

Abstract
The DNA mismatch repair (MMR) pathway corrects mismatched bases produced during DNA replication and is highly conserved across the tree of life, reflecting its fundamental importance for genome integrity. Loss of function in one or a few MMR genes can lead to increased mutation rates and microsatellite instability, as seen in some human cancers. While loss of MMR genes has been documented in the context of human disease and in hypermutant strains of pathogens, examples of entire species and species lineages that have experienced substantial MMR gene loss are lacking. We examined the genomes of 1,107 species in the fungal phylum Ascomycota for the presence of 52 genes known to be involved in the MMR pathway of fungi. We found that the median ascomycete genome contained 49 / 52 MMR genes. In contrast, four closely related species of obligate plant parasites from the powdery mildew genera Erysiphe and Blumeria, have lost between 6 and 22 MMR genes, including MLH3, EXO1, and DPB11. The lost genes span MMR functions, include genes that are conserved in all other ascomycetes, and loss of function of any of these genes alone has been previously linked to increased mutation rate. Consistent with the hypothesis that loss of these genes impairs MMR pathway function, we found that powdery mildew genomes with high levels of MMR gene loss exhibit increased numbers of monomer repeats, longer microsatellites, accelerated sequence evolution, elevated mutational bias in the A|T direction, and decreased GC content. These results identify a striking example of macroevolutionary loss of multiple MMR pathway genes in a eukaryotic lineage, even though the mutational outcomes of these losses appear to resemble those associated with detrimental MMR dysfunction in other organisms.
Introduction
An ensemble of DNA repair pathways and cell cycle checkpoints are responsible for detecting and repairing DNA damage, ensuring faithful maintenance of the genome (Friedberg et al. 2005; Giglia-Mari et al. 2010). Among DNA repair pathways, the DNA mismatch repair (MMR) pathway is arguably one of the best characterized (Marti et al. 2002). The MMR pathway is responsible for repairing bases that were incorrectly paired during DNA replication via five steps: recognition, incision, removal, re-synthesis, and ligation (Fig. 1) (Hsieh & Zhang 2017; Fukui 2010; Marti et al. 2002). The MMR pathway is highly conserved in both bacteria and eukaryotes; cells that experience reduction or loss of function in this pathway have increased levels of mutation, as seen in cancer and drug-resistant fungal pathogen strains (Fukui 2010; Campbell et al. 2017; Billmyre et al. 2017, 2020; Dos Reis et al. 2019).

The pathway is comprised of five conserved steps: recognition of mispaired bases, incision of the DNA strand, excision of the incorrectly paired bases, resynthesis of the DNA strand, and ligation of the newly synthesized segment to the DNA strand. RFC and PCNA genes are involved with multiple steps in the pathway. We categorized RFC genes as being primarily responsible for resynthesis, though they are also involved in loading proliferating cell nuclear antigen (Surtees & Alani 2004). We categorize proliferating cell nuclear antigen (PCN1, POL30) as a recognition gene, though it has been implicated in early and late steps in the MMR pathway (Lau et al. 2002; Schofield & Hsieh 2003; Umar et al. 1996; Surtees & Alani 2004). In addition, RFC and PCNA can interact with EXO1 to modulate excision directionality (Surtees & Alani 2004).
Although DNA repair genes are generally highly conserved, certain fungal lineages have been reported to have a more limited repertoire of DNA repair genes, particularly within the phylum Ascomycota. For example, budding yeasts (subphylum Saccharomycotina) and fission yeasts (Taphrinomycotina) have fewer DNA repair genes than filamentous fungi (Pezizomycotina) (Milo et al. 2019; Shen et al. 2020). Furthermore, DNA repair genes that were lost from budding yeasts and fission yeasts are more likely to also be lost in filamentous fungi (Milo et al. 2019). One lineage that has endured extensive losses in its repertoire of DNA repair genes is the Hanseniaspora genus of budding yeasts (Steenwyk et al. 2019). Hanseniaspora species have undergone punctuated sequence evolution and have accumulated large numbers of diverse types of substitutions, including those associated with specific gene losses such as UV damage, suggesting that DNA repair is impaired by the high levels of DNA repair gene loss. These findings suggest that DNA repair genes are not universally conserved across fungi and that their loss is compatible with long-term evolutionary survival and diversification of fungal lineages.
One well-established consequence of MMR dysfunction is mutation in microsatellite regions of the genome. Microsatellites are repetitive tracts of DNA, with motifs 1-6 bp long repeated at least five times (Beier et al. 2017). Microsatellites are typically highly polymorphic between individuals and are commonly used as markers in population biology, forensics, paternity testing, and tumor characterization (Richman 2015). Due to their repetitive nature, microsatellites are prone to experiencing polymerase slippage, which is usually corrected by the MMR pathway (Ellegren 2004; Richman 2015). If the MMR pathway does not recognize these errors, as is the case in cancer, microsatellite instability (MSI) can occur (Campbell et al. 2017). MSI is defined by a hypermutable phenotype resulting from a loss of function in the MMR pathway (Boland & Goel 2010). Instability in microsatellites trends towards elongation in these regions, but contraction can also occur (Ellegren 2004).
