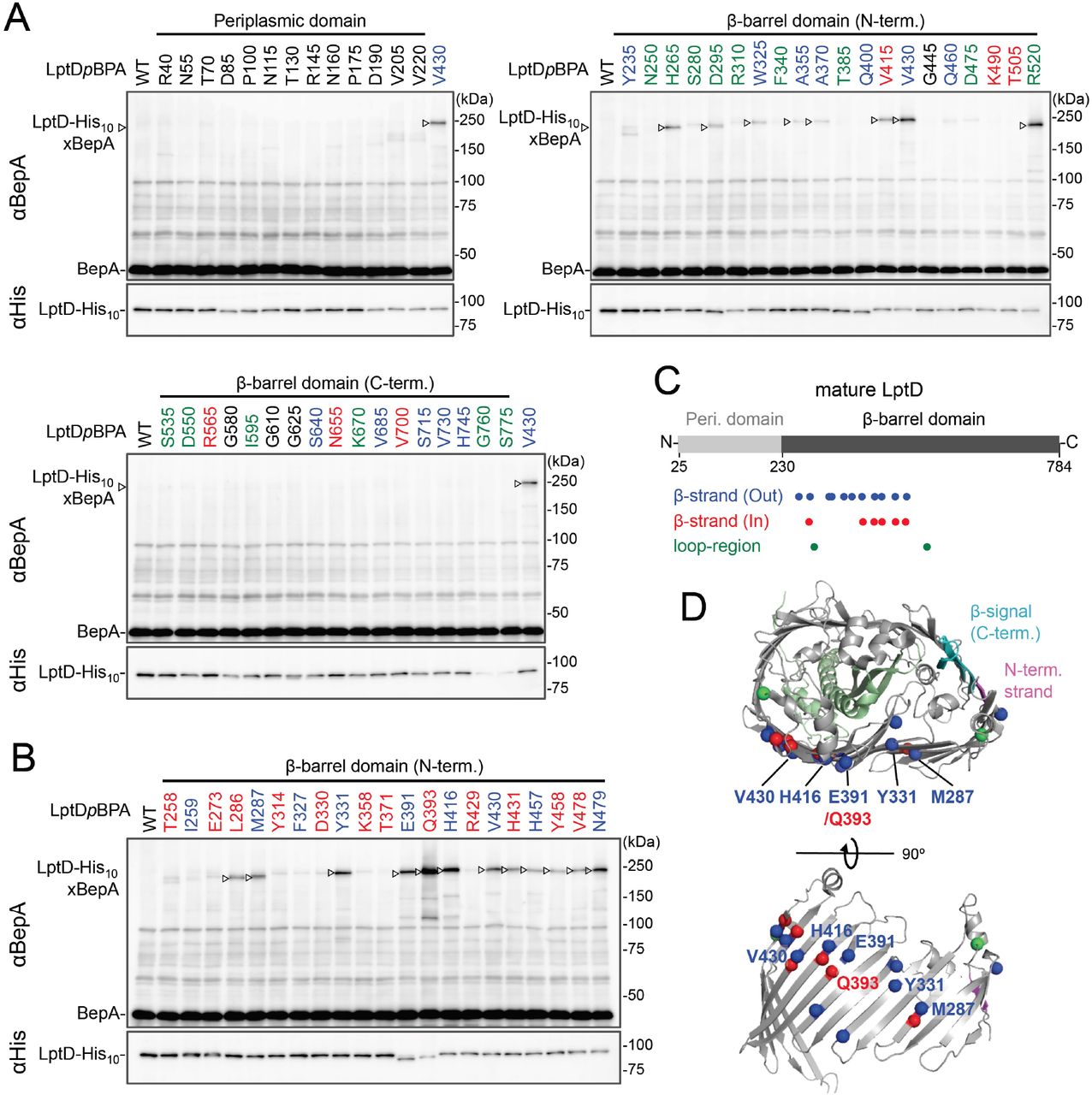
While BepA interacts with LptD to promote either its proper OM assembly or proteolytic elimination depending on the situation (20), the details of the BepA-LptD interaction, including the region(s) in LptD to which BepA binds, remain largely unknown. Thus, we performed a systematic photo-crosslinking analysis to identify the BepA-contact region in the LptD assembly intermediate LptDC. We performed photo-crosslinking experiments in cells ectopically co-expressing an LptD derivative containing a photo-reactive amino acid analog pBPA [LptD(pBPA)] and a protease-dead variant of BepA, BepA(E137Q). We first introduced pBPA at each of the 50 positions (approximately every 15 residues) in the mature part of LptD and performed photo-crosslinking analysis. Cells expressing LptD(pBPA)-His10 and BepA(E137Q) were grown and UV-irradiated, and the total cellular proteins were analyzed by immunoblotting with anti-BepA and anti-His antibodies. Under this condition, expressed LptD-His10 was considerably accumulated as LptDC (Fig. S5) irrespective of co-expression of BepA(E137Q). We detected clear crosslinking with BepA mainly in the N-terminal half of the LptD β-barrel-forming domain (Fig. 2A). We then performed a detailed photo-crosslinking analysis for the 20 additional sites in the N-terminal half of the LptD β-barrel-forming domain (Fig. 2B) and found that BepA was crosslinked at several of these sites. The residues at the BepA-cross-linkable sites were oriented both inward and outward in the mature LptD β-barrel (Fig. 2C and D). Moreover, the residue Gln-393 at which the strongest crosslinking was observed was oriented inward. These results suggest that LptD while interacting with BepA would not assume a higher order structure like closed β-barrel (See Discussion). We selected a few LptD(pBPA) derivatives that had been crosslinked with BepA as representatives and examined their functionality. They supported the growth of LptD-depleted cells when expressed from a plasmid, indicating that they were functional (Fig. S6). The above-mentioned crosslinking results thus likelyreflect a functional interaction of LptD with BepA in the normal assembly pathway.

