

ABSTRACT
Primary cilia are microtubule (MT)-based organelles that mediate sensory functions in multiple cell types. Disruption of cilia structure or function leads to a diverse collection of diseases termed ciliopathies (1–3). Mutations in the DUF3719 domain-containing protein FAM149B1 have recently been shown to elongate cilia via unknown mechanisms and result in the ciliopathy Joubert syndrome (4). The highly conserved CCRK and MAK/RCK kinases negatively regulate cilia length and structure in Chlamydomonas, C. elegans, and mammalian cells (5–11). How the activity of this kinase cascade is tuned to precisely regulate cilia architecture is unclear. Here we identify XBX-4, a DUF3719 domain-containing protein related to human FAM149B1, as a novel regulator of the DYF-18 CCRK and DYF-5 MAK kinase pathway in C. elegans. As in dyf-18 and dyf-5 mutants (11), sensory neuron cilia are elongated in xbx-4 mutants and exhibit altered axonemal MT stability. XBX-4 promotes DYF-18 CCRK activity to regulate DYF-5 MAK function and localization. We find that Joubert syndrome-associated mutations in the XBX-4 DUF3719 domain also elongate cilia in C. elegans. Our results identify a new metazoan-specific regulator of this highly conserved kinase pathway, and suggest that FAM149B1 may similarly act via the CCRK/MAK kinase pathway to regulate ciliary homeostasis in humans.
RESULTS and DISCUSSION
XBX-4 acts in the DYF-5 kinase pathway to regulate cilia morphology
The integrity of cilia present on sensory neurons in the head amphid and tail phasmid organs in C. elegans can be readily assessed by the ability of a subset of these neurons to take up lipophilic dye (12, 13). Overexpression of DYF-5 MAK [dyf-5(XS)] severely truncates cilia resulting in a failure of neurons to dye-fill (5) (Figure S1). To identify regulators and effectors of DYF-5 activity, we performed a forward genetic screen and identified mutants that restored dye-filling in dyf-5(XS) animals (see Methods) (Figure 1A). In order to assess whether suppression of the dye-filling defect was correlated with restoration of cilia length in different neuron types in isolated mutants, we examined the cilia morphology of the non dye-filling AWA olfactory neurons. Wild-type AWA cilia exhibit complex and highly arborized morphologies (14, 15) (Figure 1B); these cilia are markedly truncated and lack all branches in dyf-5(XS) animals (11) (Figure 1B-C). We isolated twenty mutants in which both the dye-filling and truncated AWA cilia morphology phenotypes of dyf-5(XS) animals were significantly suppressed (Figure 1A-C, Figure S1).

The XBX-4 DUF3719 containing protein regulates sensory cilia morphology and length.
A)Schematic of forward genetic screen to identify dyf-5(XS) suppressors (5). Dyf: dye-filling defective, nDyf: non dye-filling defective; m: mutant; EMS: ethyl methanesulfonate. Animals carrying suppressor mutations were identified based on restoration of dye-filling and AWA cilia morphology. Also see Figure S1.
B-C)Representative images (left) and quantification of AWA cilia phenotypes (right) in animals of the indicated genotypes. Rescue (B) indicates rescue with an xbx-4p::xbx-4 transgene. The gpa-4Δ6 promoter was used for AWA-specific rescue (C). AWA was visualized using gpa-4Δ6p::myr-gfp. Anterior at top left. ***: different from wild-type at P<0.001; ###: different between indicated values at P<0.001 (Kruskal-Wallis test with Bonferroni post hoc corrections for multiple comparisons). n≥30 neurons each. Also see Figure S2.
D)Structure of the xbx-4 genomic locus. The locations of the ok635 deletion and oy161 nonsense alleles, and the X-box, are indicated. Sequences encoding the DUF3719 domain are shown. Thin yellow bar indicates untranslated sequences.
E)Domains of XBX-4 and the human homologs FAM1491 and FAM149B1. Green arrows: locations of residues in FAM149B1 mutated in Joubert syndrome (4), and corresponding mutations engineered in the XBX-4 DUF3719 domain to generate xbx-4(oy162) and xbx-4(oy163) alleles. Also see Figure S3.
F)Representative images (left) and quantification of AWA cilia morphologies in the indicated genetic backgrounds. ***: different from wild-type at P<0.001 (Kruskal-Wallis test with Bonferroni post hoc corrections for multiple comparisons). n≥30 neurons each.
In all images, arrows: dendrite, yellow and white arrowheads: cilia base and cilia, respectively. Scale bar: 10 μm.