Beyond increased mutation in microsatellite regions, aberrant function of the MMR pathway is associated with genome-wide signatures of genetic instabilities (Boland & Goel 2010; Billmyre et al. 2017, 2020). MMR mutations have been implicated in the development of hypermutant and ultrahypermutant human cancers, which constitute approximately 15% of human tumors and less than 1% of tumors, respectively (Campbell et al. 2017). Interestingly, very few tumors with low mutation rates contained mutations in the MMR pathway, whereas more than a third of hypermutant tumors and virtually all the ultrahypermutants contained mutations in MMR genes (Campbell et al. 2017). Hypermutant tumors had high levels of MSI suggesting their hypermutant phenotype is due, at least in part, to MMR dysfunction (Campbell et al. 2017). Hypermutation has also been observed in fungal pathogen strains that have lost MMR pathway genes, potentially driving within-host adaptation and the evolution of drug resistance. For example, Rhodes et al. (2017) found that hypermutation caused by mutations in three MMR pathway genes, including MSH2, resulted in a rapid increase in the mutation rate of the human pathogenic fungus Cryptococcus neoformans, contributing to infection relapse. Similarly, Billmyre et al. (2017) sequenced multiple strains of the human pathogenic fungus Cryptococcus deuterogattii (phylum Basidiomycota) and found that a group of strains with mutations in the MSH2 gene experienced higher rates of mutation when compared with closely related strains harboring an intact MSH2 gene. Hypermutation in C. deuterogattii mediated rapid evolution of antifungal drug resistance (Billmyre et al. 2017, 2020).
In contrast to MMR gene loss in the microevolutionary context of genetic or infectious disease, the extent of MMR gene loss across lineages spanning multiple species remains understudied. To determine the macroevolutionary impact of MMR gene conservation and loss, we characterized patterns of MMR gene presence and absence in the fungal phylum Ascomycota (Fig. 2). We found that the MMR pathway was highly conserved across Ascomycota, with the median species having 49 / 52 MMR genes present. However, we found that Blumeria graminis and species in the powdery mildew genus Erysiphe (subphylum Pezizomycotina, class Leotimycetes), a group of obligate plant parasites, had many fewer MMR genes and a faster rate of sequence evolution than their relatives and most other fungal taxa. Specifically, Erysiphe necator has lost 10 MMR genes, Erysiphe pisi has lost 22 MMR genes, Erysiphe pulchra has lost 9 MMR genes, and Blumeria graminis has lost 6 MMR genes (Fig. 3). In contrast, species closely related to Erysiphe and Blumeria have lost only 1 – 3 MMR genes, consistent with the high degree of MMR gene conservation in the rest of the phylum. Evolutionary genomic analyses revealed that MMR gene losses in Erysiphe and Blumeria (hereafter referred to as high loss taxa or HLT) are associated with a proliferation of monomeric runs and elongation of microsatellites of all motif lengths, both of which are hallmarks of MMR pathway dysfunction. Reflecting these losses, Erysiphe and Blumeria genomes also have more pronounced mutational biases and accelerated rates of mutation. These results suggest that obligate plant parasites in the genera Blumeria and Erysiphe have diversified while lacking otherwise highly conserved MMR genes.

MMR genes are generally highly conserved across the phylum. A few model organisms and species of particular interest to medicine and agriculture are labeled as well as a representative species of the faster-evolving Hanseniaspora lineage. Genes are colored according to their function; red is recognition, orange is incision, yellow is excision, green is resynthesis, and purple is ligation. Branches are colored by subphylum; budding yeasts / Saccharomycotina (n = 332 species) are in red, fission yeasts / Taphrinomycotina (n = 14 species) are in purple, and filamentous fungi / Pezizomycotina (n = 761 species) are in green. Taxon names have been omitted from the phylogeny for visualization purposes; the phylogenetic tree with taxon names can be found in Figure S1 and Shen et al. (2020). The inset phylogenetic tree shows the HLT (in blue) and LLT (in black), with the blue box beneath highlighting them.
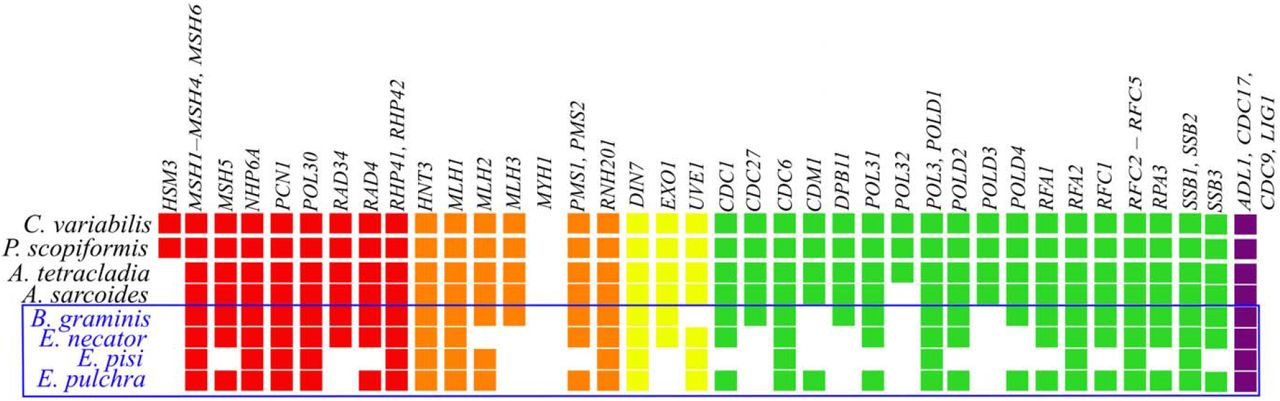
High loss taxa (HLT; shown in blue font) have lost 6 – 22 MMR genes, while low loss taxa (LLT; shown in black font) and most other species in the rest of the Ascomycota phylum have lost 1 – 3 genes. Note that the losses of EXO1, MLH2, MLH3, MSH5, PMS1, PMS2, and RFC1 are uniquely observed in HLT. Genes are colored according to their function; red is recognition, orange is incision, yellow is excision, green is resynthesis, and purple is ligation.