(A, B) In vivo photo-crosslinking between LptD and BepA. Cells of RM2243 (bepA(E137Q)) carrying pEVOL-pBpF, pMW118-bepA(E137Q, mut) and pRM294-lptD(amb)-his10 plasmids were grown at 30°C in L medium containing 0.02% and 0.5 mM pBPA until early log phase and induced with 1 mM IPTG for 3 h to express the indicated LptD(pBPA) variants. The cultures were then divided into two portions, each of which was treated with or without UV-irradiation for 10 min at 4°C. Total cellular proteins were acid-precipitated and analyzed by 7.5% Laemmli SDS-PAGE and immunoblotting with the indicated antibodies. Amino acid residues shown in red and blue indicate the ones whose side chain is pointing inward and outward, respectively. Amino acid residues shown in green indicate the ones located in the loop regions. (C) Summary of the BepA crosslinked positions in LptD. Positions where the crosslinking with BepA was clearly and reproducibly detected are indicated by colored dots. (D) Mapping of the BepA crosslinked positions on the barrel domain of LptD in the E. coli LptD–LptE structure (PDB code: 4RHB). LptD and LptE are shown in gray and light green, respectively. The N-terminal strand and the β-signal (C-terminal region) in the LptD β-barrel domain are shown in magenta and light blue, respectively. The top view of the LptD/E structure from extracellular space (upper) and the side view of the N-terminal region of LptD β-domain (lower) are shown. The positions where the crosslinking with BepA was observed were indicated by spheres colored as above.
BepA edge strand directly interacts with the Tyr-331 residue in the β7 strand of the LptD β-barrel domain
We investigated further to identify the region of LptD that interacts with the BepA edge strand. First, we examined the effects of the BepA edge strand Pro mutations (F107P and A106P) on the LptD(pBPA-BepA crosslinking. The F107P mutation significantly decreased the efficiency of the crosslinking of BepA(N105pBPA) and BepA(A106pBPA) with LptD (Fig. S7). Additionally, the A106P mutation exhibited a similar effect on the crosslinking of BepA(N105pBPA) with LptD. Based on these effects on crosslinking, we inferred that these mutations affected the interaction of the edge strand with LptD. Subsequently, we selected several LptD(pBPA) derivatives that showed relatively strong crosslinking with BepA and examined the effect of F107P and A106P mutations in the BepA edge strand on the crosslinking of LptD(pBPA) with BepA. We found that these mutations altered LptD-BepA crosslinking in a site-specific manner. Further, the amount of crosslinked products markedly decreased for LptD(Y331pBPA), but not for other mutants (Fig. 3A). These results strongly suggest that the region around Tyr-331 in the β7 strand of the LptD β-barrel domain (Fig. 2D) is crosslinked with the edge strand of BepA.

