

Abstract
Our poor understanding of the mechanism by which the peptide-hormone H2 relaxin activates its G protein coupled receptor, RXFP1 and the related receptor RXFP2, has hindered progress in its therapeutic development. Both receptors possess large ectodomains, which bind H2 relaxin, and contain an N-terminal LDLa module that is essential for receptor signalling and postulated to be a tethered agonist. Here, we show that a conserved motif (GDxxGWxxxF), C-terminal to the LDLa, is critical for receptor activity. Importantly, this motif adopts different structures in RXFP1 and RXFP2, suggesting distinct activation mechanisms. For RXFP1, the motif is flexible, weakly associates with the LDLa, and requires H2 relaxin binding to stabilize an active conformation. Conversely, the GDxxGWxxxF motif in RXFP2 is more closely associated with the LDLa, forming an essential binding interface for H2 relaxin. These differences in the activation mechanism will aid drug development targeting these receptors.
The human relaxin family peptide receptors, RXFP1 and RXFP2, are class A G protein-coupled receptors (GPCR) that are activated by the two peptide hormones of the insulin superfamily, H2 relaxin and INSL3 respectively1. H2 relaxin is also a high affinity ligand of RXFP2 although the physiological significance of this interaction in humans is unknown. The presence of a Leucine Rich Repeat (LRR) domain within the large N-terminal ectodomain further classifies the receptors as a Leucine-rich repeat containing GPCR (LGR) which includes the glycohormone-binding receptors (GHPRs) and R-spondin receptors (LGR4-6)2. An N-terminal low density lipoprotein type A (LDLa) module, connected by a 26- to 32-residue “linker”, further distinguishes RXFP1 and RXFP2 as LGR type C receptors1, 2. The LDLa, which requires a structural calcium3–6, is indispensable for activation of both RXFP1 and RXFP27, suggesting that the LDLa may be a tethered agonist. Using mutagenesis of the LDLa in whole receptor assays, we showed that residues (Leu7, Lys17) in the LDLa N-terminal region of RXFP1 appeared to be responsible for activity8, 9. In contrast, mutation of these residues in the LDLa of RXFP2 had little impact on activity, instead the C-terminal region of the LDLa of RXFP2 may be important for activity10. Nevertheless, these activity losses were modest and while the mutations could not pinpoint critical residues/regions that conclusively supported the role of the LDLa as a tethered agonist, they highlighted possible different mechanisms of activation for RXFP1 and RXFP2.
More recently, we showed that the RXFP1 linker comprises a weak-binding site for H2 relaxin that together with the strong-binding site of the LRR domain is essential for ligand-induced activation11. Indeed, mutations within the region immediately C-terminal to the LDLa (GDxxGWxxxF, conserved in both RXFP1 and RXFP2) slightly reduced H2 relaxin affinity, but in some cases almost abolished activation11. Notably, these critical residues do not appear to directly bind H2 relaxin. Rather, a central transient-helical region of the linker was identified as the ligand-binding site. In NMR experiments, a lack of 1H-1H NOEs suggested no interaction between linker and the LDLa, so we proposed that H2 relaxin binding to the linker promotes a reorientation of the LDLa to fully engage the activated receptor11.
Sequence alignment of RXFP1 and RXFP2 show that the linker of RXFP2 lacks the equivalent H2 relaxin-binding site12. Despite this, H2 relaxin binds strongly to RXFP2, through the LRR with only weak contributions to binding from the C-terminal region of the LDLa, including the GDxxGWxxxF motif12. Mutations of the conserved residues within the GDxxGWxxxF of RXFP2 induced significant losses of both H2 relaxin binding and subsequent receptor activation12. Conversely, INSL3, the cognate ligand for RXFP2, bound normally to the motif-mutant receptor, but receptor activation was completely impaired. Evidently, the linker of these receptors behaves in distinctly different manners depending on both the receptor and ligand involved.
Here we use small angle X-ray scattering (SAXS), NMR spectroscopy, and cell-based receptor assays to show that the LDLa modules and linkers associate differently for the two receptors to form distinct structures. These conformational differences lead to H2 relaxin binding at distinct sites on the LDLa-linker of RXFP1 compared to that of RXFP2. Our results suggest that the conformation of the LDLa-linker and its association with H2 relaxin modulates the structure the GDxxGWxxxF motif, and it is this motif that acts as a tethered agonist, essential for activation of these receptors.