METHODS
Curation of the set of DNA mismatch repair pathway genes
To investigate the presence and absence of MMR genes across the fungal phylum Ascomycota, we curated a dataset of MMR genes from the genomes of three fungal model organisms representing the three different subphyla: Saccharomyces cerevisiae (subphylum Saccharomycotina), Neurospora crassa (Pezizomycotina), and Schizosaccharomyces pombe (Taphrinomycotina). We used three sources to curate genes that are part of the MMR pathway: the Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.genome.jp/kegg/; Kanehisa & Goto, 2000), the Schizosaccharomyces pombe database (PomBase, https://www.pombase.org/; Lock et al., 2019; The Gene Ontology Consortium, 2019), and the Saccharomyces Genome Database (SGD, https://www.yeastgenome.org/; Cherry et al., 2012). All genes in the KEGG diagram of the MMR pathway for each species were included and the gene ontology (GO) term “mismatch repair” was used to search for the genes on SGD and Pombase (Ashburner et al. 2000). We used both computationally and manually curated genes from SGD. We began curating our set of MMR genes in S. cerevisiae, with a total of 30 MMR genes identified with KEGG and SGD. Next, we searched KEGG and Pombase for genes in S. pombe that had not been annotated as part of the MMR pathway in S. cerevisiae (n = 15). We concluded by searching for N. crassa MMR genes in KEGG which had not already been categorized as MMR genes in the other two species (n =7). KEGG listed two sequences for the N. crassa gene LIG1; however, since our sequence similarity search analyses with both sequences yielded identical patterns of loss, we present them as one gene. This approach yielded a total of 52 genes associated with MMR (Table 1).
MMR gene conservation analysis
To examine the conservation of MMR genes across Ascomycota, we implemented a sensitive probabilistic modeling approach using profile Hidden Markov Models (pHMMs) (Johnson et al. 2010) of MMR genes across the genomes of 1,107 species (Shen et al. 2020). To construct pHMMs, we first searched for putative homologs of MMR genes in the fungal RefSeq protein database using the blastp function of BLAST+, v2.8.1, with a bitscore threshold of 50 and an e-value cutoff of 1 × 10−3 (Pearson 2013). We retrieved the top 100 hits using SAMtools, V1.6 (Li et al. 2009) with the ‘faidx’ function. We used MAFFT, v7.402 (Katoh et al. 2002), with the ‘genafpair’ and ‘reordered’ parameters, a maximum of 1000 cycles of iterative refinement, the BLOSUM62 matrix, a gap opening penalty of 1.0, and the retree parameter set to 1, to align the sequences following previously established protocol (Steenwyk, Shen, et al. 2019).We then used the aligned amino acid sequences as input to the ‘hmmbuild’ function in HMMER-3.1B2 to construct each pHMM. We ran the pHMMs of the 52 proteins against all 1,107 proteomes using the ‘hmmsearch’ function. For a gene to be considered present, we set a bitscore threshold of at least 50 and an e-value threshold of less than 1 × 10−6. We used the tblastn function of BLAST+, V2.8.1 with a bitscore threshold of 50, e-value cutoff of 1 × 10−6, and 50% minimum query coverage to verify MMR gene absence using the protein sequence of the gene in question and the 1,107 Ascomycota genomes. We used the Interactive Tree of Life (iTOL), v4 (Letunic & Bork 2019) to visualize the conservation of MMR genes on the Ascomycota phylogeny and to map losses on it.
Microsatellite identification and characterization
To identify microsatellites and evaluate their numbers and lengths between genomes with substantial MMR gene loss against those with higher levels of MMR gene conservation, we used the Microsatellite Identification tool (MISA), v2.0 (Beier et al. 2017). Specifically, we compared the microsatellites of two groups of taxa, each of which contained four species. The group of high loss taxa (HLT) contains the powdery mildews Blumeria graminis, Erysiphe necator, Erysiphe pisi, and Erysiphe pulchra, which show high levels of MMR gene loss relative to other ascomycetes. The group of low loss taxa (LLT) contains four closely related species with low levels of MMR gene loss, similar to patterns seen across the rest of the phylum: Articulospora tetracladia, Ascocoryne sarcoides, Cairneyella variabilis, and Phialocephala scopiformis. The length minimums used for MISA to identify a microsatellite are as follows: 1 base pair (bp) motifs must repeat 12 times, 2 bp motifs must repeat 6 times, 3-6 bp motifs must repeat 5 times. All values used are MISA defaults, except the monomeric parameter, which was increased from the default value of 10 repeats to 12 (Beier et al. 2017; Temnykh et al. 2001). A 2-way ANOVA test was performed to test for significance in the number of microsatellites controlled by genome size of each motif length between HLT and LLT. If the 2-way ANOVA rejected the null hypothesis (α = 0.05), pairwise comparisons were made with the Tukey Honest Significant Differences (HSD) test. We performed the statistical analysis using R, v3.4.1 (https://www.r-project.org/) and made the figures using ggplot2, v3.1.0 (Wickerham 2016), and ggpubfigs, v1.0.0 (Steenwyk 2020).