(A) Effect of the BepA edge strand mutations on the crosslinking between BepA and the LptD derivatives having pBPA in the N-terminal half region of the LptD ²-barrel-forming domain. Cells of SN56 (ΔbepA) carrying pEVOL-pBpF, pMW118-bepA(E137Q, mut) and pRM294-lptD(amb)-his10 were grown, induced to express a BepA and a LptDpBPA derivative and subjected to photo-crosslinking analysis as described in Fig. 2. (B) Disulfide crosslinking between the Cys residues in the edge strand of BepA and the N-terminal half region of the LptD β-barrel-forming domain. Cells of SN56 (ΔbepA) carrying a combination of plasmids encoding WT or a Cys-introduced mutant of BepA and LptD-His10 as indicated were grown in L-medium and induced with 1 mM IPTG for 3 h to express BepA(Cys) and LptD(Cys)-His10. Total cellular proteins were acid-precipitated, solubilized with SDS-buffer containing NEM (for blocking free thiol groups) and subjected to pull-down with Ni-NTA agarose. The purified proteins were treated with or without 2-mercaptoethanol (ME) and analyzed by 7.5% Laemmli SDS-PAGE and immunoblotting with the indicated antibodies.
To further confirm the direct interaction of the BepA edge strand with the LptD β7 strand, we conducted site-specific disulfide crosslinking experiments. For this analysis, we used the above-described single Cys derivatives of BepA harboring a Cys residue at the position of Asn-105, Ala-106, or Phe-107, and derivatives of LptD having a Cys substitution at either of the six positions, including Tyr-331 at which introduction of pBPA showed clear crosslinking with BepA (Fig. 3A). The wild-type LptD protein harbors intrinsic four Cys residues that form two disulfide bonds; therefore, each of these Cys-substituted LptD mutants possessed five Cys residues in total. We confirmed that these Cys-substituted LptD derivatives accumulated normally and retained their function (Fig. S84 and B). Cells expressing a combination of BepA Cys mutants and LptD Cys mutants were grown, and total proteins were acid-denatured and dissolved in SDS containing N-ethylmaleimide (NEM; NEM was included to block free Cys residues). Then, LptD-His10 and its crosslinked products were affinity-isolated using the C-terminal His10-tag, treated with or without 2-mercaptoethanol (ME), and analyzed by SDS-PAGE and anti-BepA immunoblotting. We observed that certain combinations of BepA and LptD derivatives showed a high-molecular-mass band in electrophoresis results. Among them, thecombination of BepA(N105C) and LptD(Y331C) showed the most intense band that exhibited reaction with the anti-BepA antibody (Fig. 3B and Fig. S8B, no ME). These high-molecular-mass bands were not observed with the wild-type LptD (no additional Cys) and disappeared upon treatment with ME, suggesting that they were disulfide-crosslinked products (Fig. 3B and Fig. S8B, + ME). These results indicate that the edge strand of BepA can directly bind to several regions in the N-terminal half of the LptD β-barrel-forming domain, which includes the β7 strand containing Tyr-331.
BepA interacts with an LptD intermediate associating with the seam region of BamA on the BAM complex
We further investigated the mode of the interaction of the BepA-associated LptD with the BAM complex. It has been recently shown that an assembly intermediate of LptD (LptD4213) interacts with the seam region formed by the N- and C-terminal β-strands (β1 and β16, respectively) in the BamA barrel domain, in which the C-terminal β-signal of the LptD4213was associated with the β1 strand of the BamA seam (35). We first examined the interaction of the BepA-associated LptD intermediate with BamA and BamD by conducting in vivo photo-crosslinking experiments using the LptD derivatives with pBPA at the position of Glu-749 or Tyr-726 in addition to the position of Tyr-331. pBPA at Glu-749 and Tyr-726 residue, both of which are located near the β-signal of LptD, have been reported to be crosslinked with BamA and BamD, respectively, during the LptD assembly (Fig. 4D) (35, 36). Complementation assay results showed that either of the LptD derivatives containing one or two pBPA at Glu-749 and Tyr-726 were functional (Fig. S9). After UV-irradiation of the cells expressing LptD-His10 and the pBPA derivatives, LptD-His10 and its crosslinked products were affinity-isolated from the membrane fractions and analyzed by immunoblotting. The single pBPA derivatives indeed generated crosslinked products with the expected factors as: LptD(Y331pBPA) with BepA, LptD(D749pBPA) with BamA, and LptD(Y726pBPA) with BamD (Fig. 4A and B). With the double pBPA derivatives LptD(Y331/D749pBPA) and LptD(Y331/Y726pBPA), new crosslinked products with higher molecular sizes were observed in addition to the ones observed with single pBPA mutants (Fig. 4A and B). These results showed that the higher molecular-sized products represented BepA-LptD(Y331/D749pBPA)-BamA and BepA-LptD(Y331/Y726pBPA)-BamD crosslinked products.

