

Abstract
In this study, we developed an updated genome-scale model (GEM) of Pseudomonas aeruginosa PA14 and utilized it to showcase the broad capabilities of the GEM. P. aeruginosa is an opportunistic human pathogen that is one of the leading causes of nosocomial infections in hospital settings. We used both automated and manual approaches to reconstruct and curate the model, and then added strain-specific reactions (e.g., phenazine transport and redox metabolism, cofactor metabolism, carnitine metabolism, oxalate production, etc.) after extensive literature review. We validated and improved the model using a set of gene essentiality and substrate utilization data. This effort led to a highly curated, three-compartment and mass-and-charge balanced BiGG model of PA14 that contains 1511 genes and 2036 reactions. Even with considerable increase in model contents (genes, reactions, and metabolites), compared to the previous model (mPA14) of the same strain, this model (iSD1511) has similar prediction accuracy for gene essentiality and higher accuracy for substrate utilization assay. We assessed iSD1511 using another set of gene essentiality and substrate utilization data and computed the prediction accuracies as high as 92.7% and 93.5%, respectively. The model can simulate growth in both aerobic and anaerobic conditions. Finally, we utilized the model to recapitulate the results of multiple case studies including drug potentiation by citric acid cycle intermediates. Overall, we have built a highly curated computational model of the P. aeruginosa to decipher the metabolic mechanisms of drug resistance, and to help in the development of effective intervention strategies.
Introduction
Pseudomonas aeruginosa is a gram-negative proteobacterium that is metabolically versatile and an opportunistic human pathogen. It is one of the leading causes of nosocomial infections and is involved in multiple infections ranging from benign to fatal [1,2]. One of the well-known P. aeruginosa infections is in the lungs of cystic fibrosis (CF) patients, which can lead to high morbidity and mortality [2]. The ability of P. aeruginosa to infect and colonize host is due to their ability to resist multiple drugs (including aminoglycosides, quinolones and β-lactams), synthesize multiple virulence factors (e.g., proteases, lysins, exotoxins, etc.), and produce and survive in biofilms [1-3].
Pathogenicity of P. aeruginosa can differ between strains, and studies have shown that depending on the strains, the host response can vary widely [1]. P. aeruginosa PA14 is a hypervirulent strain of P. aeruginosa and belongs to the most common clonal group [4]. Its genome was first published in 2004, and this study showed that PA14 genome is highly similar to P. aeruginosa PAO1 genome, but PA14 also consists of two pathogenicity islands [5]. These two genomic islands contribute significantly to the virulence of PA14 [6]. Therefore, investigation of PA14 has gained particular interest in the research community.
Genome-scale metabolic models (GEMs) provide a reliable tool for systems study of bacteria including pathogens [7-9]. For the development of genome-scale models (GEMs), first reconstruction of the metabolic pathways of the organism of interest is required, and then the reconstruction can be converted to a mathematical format which can be analyzed using constraint-based modeling and flux balance analysis approaches. A GEM of Pseudomonas aeruginosa PA14 (mPA14) was published in 2017, and the model was utilized to investigate growth associated virulence factors [10]. A number of missing metabolic capabilities and standardization considerations in mPA14 motivated us to reconstruct an updated model for PA14. For instance, mPA14 does not possess multiple terminal oxidases which are known to be present in P. aeruginosa [11-14]. Likewise, phenazine-dependent reactions (including reactive oxygen species (ROS) production) were not included in the previous reconstruction [15-17]. We also sought to use human-interpretable metabolite and reaction identifiers that are standardized across a large repository of models—in particular, the BiGG standardization and modeling platform [18]. However, mPA14 was not constructed using BiGG identifiers. Therefore, in this study, we developed a BiGG [18] reconstruction of PA14 strain with 382 more genes than in the previous reconstruction. Even with considerable increase in the content, the model has demonstrated higher overall accuracy in predicting substrate utilization, and similar accuracy in gene essentiality predictions. The model can also simulate growth in anaerobic environment. Furthermore, using multiple approaches including flux sampling and flux-sum, we used the model to mechanistically explain the role of fumarate in potentiation of aminoglycoside drug, tobramycin. Overall, this GEM of Pseudomonas aeruginosa can be utilized to advance our knowledge of the metabolism, distinct respiratory capabilities, and drug tolerance mechanisms of P. aeruginosa PA14 considerably.