Estimation of mutational bias and rate of sequence evolution
To characterize the mutational spectra and estimate the rate of sequence evolution between HLT and LLT, we first identified and aligned orthologous sequences across all eight genomes. Orthologous single-copy protein sequences from genes present in all eight genomes (n = 823) were identified using the BUSCO, v4.0.4 (Waterhouse et al. 2018) pipeline and the OrthoDB, v10, Ascomycota database (Creation date: 2019-11-20) (Kriventseva et al. 2019). We hereafter refer to the 823 single-copy genes as BUSCO genes. BUSCO genes were aligned using MAFFT, v7.402 (Katoh et al. 2002), using the same settings described above. Codon-based alignments were generated by threading the corresponding nucleotide sequences onto the protein alignment using ‘thread_dna’ function in PhyKIT, v0.1 (Steenwyk et al. 2020).
To examine patterns of substitutions, we used codon-based alignments to identify nucleotides that differed in a given taxon of interest compared to C. variabilis, which was the sister taxon to a clade comprised of the other seven genomes of interest in the Ascomycota phylogeny. More specifically, we compared the character states for a species of interest to C. variabilis for each site of each alignment, tracking codon position information (i.e., first, second, or third codon position). We also determined if the substitution was a transition or transversion and examined substitution directionality (e.g., A|T to G|C or G|C to A|T) using C. variabilis as the outgroup. These analyses were completed using custom python scripts that utilize functions in Biopython, v1.70 (Cock et al. 2009).
Finally, we used the codon alignments to compare the rate of sequence evolution between HLT and LLT. Specifically, we measured the ratio of the rate of nonsynonymous substitutions to the rate of synonymous substitutions (dN/dS or ω) along the species phylogeny for each gene using the CODEML function in PAML, v4.9 (Yang 2007). For each test, the null hypothesis (Ho) was that all branches had the same ω value (model = 0); the alternative hypothesis (HA) was that all HLT branches, including the branch of their most recent common ancestor, had one ω value and all other branches had a distinct ω value (model = 2). To determine if the alternative hypothesis was a better fit than the null hypothesis (α = 0.05) we used a likelihood ratio test.
Results
MMR genes are highly conserved across the fungal phylum Ascomycota
By examining the presence of 52 MMR genes using a combination of sequence similarity search algorithms across the genomes of 1,107 fungal species, we found that the MMR pathway is highly conserved across Ascomycota (a median of 49 / 52 MMR genes per species; Fig. 2; File S1). Sixteen genes were present in all species; these included five recognition genes (MSH1, MSH2, MSH3, MSH4, and MSH6), one incision gene (MLH1), one removal gene (DIN7), five resynthesis genes (CDC6, RFC2, RFC3, RFC4, and RFC5), and all four ligation genes (ADL1, CDC17, CDC9, and LIG1). Few genes experienced extensive loss. Of the 11 most commonly lost genes, which were lost in >5% of species, two (MYH1 and UVE1) were lost in the common ancestor of Saccharomycotina, in addition to losses observed in other taxa. The remaining nine genes are unevenly distributed across functions; seven are involved in DNA resynthesis (CDC27, CDM1, POL32, POLD3, POLD4, RPA3, and SSB3), one is involved in mismatch recognition (HSM3), one is involved in incision (HNT3). These findings suggest that genes in the MMR pathway are well conserved across Ascomycota.
A comparison of our results with those reported in Milo et al. (2019) revealed similar patterns of gene presence and absence. For example, Milo et al. (2019) found that MYH1 was absent from much of Pezizomycotina, which is consistent with our results. However, we did identify a few differences (inferred losses by Milo et al. (2019) vs. inferred presence in our analyses), which suggest that our pipeline is more conservative in classifying gene losses. We surmise that these differences stem from differences in the gene detection pipelines employed and the divergent objectives of the two studies; Milo et al. (2019) aimed to identify orthologs via a reciprocal best BLAST hit approach, whereas we aimed to identify homologs using pHMMs with absences verified using TBLASTN.
Extensive loss of MMR genes in a lineage of powdery mildews
Although MMR genes are highly conserved across Ascomycota, we found that a lineage of obligate plant parasite powdery mildews have among the fewest MMR genes of the 1,107 Ascomycota species examined. Erysiphe necator has lost 10 MMR genes, Erysiphe pisi 22, and Erysiphe pulchra 9 (Fig. 3). E. necator has been previous documented to have a high rates of genome evolution (Milo et al. 2019) and genomic instability (Jones et al. 2014). Blumeria graminis, which is sister to the Erysiphe genus, has lost 6 MMR genes; previous studies reported extensive gene loss in diverse pathways in this species, generally in genes thought to be unnecessary for their biotrophic lifestyle (Spanu et al. 2010). In contrast, the closely related species Cairneyella variabilis and Phialocephala scopiformis only lack MYH1, an adenine DNA glycosylase that is lost in most filamentous fungi (Chang et al. 2001). In addition to MYH1, closely related species Articulospora tetracladia and Ascocoryne sarcoides lack HSM3, which has been lost in almost all Pezizomycotina genomes. A. sarcoides is also lacking POL32, a DNA polymerase δ subunit, which is part of a larger complex that participates in multiple DNA repair pathways, including nucleotide excision repair and base excision repair (Gerik et al. 1998). Much like the rest of the phylum, genes associated with resynthesis are lost more frequently, but Erysiphe and Blumeria have lost genes associated with all MMR functions (Table 1) except ligation. In addition, seven of the observed MMR gene losses occur nowhere else in Ascomycota: EXO1 (excision), MLH2 (incision), MLH3 (incision), MSH5 (recognition), PMS1 (incision), PMS2 (incision), and RFC1 (resynthesis). Taken together, these results suggest that HLT may have a partially functional MMR pathway.