Complementation and targeted Sanger sequencing showed that twelve of these mutations were loss-of-function mutations in the dyf-18 CCRK locus (Figure 1A). We previously showed that loss of dyf-18 restores AWA cilia morphology and dye-filling in dyf-5(XS) animals (11), indicating that DYF-18 is necessary for maximal DYF-5 activity. Seven additional mutants exhibited no dye-filling or AWA cilia morphology defects in the absence of the dyf-5(XS) transgene. These mutations may affect genes regulating dyf-5 promoter activity or dyf-5(XS) transgene expression and were not followed further (Figure 1A). The remaining oy161 mutation complemented dyf-18(ok200) for both suppression of dye-filling and AWA cilia morphology phenotypes (data not shown). While oy161 mutants alone in the absence of the dyf-5(XS) transgene exhibited only weak dye-filling defects (Figure S1), these mutants exhibited severely elongated and largely unbranched AWA cilia, similar to the phenotypes of dyf-5 and dyf-18 loss-of-function mutants (Figure 1B-C) (11).
Single nucleotide polymorphism mapping and whole genome sequencing (16) identified a nonsense mutation in the xbx-4 (X-box promoter element regulated) gene in the oy161 strain (Figure 1D) (17). Similar to oy161 mutants, animals carrying the independently isolated xbx-4(ok635) deletion mutation (17) (Figure 1D) also exhibited elongated and unbranched AWA cilia, weak dye-filling defects, and failed to complement oy161 for this phenotype (Figure 1B-C, Figure S1, and data not shown). The AWA cilia phenotype of oy161 mutants was robustly rescued upon expression of wild-type xbx-4 genomic sequences (Figure 1B-C). Moreover, expression of xbx-4 sequences under the AWA-specific gpa-4Δ6 promoter was also sufficient to rescue the oy161 mutant phenotype in AWA (Figure 1C). We conclude that oy161 is a mutation in the xbx-4 gene, and that this gene acts in the DYF-5 kinase pathway to regulate cilia morphology and length.
In addition to AWA, the cilia of the AWB and AWC amphid sensory neurons exhibit complex morphologies, whereas additional amphid sensory neurons present within a channel formed by glia contain simpler rod-like cilia (channel cilia) (14, 15). While channel cilia are slightly, albeit significantly, elongated in dyf-18 and dyf-5 mutants (5, 11, 18), complex cilia morphologies are more dramatically affected in these mutant backgrounds (11). xbx-4(ok635) mutants also exhibited elongated AWB and AWC cilia (Figure S2A), whereas the rod-like cilia of the ASH neurons exhibited more minor, but statistically significant, elongation (Figure S2B). Together, these results indicate that XBX-4 regulates cilia morphology and length in multiple amphid sensory neuron types.
xbx-4 encodes a DUF3719 domain-containing protein related to the Joubert syndrome-associated protein FAM149B1
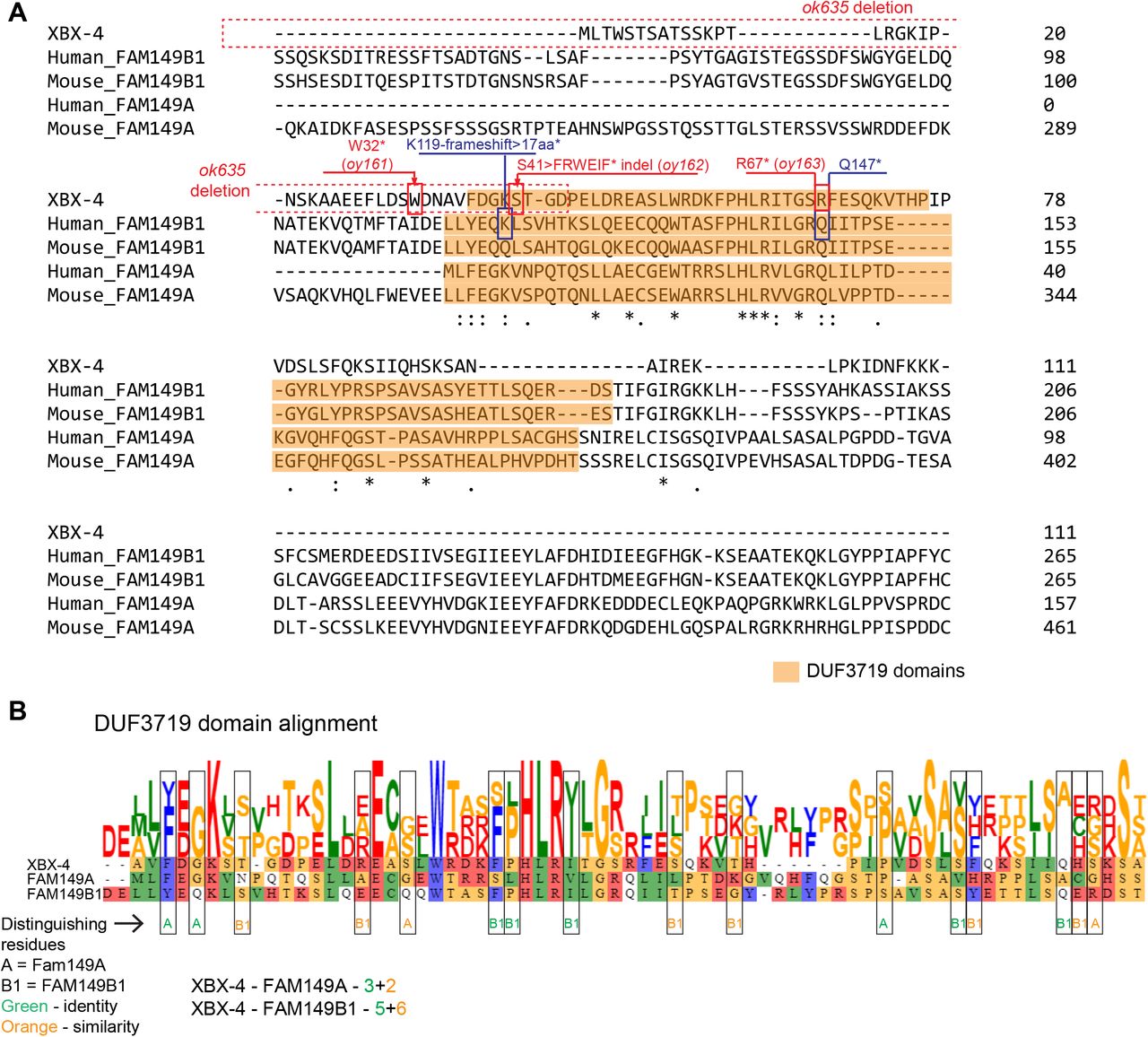