Methods
Initial Reconstruction
For the initial reconstruction, CarveMe [19] was utilized with added gap-filling function in LB media. From the previous P. aeruginosa model (iPAU1129/mPA14) [10], the biomass reaction was added along with other required reactions to simulate the growth of the model on LB media. For this process, the SEED model was first converted to a BIGG model (mPA14_BIGG). Furthermore, the bounds of non-growth associated reaction were also added from mPA14. Then, all the artificial sink and demand reactions added by CarveMe were removed by making sure that the respective metabolites can be produced or consumed by other reactions which are supported by gene evidence.
Since the initial reconstruction consisted of reactions from the universal reaction database, multiple issues in the Pseudomonas aeruginosa PA14 reactome were fixed by manual intervention. For fixing the gene-protein-reaction (GPR) associations, a custom method was devised. First, eighteen models from BiGG database and their respective protein fasta files were downloaded (details provided in supplementary text). Then, a bidirectional blast hit (BBH) was performed for all the reactions present in the model by prioritizing the strains that are taxonomically closer to PA14 strain. A stringent method was applied in this case such that only top hits in both directions were considered as the correct gene. If not, top hits were manually checked in different databases including IMG [20] and KEGG [21]. These GPRs were then used as alternatives to CarveMe GPRs. Following this, a manual check was performed for reactions whose GPR associations were derived from the BBH approach from models other than those of Pseudomonas putida. Any discrepancy between GPRs from CarveMe and those from BBH approach were checked using knowledge from IMG and/or KEGG and/or mPA14 model. Any reactions that did not have associated GPRs or with no evidence to be present in PA14 were removed in this process. Any unnecessary loops were also removed during this process. The list of removed reactions are in the supplementary table S2. Carbon substrate utilization [10] and gene essentiality [22] were used to iteratively improve and validate the model leading to the modification of biomass reaction and more changes to GPRs. In the end, any genes from mPA14 that were not associated with the model were then added.
Reaction Mass and Charge Balance
For mass and charge balance, metabolite formula and charge were assigned using iJN1462 first, and then iML1515. Next, the remaining metabolites were manually checked using multiple databases including BiGG, MetaNetX, PubChem and ModelSEED to identify the correct formula and charge of the metabolite. Finally, formula and charge of new metabolites whose information could not be found in the aforementioned databases were modified by balancing the reactions that contain only those metabolite as undetermined ones.
Manual Reconstruction
Reactions related to anaerobic metabolism, phenazine-associated metabolism, terminal oxidases and alternative terminal oxidases, thiamine metabolism, nucleotide metabolism, n-alkane metabolism, oxalate production, anaerobic quinone production, rubredoxin-based metabolism, reactive oxygen species (ROS), and Na+-translocating NADH:quinone oxidoreductases were added using annotation in KEGG and/or extensive knowledge from literature. All added reactions are listed in supplementary table S3. This reconstruction effort led to the development of the model, iSD1511.
Flux Balance Analysis
For the simulation of the model, flux balance analysis (FBA) was performed on conditions reflecting the media used in a particular experimental study. The FBA simulations were performed using COBRApy package (v. 0.18.1) [23]. For aerobic simulations, aerobic biomass reaction was used as objective function except when comparing to the anaerobic growth rates. For anaerobic growth predictions, anaerobic biomass reaction was utilized. For biomass objective function, “no growth” was assigned if biomass flux was less than 0.00001. For the computation of ubiquinone yield and growth rate comparison in aerobic and anaerobic environment, parsimonious FBA (pFBA) [24] was applied.