(A, B) In vivo photo-crosslinking of an LptD mutant having pBPA at two positions with BepA. Cells of RM2243 (bepA(E137Q)) carrying pEVOL-pBpF, pMW118-bepA(E137Q) and pRM294-lptD(amb)-his10 plasmids were grown at 30°C in L medium containing 0.5 mM pBPA until early log phase and induced with 1 mM IPTG for 3 h to express the indicated LptD(pBPA) variants. The cultures were divided into two portions, each of which was treated with or without UV-irradiation for 30 min at 4°C. Proteins of the total membrane fractions were subjected to pull-down with Ni-NTA agarose. Purified proteins were analyzed by 7.5% Laemmli SDS-PAGE by immunoblotting with the indicated antibodies. (C) Simultaneous crosslinking of LptD having Y331pBPA and E733C with the BepA edge strand and the seam region of BamA(S439C). Cells of RM3655(bamA(S439C), ΔbepA)/pEVOL-pBpF/pMW118-bepA(E137Q) carrying pRM294-lptD(E733C)-his10 or pRM294-lptD(Y331amb, E733C)-his10 were grown and induced as in A. After treatment with or without BMB and the following quenching of BMB by addition of excess cysteine, the cultures were divided into two portions, each of which was treated with or without UV-irradiation for 10 min at 4°C. Total cellular proteins were acid-precipitated, solubilized with SDS-buffer containing NEM and subjected to pull-down with Ni-NTA agarose. Purified proteins were analyzed by 7.5 % Laemmli SDS-PAGE and immunoblotting with the indicated antibodies. (D) A schematic cartoon of the interaction of the LptD assembly intermediate with BepA and BamA/D on the BAM complex.
We further conducted photo-crosslinking experiments using LptD locked on the BAM complex by using an SH-crosslinker, 1,4-bismaleimidobutane (BMB). It has been previously shown that a cysteine placed near the N-terminal β-strand of the BamA β-barrel (S439C) was crosslinked with a cysteine introduced near the β-signal of LptD (E733C) via BMB treatment (Fig. 4D; Fig. S10A) (35). We introduced a Y331pBPA mutation into LptD(E733C) and confirmed that the resultant mutant was functional (Fig. S10B). When the LptD(Y331pBPA/E733C) mutant was expressed in a strain having a chromosomal bamA(S439C) mutant gene, LptD-BepA crosslinked products were detected upon UV irradiation, whereas LptD-BamA crosslinked products was detected upon BMB treatment. When cells were first treated with BMB and then UV-irradiated, a higher mass product that reacted with both anti-BepA and anti-BamA antibodies was generated. Generation of this product depended on both BMB treatment and UV irradiation (Fig. 4C). These results demonstrated that BepA could interact with an LptD assembly intermediate associating with the seam site of BamA on the BAM complex.
Discussion
The involvement of BepA in the maintenance of structural and functional integrity of the OM was first suggested on the basis of increased sensitivities of the DbepA(yfgC) strain to several antibiotics and chemicals, which was similar to the characteristics of strains with a disruption of genes encoding proteins engaged in outer membrane biogenesis (20, 37, 38). We have previously shown that BepA is involved in the biogenesis and quality control of LptD, possibly on the BAM complex (20, 26, 27). However, the mechanism by which BepA recognizes and interacts with LptD remains elusive. This information is important to understand the mechanism of BepA to distinguish between the normal (on-) and off-pathway intermediates of LptD that are either assembled into the OM or degraded, respectively. To gain insight into the BepA-LptD interaction, we examined the role of the conserved edge strand of BepA in its function. Our results showed that the BepA edge strand participates not only in the proteolytic activity but also in the chaperone-like activity through its direct interaction with LptD (Fig. 1). The results of the photo- and disulfide-crosslinking analyses indicated that the N-terminal half of the LptD β-barrel-forming domain interacts with the BepA edge strand (Fig. 2 and 3). Additionally, we showed that BepA demonstrated interaction with an LptD assembly intermediate whose C-terminal region was associated with the seam strand of BamA on the BAM complex (Fig. 4). These observations provide useful insights into the BepA functions involved in the biogenesis and quality control of LptD.