xbx-4 was previously identified in an in silico screen for genes containing the predicted binding site (X-box) for the DAF-19 RFX transcription factor required for ciliogenesis (17, 19). Although xbx-4 expression was shown to be regulated by DAF-19, no obvious ciliary or sensory abnormalities were previously reported in xbx-4 mutant animals (17). xbx-4 encodes a novel small protein of 111 aa containing a conserved metazoan-specific 70 aa domain of unknown function 3719 (DUF3719) (Figure 1D). The oy161 mutation truncates the protein just N-terminal to the predicted DUF3719 domain, whereas the ok635 mutation deletes upstream regulatory sequences including the X-box and part of the DUF3719 domain (Figure 1D, Figure S3A). The DUF3719 domain is present in only two proteins predicted to be encoded by the human genome – FAM149A and FAM149B1 (Figure 1E, Figure S3A). Sequence analysis indicated that the DUF3719 domain of XBX-4 is more closely related to the domain present in FAM149B1 (Figure S3B). However, both human proteins are larger than XBX-4 and contain additional residues outside the DUF3719 domain that are not shared with XBX-4 (Figure 1E, Figure S3A). While the function of FAM149A is unknown, mutations in the DUF3719 domain of FAM149B1 (Figure 1E, Figure S3A) have recently been shown to underlie phenotypes associated with the ciliopathy Joubert syndrome (4). Patient fibroblasts exhibit longer cilia and altered ciliary signaling (4), although the pathway by which FAM149B1 acts to regulate cilia length and function is unknown. These observations suggest a conserved role for the DUF3719 domain in regulating cilia architecture.
Joubert syndrome-associated mutations in FAM149B1 include a 2bp deletion that results in an early frameshift and truncates ~90% of the DUF3719 domain (K119-frameshift > 17aa > STOP), and a nonsense mutation that truncates ~50% of this domain (Q147 > STOP) (Figure 1E, Figure S3A). Since the DUF3719 domain is present at the N-terminus of FAM149B1 (Figure 1E), these mutations also delete C-terminal residues of this protein that are not conserved in XBX-4. We asked whether similar truncations of the DUF3719 domain in XBX-4 also result in ciliary defects in C. elegans. Via gene editing at the endogenous xbx-4 locus, we generated the xbx-4(oy162) allele that is predicted to encode a protein in which the S41 residue is mutated to mimic the early frameshift/truncation mutation in human FAM149B1 (Figure 1E, Figure S3A). We also mutated R67 in XBX-4 (corresponding to Q147 in FAM149B1) to a STOP (oy163), resulting in loss of ~50% of the XBX-4 DUF3719 domain (Figure 1E, Figure S3A). xbx-4(oy162) animals exhibited strong AWA ciliary morphology defects resembling those in xbx-4(ok635) null mutants, whereas xbx-4(oy163) mutants exhibited similar but weaker phenotypes (Figure 1F). We infer that the DUF3719 domain is critical for XBX-4, and likely FAM149B1, function.