Substrate Utilization Screening and Gene Knockout Study
For the initial carbon source catabolic activity validation, the same dataset that Bartell et al. [10] used to validate mPA14 was utilized. For this assay, iSD1511 was simulated in minimal media containing the substrate being tested and optimized for maximum biomass production. The media composition was provided generously by Papin lab. For the initial gene essentiality validation, the same dataset [22] that Bartell et al. [10] used for mPA14 validation was also applied to iSD1511.
For substrate utilization screening and gene essentiality assessment of iSD1511, data collected from separate experimental studies were applied. For carbon source assay, the dataset by Dunphy et al. [25] containing 190 carbon sources was collected. In the collected dataset, since only general metabolite names were provided, the metabolites whose BiGG identifiers could be determined with high confidence were utilized for prediction. Hence, 131 minimal media containing substrates were compared. For assessing the gene essentiality, dataset from a recent study by Poulsen et al. [26] was used. For glucose minimal media, iron had to be added even though the media used in the study [26] presumably did not contain iron. In this dataset, core essential genes that were identified as essential in five conditions and nine different strains were defined. These core essential genes were analyzed for gene essentiality assessment.
Flux-sum Analysis
Flux-sum analysis [27] was performed on metabolites of interest. Briefly, flux-sum (Φi) for metabolite i can be computed using the following formula,
 where j is the reaction in which metabolite i participates in.
where j is the reaction in which metabolite i participates in.
For computation of yield, the flux sum was either divided by growth rate (for quinone) or substrate flux multiplied by the number of carbon atoms in the substrate (for proton motive force).
Flux Sampling
For determination of the range of possible steady-state fluxes under imposed constraints, a flux sampling approach was applied [28] in the COBRApy package [23]. For sampling, optGpSampler [29] was utilized with 10000 samplings. The growth rate was constrained to 90% of the rate predicted in FBA simulations in the respective media condition. By using the validate method of optGpSampler, only valid samples were kept for further analysis. Furthermore, autocorrelation plots and trace plotting were used for convergence analysis. To compare distributions of reaction fluxes or metabolite flux-sums between two different conditions, rank-sum tests were performed.
Addition of Unknown Reactions
For addition of unknown reactions, literature-derived reactions were used by first checking for mass and charge balance. Then, the reactions were entered in the eQuilibrator program [30] in Python (v. 3.7.7). The parameters were as follows: pH = 7.5, ionic strength = 0.25 M, temperature = 25°C, control of magnesium ion (pMg) = 3. Physiological concentrations (aqueous reactants at 1mM) were assumed for the calculation of Gibbs free energy of transformation (ΔG’m). Reactions whose ΔG’m < 0 were added to the model. These reactions were turned off for aerobic simulations but activated for anaerobic conditions.
Results and Discussion
Initial reconstruction and manual refinement
For the initial reconstruction, CarveMe was utilized to create a draft reconstruction. The draft reconstruction contained 2196 reactions and 1482 genes. Following this, the biomass reaction from the previous SEED model (mPA14_BIGG) [10] was added to the model. The model was gapfilled in LB media in order to produce all the biomass constituents. For this gap-filling process, the reactions from mPA14_BIGG or from other BiGG models were added until the model was able to simulate growth in LB media.
Using a semi-automated approach, eighteen different BiGG models were chosen to perform a bidirectional blast hit for each of the reactions present in the model (more details in supplementary text). Then, the reactions were manually checked to find any discrepancy between the GPRs derived from CarveMe and the semi-automated approach. Any discrepancy was resolved using the knowledge from other databases (e.g., IMG, KEGG). Any reaction that was most likely absent in the organism was removed. A list of reactions removed from the model is provided in the supplementary table S2.
Using carbon substrate essentiality [10] and gene essentiality data [22], more knowledge gaps were iteratively filled using either the annotations or literature-derived information. A list of reactions that were added to the model is provided in the supplementary table S3. GPRs were also fixed using the gene essentiality data. Likewise, biomass reaction was also modified during this process in order to better reflect the gene essentiality data.