Our previous results have shown that BepA interacts with the BAM complex via its C-terminal TPR domain partly inserted into the periplasmic ring-like structure of the BAM complex (24, 32). In this study, we observed that BepA showed crosslinking with the N-terminal half of the LptD β-barrel-forming domain but not with the C-terminal half. This observation was consistent with the localization of LptD on the BAM complex; the N-terminal region was localized at/near the periplasmic surface of the BAM complex, and the C-terminal region was inserted deep into the BAM complex’s interior. The results of the Cys crosslinking experiments showed that several positions, including Tyr-331, in the N-terminal half of the LptD β-barrel-forming domain interacted with the edge strand. The recently solved structures of BepA (24, 25) showed that the active site region including the edge strand was located inside the BepA molecule, leading to the suggestion that structural changes in BepA including the movement of the α6 and α9 loops covering the active site/edge strand were necessary to enable access of a substrate to the active site/edge strand region (Fig. S11). However, even after such structural changes, the edge strand should be located at a recessed position. This suggests that the regions around the BepA-crosslinked positions in the LptD assembly intermediate do not form an extensive ²-sheet structure as found in the mature LptD to gain access to the edge strand of BepA (Fig. S11). Furthermore, pBPA at positions of both the inwardpointing and outward-pointing residues in the mature ²-barrel domain of LptD was crosslinked with BepA. Collectively, these results support the hypothesis that the BepA-interacting region of the LptD intermediate is largely unstructured. The unstructured nature of these proteins is in fact helpful to accommodate in or around the narrow space inside the BAM complex. The TPR domain of BepA has also been shown to contacts with LptD to promote its biogenesis and degradation (26). Currently, we have no information available on the part of LptD that interacts with the TPR domain. The TPR domain may act either together with the edge strand at the same step or independently at other steps during LptD assembly and degradation.
Further, our results suggest that the proper interaction of LptD with the edge strand of BepA is important for the promotion of its assembly as well as degradation by BepA (Fig. 1 and 5A). This finding was unexpected as it raised a question of how degradation of the normally assembling LptD intermediate can be avoided despite its interaction with the edge strand near the protease active site. In the BepA structures, the conserved His-246 residue is coordinated to a zinc ion in the active site to block the activation of a water molecule necessary for the catalysis of the proteolytic reaction (24, 25). We have recently reported that His-246 acts as a switch to regulate the proteolytic activity of BepA (27). One possible model for the discrimination of LptD intermediates by BepA for assembly or degradation would be as follows (Fig. 5A). In both assembly and degradation pathways of LptD, an unknown signal(s), such as a specific interaction of BepA with the substrate and/or the BAM complex, induces a structural change in BepA (dislocation of the α6 loop covering the active site) to expose its active site and to enable the interaction between the LptD polypeptide and the edge strand, whereas His-246 continues to repress the degradation of LptD by BepA. This interaction is necessary for both proper assembly and degradation of LptD. The transient interaction of normally assembling LptD with the “protease activity-repressed” state of BepA would provide sufficient time for the progression of the maturation processes of LptD (including association with LptE), resulting in the final release of mature LptD from BamA. In contrast, during the prolonged stay of stalled or misassembled LptD molecules, BepA undergoes a further structural change including the movement of the α9 loop, stochastically or induced by a specific signal(s), to remove His-246 from the active site zinc, leading to the degradation of the molecules that cannot be subjected to maturation successfully. Notably, it has been reported that BepA expression can result in the degradation of a mutant form of LptD (LptD4213) that probably mimics a late-state assembly intermediate of LptD (22) and possesses a substantial degree of a higher order (²-barrel-like) structure. The mechanism by which LptD4213 is degraded by BepA remains unclear, but its N-terminal region may interact with BepA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) A schematic cartoon of the substrate recognition by BepA at its active site. See the text for details. (B) An overview of the proposed LptD assembly process and BepA-mediated discrimination of the assembling and stalled LptD species. See the text for details.