While select Erysiphe taxa have lost more genes than any other species, there are other species with moderate to high levels of MMR gene loss across the phylum. A total of 186 species have lost 6 or more genes across Ascomycota: 4 species in subphylum Taphrinomycotina, 161 in Saccharomycotina, and 21 in Pezizomycotina. The disproportionate number of Saccharomycotina and Taphrinomycotina species is consistent with our knowledge that organisms in these lineages have, on average, a smaller number of DNA repair genes compared to Pezizomycotina (Milo et al. 2019). MMR gene loss in certain Saccharomycotina lineages, such as in some species from the genera Hanseniaspora (Steenwyk, Opulente, et al. 2019), Tetrapisispora, and Dipodascus, is comparable to the loss observed in HLT; however, only 18 other species in Ascomycota showed MMR gene loss to the same degree as any Erysiphe species. In general, species with elevated levels of gene loss primarily lost genes noted as commonly lost earlier in this paper (see “MMR genes are highly conserved across the fungal phylum Ascomycota”), with occasional losses in other genes.
There was a striking discrepancy between the presence and absence of MMR genes inferred by pHMM versus TBLASTN in HLT that was not observed in other species. When measured by pHMM, E. pulchra lost 42 MMR genes, as opposed to 9 when confirmed with TBLASTN, and E. necator and E. pisi lost 51 MMR genes, as opposed to 10 and 22, respectively. B. graminis lost 9 MMR genes by pHMM and 6 when absences were verified with TBLASTN. In the closely related species C. variabilis, P. scopiformis, A. tetracladia, and A. sarcoides, genes deemed absent by HMM also did not have a match for TBLASTN. This is unexpected, given that pHMMs are more sensitive in sequence similarity searches and typically outperform TBLASTN when detecting genes on an evolutionary timescale (Yoon 2009). Given that the inputs in the pHMM-based searches were gene annotation predictions and the inputs in the TBLASTN-based searches were genome assemblies, it is possible that the genes missed by pHMMs are genes that have not been annotated. Many of the TBLASTN hits are not located near a start codon, suggesting that they may have been pseudogenized.
High MMR gene loss taxa show increased number and length of microsatellites
Examination of microsatellites revealed microsatellite expansions in HLT in comparison to LLT. Specifically, we found statistically significant increases in the number and length of microsatellites in HLT compared to LLT (Fig. 4A, Tables 2, 3, S1, and S2). Overall, after controlling for genome size, HLT had significantly more microsatellites than LLT (F = 34.83; p < 0.001; ANOVA; Tables 3 and S1). This effect was driven by the very large increase in the number of homopolymer runs in Erysiphe and Blumeria (Fig. 4C) (p < 0.001; Tukey HSD; Table S1). There was no statistically significant difference between the groups in the numbers of microsatellites with a 2-6 bp motif length (Table S1). HLT showed significantly higher average microsatellite lengths at every motif size than LLT (Fig. 4B) (p < 0.01 for 1 bp, p < 0.001 for all other motif lengths; Wilcoxon rank sum test; Table S2). HLT have an increased number of monomeric runs (after controlling for genome size) and an increase in length of microsatellites of all motif lengths, suggesting that the MMR pathway’s function is compromised in these species.
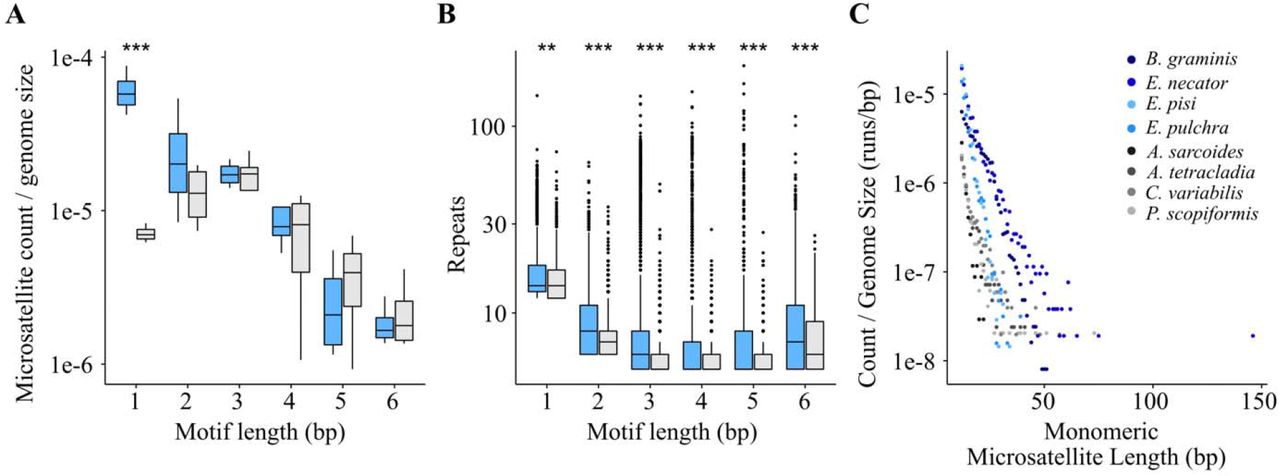
(A) Examination of microsatellites in HLT and LLT (gray bars) showed a significant increase in the number of monomeric runs in HLT (p < 0.001; ANOVA, Tukey HSD; Tables 3 and S1). (B) Microsatellites of each motif length are significantly longer in HLT (p < 0.01 for 1 bp, p < 0.001 for all other motif lengths; Wilcoxon rank sum test; Table S2). (C) Monomeric runs are longer and more numerous in HLT than LLT (Tables S1 and S2).