XBX-4 regulates axonemal MT stability
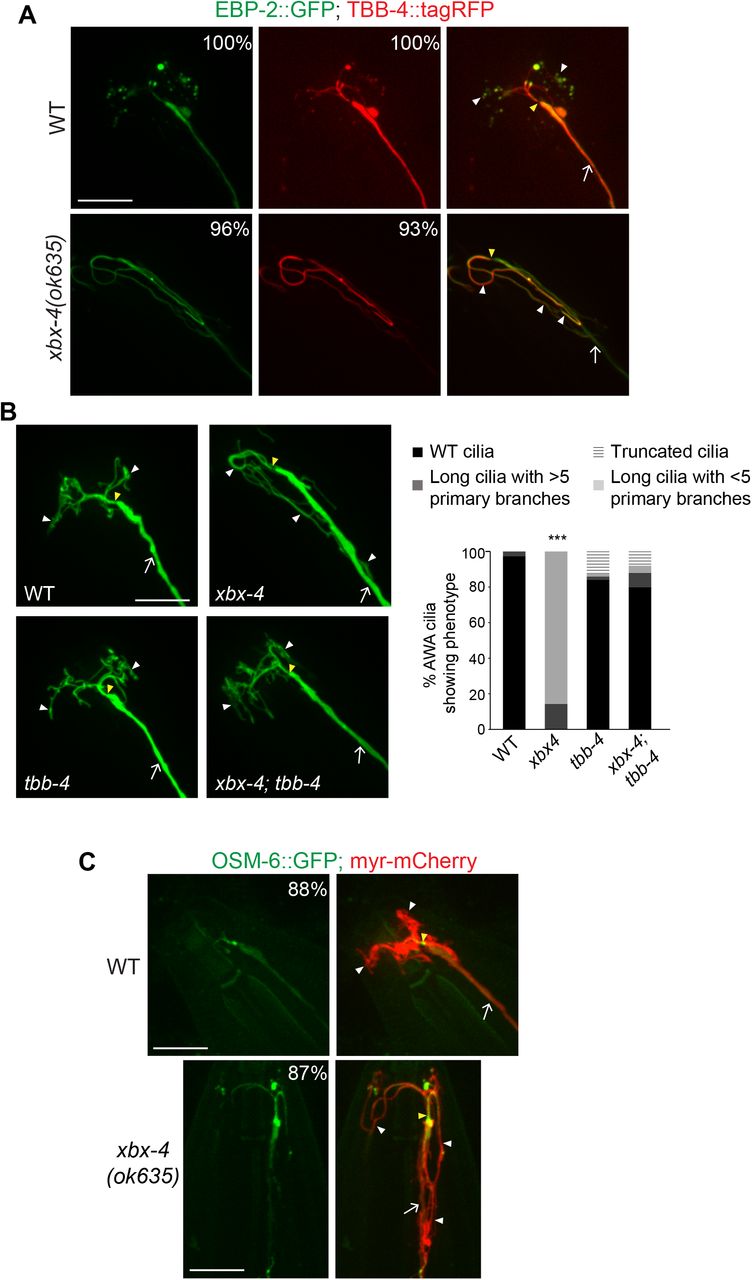
The mechanisms by which FAM149B1 regulates cilia length in humans are unknown. The possible related functions of XBX-4 and FAM149B1 provide an opportunity to use the C. elegans system to characterize the pathways in which these proteins act to regulate cilia structure and function. We previously showed that the elongated AWA cilia phenotype in dyf-18 and dyf-5 mutants arises in part due to increased axonemal MT stability in these mutant backgrounds (11). While the end-binding protein EBP-2 is localized to the primary proximal stalk and distal tips of the branches in wild-type AWA cilia, this protein is present along the entire elongated AWA cilium and not enriched at the distal ciliary endings in dyf-18 and dyf-5 mutants (11). We also showed that destabilizing temperature-sensitive missense mutations in the tbb-4 and tba-5 tubulin genes (20) suppress the elongated cilia and loss of branching phenotype of dyf-18 mutants at 15°C (11). Given the similar ciliary phenotypes of xbx-4, dyf-5 and dyf-18 mutants, we tested whether AWA ciliary axonemal MTs are similarly affected in xbx-4 mutants.
As in dyf-5 and dyf-18 mutants (11), both EBP-2::GFP and TBB-4::tagRFP were localized along the entire AWA axoneme in xbx-4(ok635) mutants (Figure 2A). Moreover, the tbb-4(sa127) missense mutation significantly suppressed the elongated cilia phenotype of xbx-4(ok635) mutants such that a large majority of xbx-4(ok635); tbb-4(sa127) mutants exhibited wildtype-like branched AWA cilia (Figure 2B). As in dyf-18 mutants but distinct from the localization patterns in wild-type animals (11), the ciliary intraflagellar transport B (IFT-B) protein OSM-6::GFP was localized at low levels in the elongated AWA cilium in xbx-4 mutants (Figure 2C). We conclude that similar to our previous observations in dyf-18 and dyf-5 mutants, the elongated AWA cilia phenotype in xbx-4 mutants arises in part due to increased axonemal MT stability.

Axonemal microtubule dynamics may be altered in xbx-4 mutants.
A,C)Representative images of AWA cilia in the indicated genetic backgrounds. Numbers at top right indicate the percentage of neurons showing the phenotype. AWA was visualized using gpa-4Δ6p::myr-mCherry (C). EBP-2::GFP, TBB-4::tagRFP (A) and OSM-6::GFP (C) were expressed under gpa-4Δ6 regulatory sequences. n≥30 neurons each.
B)Representative images (left) and quantification (right) of AWA cilia morphology in the shown genetic backgrounds. Alleles used were xbx-4(ok635) and tbb-4(sa127). Animals were grown at 15°C. AWA was visualized using gpa-4Δ6p::myr-gfp. ***: different from wild-type at P<0.001 (Kruskal-Wallis test with Bonferroni post hoc corrections for multiple comparisons). n≥30 neurons each.