BepA possibly facilitates association of LptD with LptE on the BAM complex as the phenotypes caused by the disruption of BepA, including drug sensitivity and retarded disulfide rearrangement in LptD, can be suppressed by the overproduction of LptE (20). We have previously detected two species of LptDC; one is not associated with LptE and observed only in an extremely early phase of its membrane assembly, and the other is associated with LptE and formed at a later phase (31). It is likely that the LptDC molecule that was simultaneously crosslinked with BepA and BamA was not associated with LptE, as it was accumulated in the LptE-limiting condition; therefore, it would represent the former species of LptDC mentioned above. We assume that this LptD intermediate may possess a partially folded structure in which the BamA-associating C-terminal region of the barrel domain has a certain degree of a higher order (²-sheet) structure, but the BepA-associating N-terminal region is largely unstructured. Such a state of LptD may be favorable for the association of LptE with LptD (Fig. 4D). Interestingly, Tyr-331 of LptD that contacst with the edge strand of BepA is located at the end of the LptE-surrounding region of the LptD β-barrel in the mature LptD/E complex (Fig. 2D). The interaction with the edge strand of BepA may pin the partially folded structure of LptD transiently and facilitate the association between LptD and LptE. It might also assist formation of the β-sheet structure in the β barrel forming domain of LptD. Although it is possible that BepA actively promotes the LptD–LptE association by interacting with both LptD and LptE, it is also possible that BepA plays a passive role by maintaining an appropriate structure of LptD for association with LptE without showing direct interaction with LptE.
The biogenesis of LptD has been well studied. LptD has been a focus of OM protein research because it provides important information on its essential cellular function and it can be used as a model for OM protein insertion into the OM by the BAM complex. Based on the results obtained in the previous and current studies, we propose a model of the BepA-assisted biogenesis of LptD (Fig. 5B): (i) After synthesis in the cytoplasm, LptD is translocated to the periplasm through the SecYEG translocon, during or just after which the Cys residues are oxidized by DsbA to form LptDC (19, 20). It is then targeted to the BAM complex with the aid of periplasmic chaperones including SurA (39, 40); (ii) The C-terminal β-signal region of LptD is inserted into the BAM complex possibly through the periplasmic ring-like structure formed by the BamA POTRA domains and the lipoprotein subunits of the BAM complex, and it establishes interaction with BamD and BamA (35); (iii) Although details of the exact juncture at which interaction occurs are unknown, BepA interacts with the largely unstructured N-terminal half of the LptD β-barrel-forming domain whose C-terminal β-signal region is associated with the seam region of BamA. This helps the association of LptE with LptD; (iv) Then, the folding (ß-sheet formation) of the unstructured N-terminal region of the LptD β-barrel domain occurs to form a premature form of LptD with a substantially folded β-barrel domain and LptE within it, like LptD4213 (22, 35); (v) The β-barrel domain of LptD β-barrel is finally released from BamA and closed to form the mature LptD/E complex. The isomerization of two disulfide bonds in LptD (LptDC to LptDNC) should occur at a later step after the association of LptD and LptE (22, 31). The LptD intermediates that are stalled at certain steps in the above-mentioned processes as a result of misfolding are eliminated by the action of several peptidases including DegP (in the periplasm) and BepA /YcaL (on the BAM complex) (23).
Our study reports several new findings on the interaction of BepA with LptD and BamA on the BAM complex where BepA plays a crucial role in the biogenesis and degradation of LptD. Further, we proposed a model explaining the role of BepA in these processes. Nonetheless, there are many questions that warrant further investigation. It would be especially important to elucidate the mechanism by which the substrate gains access to the active site buried inside the BepA molecule and the manner in which the switching of BepA from the state with chaperone-like function to that with protease function occurs. It is possible that there are signals that arise from the BAM complex and/or from (stalled) LptD to induce this structural/functional conversion of BepA. Further study, including structural and biochemical analysis of the BepA-LptD-BAM complex, is necessary to substantiate our model and to elucidate the molecular details of BepA functions. Cell surface proteins are suitable drug targets as they are more easily accessible form the external milieu than cytoplasmic proteins. These studies can provide a basis for the development of new drugs targeted to BepA/LptD/BAM.