High loss taxa show mutational biases
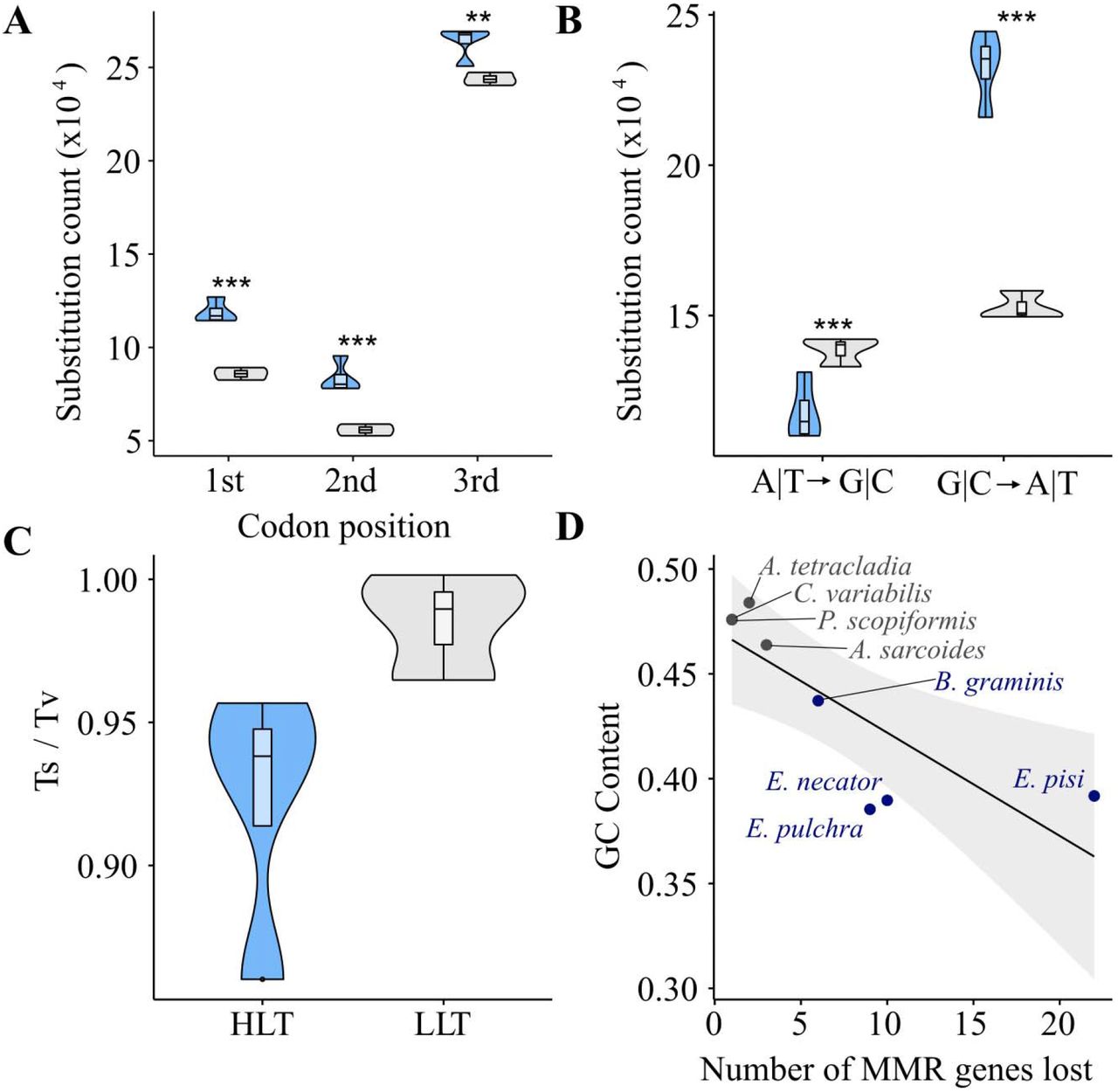
By examining patterns of substitutions among HLT and LLT we found that HLT displayed stronger mutational biases associated with impaired DNA repair pathway function in comparison to LLT. For example, significantly more substitutions were observed at all codon positions in HLT vs. LLT (Fig. 5A) (p < 0.01; Tukey HSD; Table S3) and a significant bias towards substitutions in the A|T direction (Fig. 5B) (p < 0.001; Tukey HSD; Table S4). HLT also had a lower ratio of transitions to transversions (0.92 ± 0.04) than LLT (0.99 ± 0.02), though this is not statistically significant (Fig. 5C) (p = 0.06; Wilcoxon rank sum exact test; Table S5). Additionally, HLT had lower GC content (HLT: 40.10 ± 0.02% vs. LLT: 47.49 ± 0.01%). Linear regression revealed a significant decrease (F = 11.7, p = 0.01; Table S6) in GC content of the genomes as the number of MMR genes lost increases (Fig. 5D).