In all images, arrows: dendrite, yellow and white arrowheads: cilia base and cilia, respectively. Anterior at top left. Scale bar: 10 μm.
XBX-4 acts upstream of DYF-18
The similar ciliary phenotypes of xbx-4, dyf-18, and dyf-5 loss of function mutants, together with the observation that mutations in either dyf-18 or xbx-4 suppress the AWA ciliary phenotype of dyf-5(XS) animals to a comparable extent (Figure 1B-C) (11), suggest that XBX-4 acts in, or in parallel to, the DYF-18 and DYF-5 kinase pathway to regulate AWA cilia morphology.
We first tested whether loss of xbx-4 enhances the AWA cilia phenotype of dyf-5 or dyf-18 single mutants. However, the AWA cilia morphology defects of xbx-4 dyf-18 or dyf-5; xbx-4 double mutants were no more severe than those of dyf-18 or dyf-5 single mutants alone (Figure 3A). Moreover, the AWA cilia morphology defects of dyf-5; xbx-4 dyf-18 triple mutants were also not significantly different from those of dyf-5; dyf-18 double mutants (Figure 3A). Although these observations suggest that XBX-4 acts in a linear pathway with DYF-18 and DYF-5, it remains possible that AWA cilia length was not further enhanced in the double and triple mutants due to a ceiling effect.

XBX-4 acts upstream of DYF-18 to regulate DYF-5.
A-C)Representative images (left) and quantification (right) of AWA cilia morphology in the indicated genetic backgrounds. AWA was visualized using gpa-4Δ6p::myr-gfp. Alleles used were dyf-18(ok200) and xbx-4(ok635). KD: kinase-dead. ***: different from wild-type at P<0.001, ###: different from indicated at P<0.001; ns: not significant (Kruskal-Wallis test with Bonferroni post hoc corrections for multiple comparisons). n≥30 neurons each.
In all images, arrows: dendrite, yellow and white arrowheads: cilia base and cilia, respectively. Anterior at top left. Scale bar: 10 μm.
To address this issue, we next examined AWA cilia in dyf-5(XS) animals mutant for both xbx-4 and dyf-18. We previously proposed that DYF-18 is necessary to maximally activate overexpressed DYF-5 and promote cilia truncation (11). In dyf-18 mutants, DYF-5(XS) activity is reduced but not abolished, such that the AWA cilia morphology of dyf-5(XS); dyf-18 double mutants is similar to that in wild-type animals (11). If XBX-4 acts in parallel to DYF-18 to regulate DYF-5, loss of both xbx-4 and dyf-18 together should further reduce DYF-5(XS) activity possibly phenocopying dyf-5(lof) mutants. However, we found that AWA cilia phenotype of the dyf-5(XS); xbx-4 dyf-18 triple mutants was indistinguishable from that of dyf-5(XS); dyf-18(ok200) double mutants (Figure 3B). These results suggest that XBX-4 likely acts upstream or downstream of DYF-18 to regulate DYF-5 activity in AWA. Since CCRK directly phosphorylates MAK in vitro and in HEK293 cells (21, 22), we suggest that XBX-4 acts upstream of DYF-18.
To further test this hypothesis, we asked whether overexpression of DYF-18 is sufficient to suppress the AWA ciliary phenotype of xbx-4(ok635) mutants. Overexpression of dyf-18 in ciliated neurons via the bbs-8 promoter resulted in significantly shorter and more branched AWA cilia in xbx-4 mutants, closely resembling the morphology of wild-type cilia (Figure 3C). In contrast, overexpression of a kinase-dead DYF-18 had little or no effect on the AWA cilia phenotype of xbx-4 mutants (Figure 3C). These results further support the notion that XBX-4 functions upstream of, and regulates, DYF-18 function.
XBX-4 acts via DYF-18 to regulate DYF-5 localization
To begin to investigate how XBX-4 might modulate DYF-18, we first examined the expression pattern and subcellular localization of XBX-4. An xbx-4 cDNA tagged with GFP-encoding sequences at the C-terminus and driven under xbx-4 upstream regulatory sequences was expressed in the majority of ciliated sensory neurons in the head (Figure 4A) (17). An XBX-4::GFP fusion protein expressed specifically in AWA showed diffuse localization throughout the soma and processes (Figure 4A) and robustly rescued the AWA cilia phenotype of xbx-4 mutants (Figure 1C). In AWA, XBX-4::GFP was present throughout the neuron including in the main proximal ciliary stalk with weaker expression in the distal ciliary branches (Figure 4A). This localization pattern resembled that of DYF-18::GFP in AWA, but was distinct from that of GFP::DYF-5 which is primarily enriched in the proximal stalks of AWA cilia (11). A rescuing GFP::XBX-4 protein did not undergo IFT (Figure S4A-B), and localization of this protein appeared unaltered in dyf-18 or dyf-5 mutants (Figure S4C).