Materials and methods
Bacterial strains, plasmids, and media
Escherichia coli K12 strains and plasmids used in this study are listed in Tables S1 and S2, respectively. Details of the strain and plasmid construction and media are described in Supplementary Materials and Methods.
Antibodies
Penta-His HRP conjugate was purchased from QIAGEN (Hilden, Germany). Anti-BepA (20), anti-LptD (20) and anti-BamA (41) antibodies were described previously.
Immunoblotting and pulse-chase analysis
Immunoblotting analysis was carried out essentially according to the previously described procedures (26). Details of the pulse-chase analysis are described in Supplementary Materials and Methods.
In vivo photo-crosslinking analysis
For the experiments in Fig. 2A, B and 3A, cells were grown at 30°C in L medium containing 0.5 mM pBPA and 0.02% arabinose until early log phase and induced with 1 mM IPTG for 3 h. While a half volume of the cell cultures was UV-irradiated for 10 min, the other half was kept on ice as non-UV-irradiated samples, as above. Total cellular proteins were precipitated with 5% TCA, washed with acetone, and solubilized in SDS-sample buffer containing ME. The samples were subjected to SDS-PAGE and immunoblotting analysis.
For the experiments in Fig. 1D and Fig. S7, cells were grown at 30°C in L medium containing 0.5 mM pBPA and 0.02% arabinose until early log phase and induced with 1 mM IPTG for 1 h. A half volume of the cell cultures was put on a petri dish and UV-irradiated at 4ºC for 10 min using B-100AP UV lamp (365 nm; UVP, LLC., Upland, CA), at the distance of 4 cm. The other half was kept on ice as non-UV-irradiated samples. Membrane fractions were prepared by sonical cell disruption and the following ultracentrifugation, and solubilized with SDS-buffer (50 mM Tris-HCl (pH 8.1), 1% SDS, 1 mM EDTA). After 33-fold diluted with Triton-buffer (50 mM Tris-HCl (pH 8.1), 150 mM NaCl, 2% Triton X-100, 0.1 mM EDTA) and clarification, samples were subjected to pulldown with Ni-NTA Agarose (QIAGEN). The isolated proteins were solubilized in SDS-sample buffer (62.5 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 5 mg/mL bromophenol blue) containing ME and analyzed by SDS-PAGE and immunoblotting analysis.
For experiments in Fig. 4A and B, cells were grown at 30°C in L medium containing 0.5 mM pBPA and 0.02% arabinose until early log phase and induced with 1 mM IPTG for 3 h. While a half volume of the cell cultures was UV-irradiated for 10 min, the other half was kept on ice as non-UV-irradiated samples, as above. Membrane fractions were prepared as above and solubilized in SDS-buffer. After dilution with Triton-buffer and clarification, samples were subjected to pull-down with Ni-NTA Agarose. The isolated proteins were solubilized in SDS-sample buffer containing ME and analyzed by SDS-PAGE and immunoblotting analysis.
BMB crosslinking combined with photo-crosslinking
Cells were grown at 30°C in L medium containing 0.5 mM pBPA and 0.02% arabinose until early log phase and induced with 1 mM IPTG for 3 h. BMB crosslinking was performed essentially according to the previously described procedures (35). After quenching the BMB crosslinking, cells were treated with or without UV-irradiation for 30 min at 4°C. Total cellular proteins were precipitated with 5% TCA, washed with acetone, and suspended in SDS-sample buffer containing 12.5 mM NEM for blocking the free Cys residues. After dilution with Triton-buffer and clarification, samples were subjected to pull-down with Ni-NTA Agarose. The isolated proteins were suspended in SDS-sample buffer containing ME and analyzed by SDS-PAGE and immunoblotting analysis.
Author Contributions
R.M. and Y.A. designed research; R.M., T.W. and K.Y. performed research; R.M., T.W. and Y.A. analyzed data; R.M. and Y.A wrote the paper.
Acknowledgments and funding sources
We thank our laboratory members and S. Narita for discussion and helpful advice. This work was supported by the Japan Society for the Promotion of Science KAKENHI Grants 18H06047, 19K21179 and 20K15715 (to R.M.), and 15H01532 and 18H023404 (to Y.A.), and research grants from the Institute for Fermentation Y-2020-02-027 (to R.M.). and from the Nagase Science and Technology Foundation (to Y.A.).
References