(A) HLT (blue bars / fonts) show increased counts in base substitution at every codon position when compared to LLT (gray bars / fonts) (p < 0.01; ANOVA, Tukey HSD; Table S3). (B) HLT show significant mutational bias towards mutation in the A|T direction, while this trend is not significant in LLT (p < 0.001; p = 0.27; ANOVA, Tukey HSD; Table S4). (C) HLT show a decreased ratio of transitions to transversions when compared to LLT, though this difference is not statistically significant (p = 0.06; Table S5). (D) Genome GC content decreases with increasing MMR gene loss (adjusted R2 = 0.6045; p = 0.01; linear regression; Table S6).
High loss taxa have experienced accelerated rates of sequence evolution
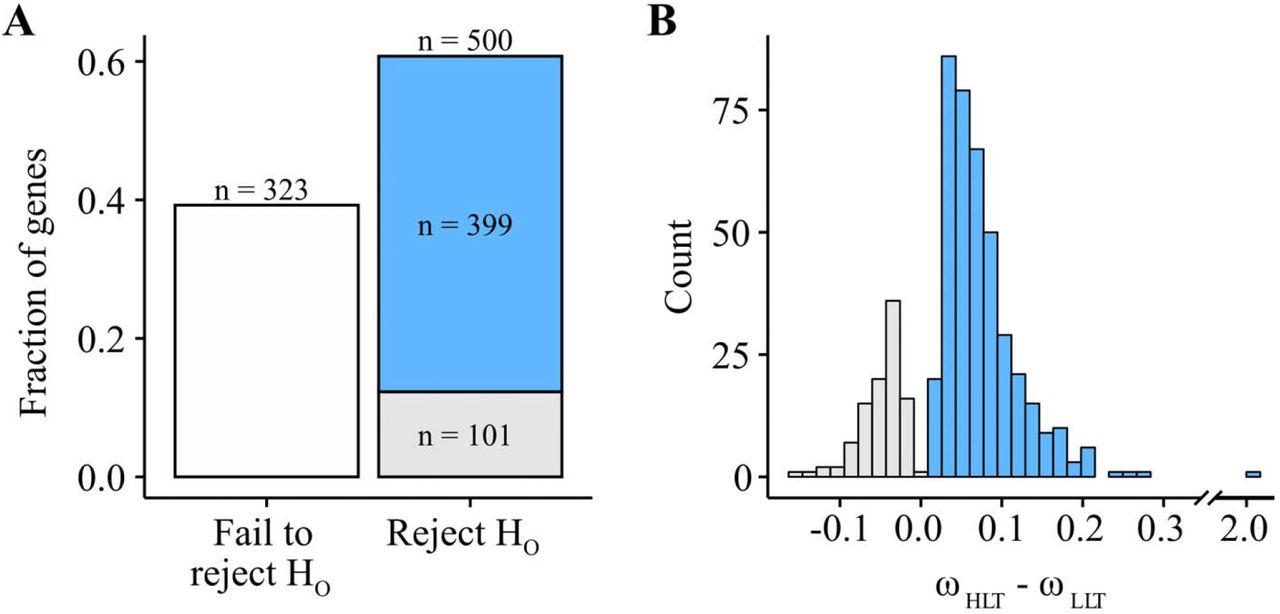
To test if the rate of evolution of HLT differed from that of LLT, we performed ω-based branch tests. Our null hypothesis was that all branches of the phylogeny for our selected eight species had the same rate of evolution, while our alternate hypothesis posited that HLT branches, including the branch of their most recent common ancestor, experienced a different rate of sequence evolution than LLT branches. We found that 60.75% of genes rejected the null hypothesis (α = 0.05; n = 500) and 39.25% failed to reject the null (n = 323) (Fig. 6A; File S2). Of the genes which rejected the null hypothesis, 79.80% (n = 399) experienced higher rates of substitution in HLT, which constitutes 48.48% of all genes tested (Fig. 6A). Among the genes that rejected the null hypothesis, the difference between the ω values for the HLT (median ω = 0.0899) and the LLT (median ω = 0.0567) showed accelerated rates of substitution for HLT branches (Fig. 6B). These results suggest that MMR gene loss is associated with a genome-wide signature of accelerated mutation rates.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Most (60.75%; n = 500) BUSCO genes reject the null hypothesis that HLT branches experience the same rate of substitution as LLT branches. 48.48% of BUSCO genes support a higher rate of evolution for HLT (n = 399; in blue), 12.27% support a higher rate of evolution for LLT (n = 101; in grey), and 39.25% (n = 323) fail to reject the null hypothesis that the rate of substitution is uniform across HLT and LLT branches (in white). Among genes that support the alternative hypothesis, 79.80% (n = 399) support that Erysiphe and Blumeria evolve more quickly than LLT. (B) Among genes which reject the null hypothesis, the distribution of differences between ω values for HLT and LLT branches show elevated substitution rates in HLT.