XBX-4 regulates DYF-5 localization in AWA.
A)Representative images of XBX-4 localization in ciliated sensory neurons. XBX-4::GFP was expressed under its own promoter or under AWA-specific gpa-4Δ6 regulatory sequences. Images show expression in the entire neuron(s) (left) and in the distal dendrites and cilia (right). Images at left and right are from different animals. Numbers at top right indicate percentage of neurons exhibiting the phenotype. AWA was visualized using gpa-4Δ6p::myr-tagRFP. Anterior at left (left) or at top (right). n≥30 neurons each.
B)Representative images of DYF-18::GFP localization in AWA in WT and xbx-4 mutants. Expression in AWA was driven under the gpa-4Δ6 promoter. AWA was visualized using gpa-4Δ6p::myr-tagRFP. Asterisks indicate autofluorescence in the pharynx. Numbers at top right indicate the percentage of neurons showing the phenotype. Anterior at top left. n≥30 neurons each.
C)Localization of GFP::DYF-5 expressed in AWA in the indicated genetic backgrounds. Brackets indicate the distal tip of the AWA cilium. Numbers at top right indicate the percentage of neurons showing the phenotype. Anterior at top left. n≥30 neurons each.
In all images, arrows: dendrite, yellow and white arrowheads: cilia base and cilia, respectively. Scale bar: 10 μm.
D)Schematic of known regulators and interactors of the CCRK and MAK kinase pathway in different organisms. See text for details and references.
We next tested the hypothesis that XBX-4 regulates DYF-18 localization and/or activity. Loss of xbx-4 had no effect on the level or localization of DYF-18::GFP expressed specifically in AWA (Figure 4B). DYF-18 activity can be assessed by examining localization of the downstream DYF-5 kinase. While GFP::DYF-5 localization is restricted to the proximal stalk of wild-type AWA cilia, this fusion protein is instead present throughout the elongated and unbranched AWA cilia with enrichment at the distal tip in dyf-18 mutants (Figure 4C) (11). We found that the localization pattern of GFP::DYF-5 in AWA cilia in xbx-4 mutants was similar to that in dyf-18 mutants (Figure 4C). Loss of both xbx-4 and dyf-18 together did not further alter GFP::DYF-5 localization (Figure 4C). We infer that XBX-4 regulates DYF-18 activity to control DYF-5 localization and AWA cilia architecture.
In summary, our results identify XBX-4 as a new metazoan-specific regulator of the highly conserved DYF-18 CCRK and DYF-5 MAK kinase cascade implicated in the regulation of cilia length and morphology across species (Figure 4D). Given the related sequences and phenotypic consequences of mutations in the DUF3719 domains of XBX-4 and FAM149B1, our observations raise the possibility that FAM149B1 also acts via the CCRK/MAK pathway to maintain cilia length in humans. Despite the functional conservation of the core CCRK and MAK regulatory pathway in ciliary length control, this pathway appears to interact with, and be regulated by, diverse molecules in different species. For instance, the LF1 and LF3 proteins interact with LF2 CCRK in Chlamydomonas, and lf1 and lf3 mutants also exhibit elongated flagella (8, 23, 24). However, LF1 and LF3 homologs are not present in mammals or C. elegans, and conversely, the Chlamydomonas genome does not encode proteins containing the DUF3719 domain. The FAP20 protein was identified as a CCRK interactor in human interactome studies (25, 26), but this protein appears to have distinct ciliary roles in different species and cilia types (27–29). CCRK also directly interacts with the Broad-minded (BROMI) protein in vertebrates, and BROMI was shown to regulate CCRK stability (30, 31). BROMI in turn interacts with FAM149B1 (4). The Chlamydomonas genome does not encode a Bromi homolog, and mutations in a homolog in C. elegans do not affect cilia (A.K.M. and P.S., unpublished). The concept of interconnected functional modules in cellular networks is well-established (32, 33). We propose that the CCRK and MAK kinases together form a functional module whose output is tuned by diverse regulatory inputs to permit precise shaping of cilia morphologies in a species-, cell-, and context-dependent manner (Figure 4D).
A characteristic of ciliopathies such as Joubert Syndrome is their pleiotropy and variable expressivity across cell and tissue types (2, 34, 35). Specific mutations in ciliary genes affect different cells and tissues to different extents, highlighting the context-specific roles of these conserved proteins. Thus, while mutations in dyf-5, dyf-18 and xbx-4 result in relatively minor effects on cilia length in channel cilia in C. elegans possibly due to anatomical constraints (5, 11, 18) (this work), AWA and other complex wing cilia are dramatically elongated in these mutants (11) (this work). Moreover, while DYF-18 also regulates the localization of the CDKL-1 kinase in amphid channel cilia to influence cilia length (36), AWA cilia morphology is unaffected in cdkl-1 mutants (A.K.M. and P.S., unpublished). C. elegans is now well-established as an excellent experimental system in which to explore the roles of genes implicated in ciliopathies (37–39). Examining the roles of ciliopathy-associated genes across diverse ciliated species and cell types will allow for a more complete understanding of the underlying pathways, and provide insights into how cell type-specific modulation of conserved ciliogenic modules drives structural and functional specialization of cilia in development and disease.