Following these changes to the model, any extra genes in the mPA14 were added to the model. This process led to the addition of reactions related to alginate production, rhamnolipid production, pyochelin production, etc. Only 43 genes from mPA14 [10] are missing in iSD1511. The list of the genes and the reasons for not including them are given in the supplementary table S4. Finally, the reactions were checked for mass and charge balance. First, information from well-curated BiGG models (iJN1462 [31] and iML1515 [32]) were applied leading to only 112 metabolites that required additional manual curation. The formula and charges of those metabolites were changed either using information from metabolite databases (BiGG, MetaNetX, PubChem or ModelSeed) or manually by mass-and-charge balancing the reactions.
Literature derived addition of reactions
For this step, knowledge derived from literature was extensively used to make the reconstruction of P. aeruginosa PA14 more strain-specific. The reactions pertaining to multiple terminal oxidases, reactive oxygen species elimination, n-alkane degradation, phenazine-associated metabolism, oxalate production, rubredoxin-based metabolism and Na+-translocating NADH:quinone oxidoreductases were added. After this step, the model possessed 1511 genes which is close to the number of genes in the latest E. coli M-model [32]. The model is hence named iSD1511. It contains 2036 reactions and 1643 metabolites. Furthermore, iSD1511 is a mass-and-charge balanced model with three different compartments.
The reconstructions iSD1511 and mPA14_BIGG were compared, and iSD1511 was found to contain significantly higher number of genes, reactions and metabolites than mPA14 (Figure 1a). iSD1511 contains 425 more unique genes than mPA14. 43 genes from mPA14 were excluded (supplementary table S4). Overall, 382 more genes are present in iSD1511 than in mPA14. Furthermore, when the top KEGG pathways between the two models were compared for gene content, the number of genes in every pathway was higher in iSD1511 than in mPA14 (Figure 1b). The top 20 KEGG pathways of the new metabolic reconstruction were then examined (Figure 1c). The greatest number of genes belong to the KEGG pathway “Metabolic Pathways.” Interestingly, other significant pathways were observed to be associated with metabolism of secondary metabolites, biosynthesis of antibiotics, metabolism in diverse environments indicating a reconstruction of a metabolically versatile organism (Figure 1c).

a. When compared with mPA14, the latest model iSD1511 has considerable increase in the reaction, gene and metabolite content. b. The top KEGG pathways between both reconstructions were compared, and iSD1511 evidently has higher gene content in every top KEGG pathway. c. The top 20 KEGG pathways of iSD1511.
Model validation
For model validation, two tests were performed: 1) substrate utilization and 2) gene essentiality. The model could simulate growth on both LB and SCFM (Synthetic Cystic Fibrosis Media) media. For the utilization of substrate in minimal media, the same dataset [10] that was used for validating mPA14 was applied. Comparing experimental and predicted growth data on different substrates, a 94.3% accuracy was computed for iSD1511. This is a significant improvement from mPA14 model (∼79.3%). When the predicted growth yield on glucose minimal media was compared with that of P. aeruginosa PAO1 [33], it was found to be within <0.01%.
For validation using gene essentiality, previously published data was used [22], and predictions were performed in LB media. When only the genes common between iSD1511 and mPA14 (1086 genes) were compared, iSD1511 had an overall accuracy of 90.6% with recall 64.2% (Figure 2a). Therefore, iSD1511 retaining similar accuracy was able to achieve a ∼8% higher recall than the previous model. When all the genes in iSD1511 were considered, an increase in 1% overall accuracy was achieved (see the supplementary figure S1).

a. The predictions for gene essentiality were made using both iSD1511 and mPA14 models. The dataset used is the same as that used for mPA14 validation [10]. Only common genes were included for this comparison. The overall accuracy of iSD1511 was found to be similar to mPA14 even when 425 more unique genes were present in iSD1511. b. iSD1511 was then used for predicting core essential genes determined in a separate study [26]. The model prediction accuracy was found to be > 90% in three different media conditions indicating a highly predictive model. c. Using iSD1511, carbon substrate utilization was predicted for a new set of data [25]. The prediction accuracy was computed to be 93.5% which is a significant improvement over previous model.