Discussion
Using sequence similarity searches, we examined the conservation of the MMR pathway across 1,107 ascomycete species. The near universal conservation of the vast majority of MMR genes across the phylum confirms this pathway’s known critical role for DNA maintenance (Fukui 2010; Schofield & Hsieh 2003; Kunkel & Erie 2005). However, we also discovered that a lineage of Erysiphe and Blumeria powdery mildews, named HLT, have experienced significant MMR gene loss (Fig. 3). HLT exhibit increases in monomer count, microsatellite length, mutational biases, and rate of evolution (Figs. 4, 5, and 6), suggesting that the function of their MMR pathway may be impaired. While DNA repair mechanisms are present in all eukaryotes and are highly conserved, there is mounting evidence of exceptions to this rule in the fungal kingdom (Steenwyk, Opulente, et al. 2019; Steenwyk 2021). The increased MMR gene absence observed in HLT correlates with changes in the microsatellite compositions of their genomes. The significant difference between HLT and LLT in the number of monomeric runs is consistent with mutational patterns present in human cancers and MMR deficient yeast (Arzimanoglou et al. 1998; Lang et al. 2013). Monomeric runs are the most prone to replication fork slippage and are used to diagnose MSI in tumors (Richman 2015). In addition to an increase in the number of monomeric runs (Fig. 4A), HLT showed significantly longer microsatellites for each motif length than LLT (Fig. 4B), which suggests impaired MMR function and increased replication fork slippage.
Examination of HLT genomes revealed mutational signatures suggesting that the MMR pathway has been impaired by the observed gene losses. Patterns of substitutions suggest the loss of MMR genes leads to increased substitution rates (Fig. 5A) and lower GC content (Fig. 5D). More specifically, the prominent A|T bias of substitutions in the HLT is likely driven by the known A|T bias of mutations previously observed in bacteria and eukaryotes, including S. cerevisiae (Hershberg & Petrov 2010; Keightley et al. 2009; Zhu et al. 2014; Lynch 2010). Furthermore, GC content decreases in proportion to the number of MMR genes lost in the HLT and LLT, which was also observed among Hanseniaspora, a lineage of budding yeasts that have lost diverse DNA repair genes (Steenwyk, Opulente, et al. 2019). There is no significant difference between the transition to transversion (Ts/Tv) ratios of the HLT (0.92 ± 0.04) and LLT (0.99 ± 0.02), though the trend follows what we would expect for HLT having less efficient DNA repair. Both lineages exhibit near-neutral Ts/Tv ratios (Zhu et al. 2014; Lynch et al. 2008). Examination of ω values suggests that faster rates of sequence evolution in HLT compared to the LLT may be associated with MMR gene loss. Long branches, which reflect more substitutions per site, have been previously reported elsewhere for E. necator (Milo et al. 2019), providing independent support to our findings.
Loss of function in the MMR pathway may be advantageous in certain environments or under certain lifestyles. For example, strains of human pathogens with impaired MMR function are found in environments where antifungal drugs are present. Some of these strains have evolved drug resistance, so the elevated mutation rate generated by MMR gene loss may be adaptive under certain stressful situations (Billmyre et al. 2017, 2020; Rhodes et al. 2017). Loss in separate DNA repair pathways is also present in other parasites and may contribute to elevated mutation rates. Organisms with parasitic lifestyles tend to evolve more rapidly than free-living organisms; while these mechanisms are unknown, previous work has identified that the loss of the classical nonhomologous end joining (C-NHEJ) pathway is common in this lifestyle and may even be a contributing factor (Nenarokova et al. 2019). Previous studies of genome structure in E. necator have found genome expansion largely driven by transposable elements and suggest that genome instability, particularly in copy number variants, can mediate rapid evolution of fungicidal resistance (Jones et al. 2014). The evolution of fungicide resistance in powdery mildews has massive implications for agriculture; major crops are impacted by these pathogens and some are able to quickly evolve resistance to antifungal chemicals, with resistance evolution accelerated by increased use (Jones et al. 2014; Vielba-Fernández et al. 2020). More broadly, genome instability among HLT taxa reflects their parasitic lifestyle, which is associated with gene loss and plastic genomic architecture (Schmidt & Panstruga 2011). Gene loss in primary and secondary metabolism, enzymes acting on carbohydrates, and transporters, has been documented in Blumeria graminis, as well as massive expansion in retrotransposons and genome size, reflecting extreme genomic changes associated with its parasitic lifestyle (Spanu et al. 2010). Given their extreme genomic changes and importance to agriculture, Blumeria and Erysiphe may be novel models to study the outcome and evolutionary trajectory of sustained loss of MMR pathways.
Funding
M.A.P. was partially supported by the Vanderbilt Undergraduate Summer Research Program and the Goldberg Family Immersion Fund. J.L.S. and A.R. were supported by the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study Program. A.R.’s laboratory received additional support from the Burroughs Wellcome Fund, the National Science Foundation (DEB-1442113), and the National Institutes of Health/National Institute of Allergy and Infectious Diseases (R56AI146096). X.-X.S. was supported by the National Natural Science Foundation of China (no. 32071665) and the Young Scholar 1000 Talents Plan.
Data availability statement
Supporting statistical analysis, the Ascomycota phylogeny with species names, and 2 supplementary data files (MMR gene presence/absence matrix and ω output) are available via figshare at https://doi.org/10.6084/m9.figshare.14410994.v1. The data supporting the phylogeny of Ascomycota are available at https://doi.org/10.6084/m9.figshare.12751736.v4.
Acknowledgements
We thank Qianhui Zheng for performing exploratory analysis on the HLT and LLT genomes.
References