METHODS
Growth of C. elegans
C. elegans strains were grown on E. coli OP50 bacteria using standard procedures. The wild-type strain was C. elegans Bristol N2. Double mutant strains were generated using standard genetic methods, and the presence of the desired mutations was verified by PCR-based genotyping and/or sequencing. Animals were maintained with plentiful food for at least two generations at 20°C before analyses unless indicated otherwise. All strains used in this work are listed in Table S1.
Generation of transgenic strains
DNA constructs were injected at 5-30 ng/μl. unc-122p::gfp or unc-122p::mCherry injected at 15-30 ng/μl were used as coinjection markers. All shown data are from two independent transgenic lines each unless indicated otherwise. Transgenes expressed from the same extrachromosomal array were examined in wild-type and mutant animals.
Isolation of dyf-5(XS) suppressors
dyf-5(XS) suppressors were identified in a forward genetic screen. Animals from the strain PY11440 (Table S1) carrying stably integrated dyf-5(XS) and gpa-4Δ6p::myr-gfp transgenes were mutagenized with EMS using standard procedures, and 8-12 P0 animals were placed on each of 8 NGM growth plates. Subsequently, 4-6 F1 animals from each P0 plate were individually cloned out onto 10 NGM growth plates, and their F2 progeny examined for dye-filling as described below. Animals identified as defective in dye-filling were recovered from the imaging slide and their progeny were examined for their AWA cilia morphology phenotype. Twenty mutants were isolated in which both the dye-filling and AWA cilia morphology defects of dyf-5(XS) were significantly restored.
Cloning of xbx-4(oy161)
The oy161 allele identified in the forward genetic screen was outcrossed to N2 and segregated from the dyf-5(XS) transgene. The unbranched and long AWA cilia phenotype of oy161 mutants was used to map and identify the affected gene. oy161 was mapped to LG I using the EG8040 and EG8041 fluorescent mapping strains (40). To identify the causal mutation, oy161 animals carrying the stably integrated gpa-4Δ6p::myr-gfp transgene were crossed to the polymorphic Hawaiian CB4856 strain. Around 60 F2 progeny of this cross exhibiting elongated and unbranched AWA cilia were identified (homozygous mutants) using a compound fluorescence microscope, genomic DNA was isolated from their pooled progeny, and subjected to Illumina (paired end 150 bp) sequencing (20X coverage, Beijing Genomics Institute) (16).
The density of unique and polymorphic single nucleotide polymorphisms was assessed using online tools (CloudMap) on usegalaxy.org server as previously described (41). The generated chromosomal level frequency plots (bins of 1 Mb and 0.5 Mb) for pure parental alleles showed a single peak of unique variants on chromosome 1. Within the middle of this peak a nonsense mutation in xbx-4 was identified (oy161). An independently obtained deletion allele (ok635; CGC) displayed similar AWA ciliary phenotypes and failed to complement oy161.
Dye-filling
Animals were washed from culture plates into 1.5 ml tubes, and pelleted and resuspended in 100-200 μl of M9 with 10 μg/ml DiI (Life Technologies). Tubes were gently rocked for 2 hrs and animals were subsequently washed with M9 and placed on seeded NGM plates. Dye-filling was assessed after 20 mins under a fluorescent dissecting microscope.
Microscopy
Cilia were imaged by anesthetizing animals with 10 mM tetramisole hydrochloride (Sigma) and mounted on agarose pads (2-10%). Animals were imaged using a 100X objective on a spinning disk confocal microscope (Zeiss Axio Observer with a Yokogawa CSU-22 spinning disk confocal head). Confocal sections were obtained in 0.25 μm steps in the z-axis and maximum intensity projections were obtained using SlideBook 6.0 software (3i, Intelligent Imaging Innovations). To visualize cilia, images were adjusted in ImageJ (NIH) for brightness and contrast.