Predictive assessment of iSD1511
Following the validation step, the model was assessed using new sets of data. For the gene essentiality data, 321 core essential genes identified in a recent transposon insertion sequence study were compared with predictions [26]. Since the core essential genes were identified in three different media conditions--LB, SCFM and glucose minimal (M9) media, gene essentiality was computed for all three conditions. For LB, SCFM and glucose minimal media, overall accuracies were 92.7%, 92.6% and 90.5%, respectively. The range of precision was between 52.3% (glucose) and 65.4% (LB) whereas the highest and the lowest recall values were 59.2% (glucose and SCFM) and 58.6% (LB), respectively (Figure 2b).
For substrate utilization assessment, dataset from a study conducted by Dunphy et al. [25] was used. In this assay, the model was simulated for growth on 131 minimal media containing different substrates. Several discrepancies were identified between the previous set of substrate utilization data and the new one leading to eight of the false predictions. Therefore, those cases were removed from the analysis. With 123 substrates, the model was able to simulate growth on substrates with an accuracy of 93.5% (figure 2c).
Anaerobic growth
P. aeruginosa is well-known to utilize nitrate and nitrite for growth in the absence of oxygen as terminal acceptor. When the model was first simulated in anaerobic condition, it was unable to predict any growth. Two main biomass constituents caused this issue: 1) thiamine diphosphate (THMPP) and 2) ubiquinone-9 (UQ9).
For the production of THMPP, the reaction catalyzed by glycine oxidase requires oxygen for the conversion of glycine to iminoglycine. Since no alternative pathway could be identified in P. aeruginosa by literature search or annotation-based methods, this metabolite was removed, and the biomass reaction was adjusted. For UQ9, a recent study [34] has demonstrated that UQ9 can be produced anaerobically by an alternate pathway within the aerobic UQ9 biosynthesis chain. In this pathway, the biosynthetic proteins are shared except for the proteins that catalyze the three reactions associated with oxidation of intermediate metabolites. In anaerobic condition, an alternative oxidizing molecule is involved. Since prephenate was identified as a possible alternative in a recent literature review [35], prephenate-based reactions were added for the aforementioned three reactions. The hypothetical reactions were mass- and charge-balanced, and their ΔG’m were computed using eQuilibrator program to determine the feasible reactions which were then added to the model. These reactions were turned on only for anaerobic simulations.
The model was then able to simulate growth in anaerobic condition in rich media using nitrate and nitrite as terminal acceptors. For comparison between aerobic and anaerobic conditions, pFBA was applied, and anaerobic biomass reaction was used as objective function. The model clearly suggested that the biomass production yield on nitrate and nitrite is lower than that in aerobic condition (Figure 3). Furthermore, the UQ9 production was computed between aerobic and anaerobic (nitrate) conditions to examine whether the model can accurately predict the difference in the amount of cofactor produced in these conditions. For this analysis, the reactions pertaining to ubiquinone-8 (UQ8) were shut down, and the flux-sum was computed for UQ9, and was divided by the computed growth rate. The model was clearly able to predict that the UQ9 production is higher in anaerobic condition than in aerobic condition which has been experimentally demonstrated (Figure 3)[34].
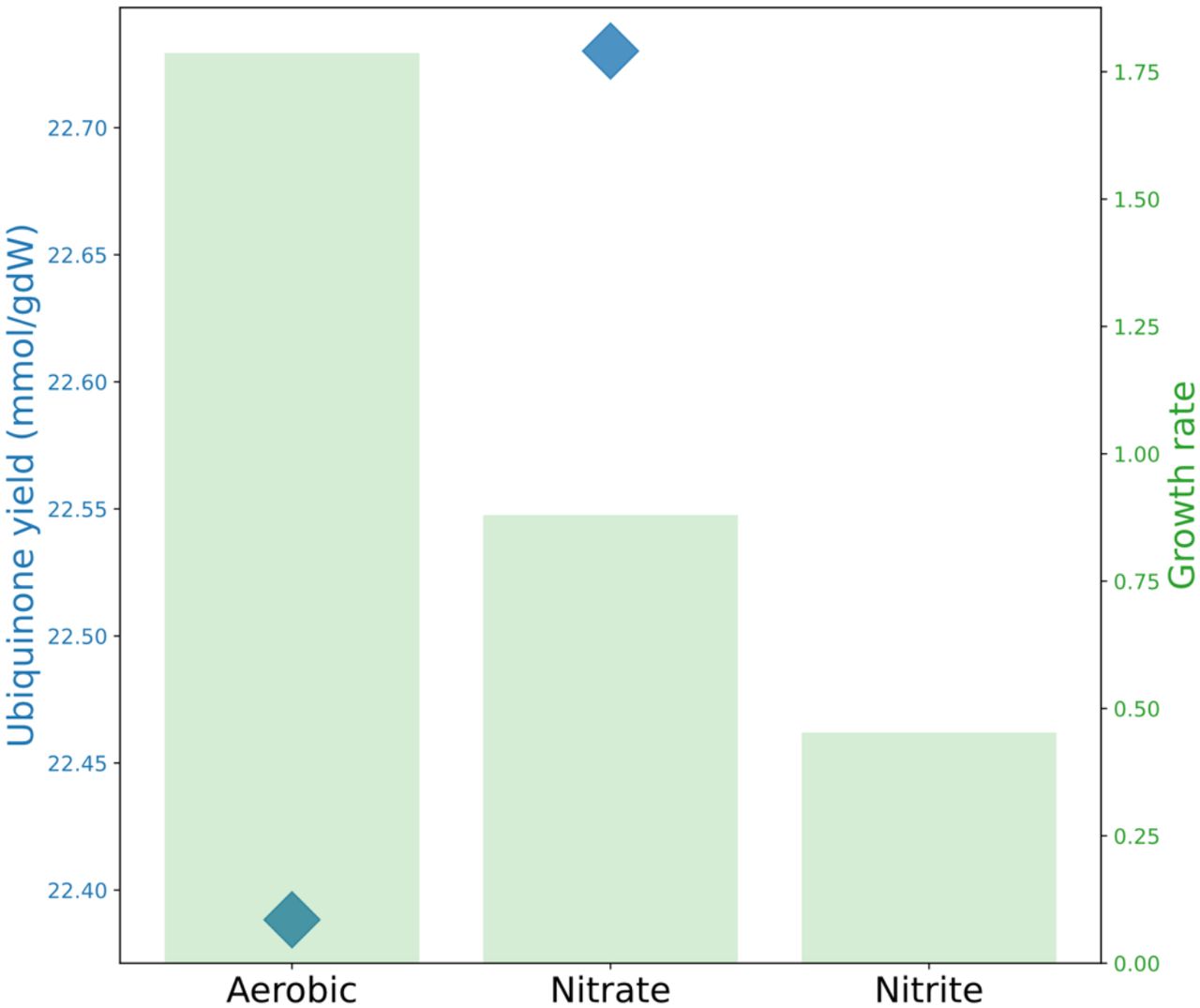
The model simulates growth rate (green bars) in media with different terminal electron acceptors including nitrate and nitrite. Likewise, the model predicts that during growth in anaerobic conditions, ubiquinone (UQ9) production (blue diamonds) increases compared to that in aerobic conditions.
Glyoxylate shunt decreases the TCA flux leading to lower respiration rate and lower proton motive force
Since P. aeruginosa is known for drug tolerance, a study by Meylan et al. [36] showed that the addition of fumarate along with tobramycin leads to greater uptake of the drug whereas glyoxylate protects the cells from tobramycin. The study demonstrated that addition of glyoxylate diverts the flux away from the TCA cycle into the glyoxylate shunt. Furthermore, the authors also proposed that glyoxylate might inhibit the activity of α-ketoglutarate dehydrogenase which could cause the lower flux through lower TCA cycle. The investigators demonstrated that the lower uptake of drug tobramycin in media containing glyoxylate and the drug was possibly due to the lower respiration rate and proton motive force activity as a result of lower flux in the TCA cycle.