Generation of xbx-4(oy162) and xbx-4(oy163) alleles
xbx-4(oy162) and xbx-4(oy163) were generated via CRISPR-Cas9-mediated gene editing as described (42). crRNA and homology oligo donors were obtained from Integrated DNA Technologies (IDT). The edited sequences were designed to include EcoRV and NheI restriction sites. Animals were injected with Cas9-crRNA RNP complexes along with the oligo homology donor and a unc-122p::gfp co-injection marker. Animals transgenic for the coinjected marker were isolated and their progeny analyzed for the presence of the desired mutations by sequencing. xbx-4(oy162) crRNA: 5’-TTCGATGGGAAAAGTACTGGGTTTTAGAGCTATGCT-3’ xbx-4(oy162) oligo donor: 5’-GTTTCTGGATTCATGGGATAACGCGGTTTTCGATGGGtAAAGTACTGGTGgtgggtggcttttt ttttgaaaatagtaat-3’ xbx-4(oy163) crRNA: 5’-CTGAGATTCAAAACGGGATCGTTTTAGAGCTATGCT-3’ xbx-4(oy163) oligo donor: 5’-TATGGAGGGACAAATTTCCTCATTTAAGGATTACCGGAagCtagcTTGAATCTCAGAAA GTGACGgtaataagaatttttact-3’
Molecular biology
To generate xbx-4p::xbx-4, xbx-4 coding sequences were amplified from genomic DNA and cloned into the pPD95.77 expression vector along with 1293 bp upstream and 944 bp downstream xbx-4 genomic sequences using Gibson assembly (NEBuilder® HiFi DNA Assembly Master Mix). N- and C-terminal GFP-tagged XBX-4 was generated by inserting GFP coding sequences at the appropriate location using PCR and Gibson cloning. gfp::xbx-4 and xbx-4::gfp sequences were expressed under the xbx-4 upstream or gpa-4Δ6 promoters. The bbs-8p::dyf-18::gfp (PSAB1121) and bbs-8p::dyf-18 (KD)::gfp (PSAB1124) constructs for overexpression have been described previously (11).
Statistical analyses
Statistical analyses were performed using the SPSS 25 statistical analyses package (IBM). Phenotypic categories of cilia were defined as ordinal variables. The nonparametric Kruskal-Wallis test was used for data with non-normal distributions. When necessary, posthoc corrections for multiple comparisons were performed; the test used is indicated in each Figure Legend. Data are shown from at least 2-3 biologically independent experiments. Neither data acquisition nor analyses were blinded. Total numbers of analyzed cilia and significance values are indicated in each Figure Legend.
Supplemental Figure Legends

Mutations in xbx-4 restore dye-filling to dyf-5(XS) mutants.
Percentage of animals exhibiting dye-filling in six head amphid sensory neuron pairs in the indicated strains. nDyf: no dye-filling defect; partial Dyf: dye-filling observed in a subset of neurons; Dyf: no dye uptake; ectopic Dyf: dye uptake observed in additional head neurons (the dendritic bundle also appears unfasciculated in these animals). n≥300 animals each.

XBX-4 regulates length and morphology of multiple sensory cilia.
A)Representative images of the cilia of the indicated sensory neurons in WT and xbx-4(ok635) mutants. AWC, AWB and ASH/ASI were visualized using odr-1p::rfp, str-1p::mCherry and sra-6p::gfp, respectively. Numbers at top right indicate the percentage of neurons showing the phenotype. Anterior at top left. n≥30 neurons each.
B)Quantification of ASH cilium length. Each dot is the length of a single ASH cilium. Horizontal bars indicate 75th, 50th, and 25th percentiles; error bars are 95th and 5th percentiles.

XBX-4 contains a conserved DUF3719 domain.
A)Alignment of C. elegans XBX-4 and human and mouse FAM149A and FAM149B1 protein sequences. The DUF3719 domain in each protein as predicted by Interpro (https://www.ebi.ac.uk/interpro/) is shaded orange. The extent of the ok635 deletion and locations and lesions of xbx-4 mutations are indicated in red. Identified mutations in Joubert syndrome patients (4) are indicated in blue.
B)Alignment of C. elegans and human DUF3719 domains (https://toolkit.tuebingen.mpg.de/tools/clustalo). To assess homology between XBX-4 and either of FAM149A or FAM149B1 DUF3719 domains, residues that are conserved between either XBX-4 and FAM149A, or XBX-4 and FAM149B1 (distinguishing residues) are indicated by boxes and color coded for identities or similarities.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DYF-18 and DYF-5 do not regulate XBX-4 localization.
A)GFP::XBX-4 rescues the AWA cilia morphology phenotype of xbx-4(ok635) mutants. Representative images and quantification of wild-type or xbx-4(ok635) mutants expressing a xbx-4p:: gfp::xbx-4 transgene. AWA was visualized using gpa-4Δ6p::myr-gfp. Anterior at top left. ***: different between indicated at P<0.001 (Kruskal-Wallis test with Bonferroni post hoc corrections for multiple comparisons). n≥30 neurons each.
B)Two representative kymographs of GFP::XBX-4 movement in channel cilia.
C)Representative images of GFP::XBX-4 localization in the indicated genetic backgrounds. GFP::XBX-4 and myr-tagRFP were expressed under the xbx-4 and gpa-4Δ6 promoters, respectively. Numbers at top right indicate the percentage of neurons showing the phenotype. n≥20 neurons each.
In all images, arrows: dendrite, yellow and white arrowheads: cilia base and cilia, respectively. Anterior at top left. Scale bar: 10 μm.
Strains used in this work.
Plasmids used to generate strains in this work.
Acknowledgements
We thank the Caenorhabditis Genetics Center and Shohei Mitani (National BioResource Project, Japan) for strains. We thank the Sengupta lab for advice and Hannah Lawson, Lauren Tereshko, Inna Nechipurenko and Oliver Blacque for comments on the manuscript. This work was funded in part by the NIH (R35 GM122463 – P.S.).
REFERENCES