For this analysis, iSD1511 was simulated on minimal media containing glyoxylate and fumarate by optimizing for the biomass production. Since glyoxylate was not predicted to be a growth-inducing substrate in the substrate utilization assessment step, an artificial glyoxylate uptake reaction was added to the model. A flux sampling analysis was performed by constraining the biomass flux to 90% of the estimated growth (by FBA) on both fumarate and glyoxylate minimal media containing low amount of citrate. For proton flux calculation, a flux-sum analysis was performed. When the median fluxes of the reactions pertaining to TCA cycle and glyoxylate cycle were compared, the model predictions agreed with Meylan et al. [36], and predicted that the glyoxylate flux shunt indeed drives the flux away from TCA cycle. Likewise, the simulations also indicate that the glyoxylate flux is diverted towards the reactions catalyzed by malic enzymes in order to recycle the necessary cofactors— NADH and NADPH. Unlike the Meylan study, the model predicted higher flux activity through pyruvate dehydrogenase in glyoxylate minimal media. Moreover, the production of oxalate and glycolate were not confirmed by the model predictions. Instead, the glyoxylate flux appeared to be diverted toward the reaction catalyzed by glyoxylate carboligase (Figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Flux sampling analysis was performed in minimal media containing fumarate and glyoxylate to identify the flux distributions of TCA cycle reactions. According to the analysis, TCA cycle is significantly upregulated in fumarate than compared to glyoxylate. The asterisk for reversible succinyl-CoA synthetase reaction is to note that the reaction flux is negative. In glyoxylate, the flux from acetyl-CoA is shunted towards the glyoxylate cycle as shown in the figure. Then, the flux goes through the reactions catalyzed by malic enzymes to regenerate cofactors— NADH and NADPH. This leads to lower TCA flux, decreased oxygen uptake rate and proton motive force. All the sampling results (boxes with flux distributions where green: glyoxylate and blue: fumarate) are for reaction fluxes except for proton for which flux-sum yield was computed from the sampling data. Arrow colors indicate median fluxes being higher in one condition versus the other such that green: glyoxylate, blue: fumarate, black and bold: similar, and black and dotted: low flux in both conditions.
Flux sampling using the model also predicted that the oxygen uptake rate is higher in fumarate than in glyoxylate. Finally, proton motive force between the two conditions was compared based on the flux-sum of periplasmic proton. The median proton flux-sum was higher in fumarate than in glyoxylate (p < 0.05). Overall, the model was able to recapitulate the key metabolic responses thought to underlie the potentiation of tobramycin by metabolite supplementation, and provides means to study other metabolites that can potentiate drug susceptibility.
Conclusion
In this study, we have developed a genome-scale model containing the greatest number of genes for Pseudomonas species in the BiGG database. The reconstruction was carried out using a combination of automated and semi-automated approaches which led to a highly curated and strain-specific model of P. aeruginosa PA14. For the systems research of this pathogen, the developed GEM is a highly predictive and reliable model with accuracies of ∼93% and ∼93.5% for predictions of gene essentiality and substrate utilization, respectively. Likewise, the model is able to simulate growth under multiple environmental conditions, which reflects the metabolic diversity of the strain. Guided by the model simulations, we were able to predict that anaerobic biosynthesis of ubiquinone-9 is required for the growth of PA14, and a recent study [34] corroborated this hypothesis. Finally, the model was able to recapitulate the potentiation of an antibiotic by metabolite supplementation [36]. The model correctly differentiated between metabolites that increased drug uptake versus those that did not, and offered mechanistic explanations for these responses. Namely, supplementing glyoxylate diverted flux away from the TCA cycle, lowering the respiration rate, proton motive force (PMF) that consequently lead to lower drug uptake. Supplementing fumarate had the opposite effect of increasing PMF (and ultimately drug uptake). These results suggest that our model may be used to design antimicrobial strategies based on metabolic mechanisms, including metabolite supplementation, against P. aeruginosa.
Acknowledgements
This work was supported by Queen’s University and the Natural Sciences and Engineering Research Council of Canada (NSERC) [RGPIN-2020-06325].
References