Results
The LDLa module is essential for both RXFP1 and RXFP2
At high concentrations H2 relaxin weakly dimerises (KD ∼10 µM) which can be prevented by amidation of the C-termini of H2 relaxin13. Therefore, we recombinantly expressed and purified 15N-labeled RXFP1 LDLa module and its 32-residue linker (RXFP1(1-72)) and performed a 2D 1H-15N HSQC-monitored titration with amidated H2 relaxin (hereon referred to as relaxin-NH2). Compared to previous titrations with H2 relaxin11 we observed significantly larger chemical shift changes for similar residues on the linker, Asp51-Thr61 (Fig. 1a, Table 1), with a 3-fold stronger affinity (85 ± 10 μM) than H2 relaxin (200 ± 10 µM)11. We also prepared a similar construct of 15N-labelled RXFP2 LDLa module and its 26-residue linker (RXFP2(1-65)) and titrated it against relaxin-NH2. Smaller chemical shift perturbations were observed for residues including Cys26, Asp30 and Glu38, from the C-terminal end of the LDLa, and Asp43-Ile50, from the linker giving an affinity of 205 ± 4 µM (Fig. 1b, Table 1), slightly stronger than previously reported for B5-29 [B-K9R,A-K9/17R] H2 relaxin (330 ± 10 µM)12. These findings support the proposal that relaxin has auxiliary binding sites in the LDLa and linker but involves unique binding modes and residues for each receptor.

Plots of changes in the average 1HN and 15N chemical shifts differences (Δδ ppm) for titrations with relaxin-NH2 of (a) 15N-labelled RXFP1(1–72) and (b) 15N-labelled RXFP2(1–65) in the presence (black circles) and absence (red circles) of 10 mM CaCl2. For comparison in blue circles a similar titration with relaxin-NH2 of (a) 15N-labelled RXFP1(1-40) and (b) 15N-labelled RXFP2(1-41) (with calcium) is shown. Resonances of residues most significantly affected by relaxin-NH2 are labelled. Experiments were conducted at pH 6.8 and 25 °C with 25 μM protein and titrated with 20 equivalents of relaxin-NH2.
Binding affinities determined by saturation binding (Xcrvfit)
The indispensable role of the LDLa in ligand-mediated activation of both RXFP1 and RXFP2 has been demonstrated14. However, the chemical shift changes observed (Fig. 1) on titration with relaxin-NH2 suggest that the LDLa in RXFP2 and not RXFP1, has a role in relaxin binding. As the LDLa modules ligate calcium to stabilize their structures3–6 we investigated the importance of the folded LDLa in relaxin binding by titrating 15N-labelled RXFP1(1-72) and RXFP2(1-65) against relaxin-NH2, but in the absence of calcium. For RXFP1(1-72), similar key residues in the linker, Asp51-Thr61, showed chemical shift perturbations, but with a 4-fold decrease in affinity (Fig. 1a, Table 1). For RXFP2(1-65), no significant chemical shift changes in either the LDLa or linker were observed (Fig. 1b, Table 1) suggesting no binding and demonstrating that the fold of the LDLa is essential for binding of relaxin to RXFP2. To further investigate if the folded LDLa alone presents a binding interface for relaxin, we titrated 15N-labelled RXFP1(1-40) and RXFP2(1-41) (LDLa module alone without linker) with relaxin-NH2 and found that neither showed an interaction (Fig. 1). These data suggest that for RXFP1 the folded LDLa allosterically modulates the affinity of relaxin for the linker; while for RXFP2, the folded LDLa and the linker region Asp43-Ile50 form a structure essential for relaxin binding.
Previously we showed that the LDLa-linker of both RXFP1 and RXFP2 can directly interact with exoloop-1 and -2 (EL1, EL2) of the transmembrane domain (TMD)11, 12, 15. These experiments used peptide mimetics of the exoloops, ssRXFP1 (EL1(475-486)/EL2-RXFP1) and ssRXFP2 (EL1(475-486)/EL2-RXFP2), thus expressing partial exoloop-1 and full-length exoloop-2 with the disulfide that is critical for receptor structure and function16. As these experiments were conducted in the presence of calcium, hence a structured LDLa, we tested the importance of a folded LDLa module, by titrating 15N-labelled RXFP1(1-72) and RXFP2(1-65), in the absence of calcium, with ssRXFP1 and ssRXFP2 and observed a complete lack of interaction (Supplementary Fig. 1). These data suggest that while the linker region contains all the key residues, it requires structured LDLa modules to effectively interact with H2 relaxin and the TMD. Hence for both receptors, RXFP1 and RXFP2, it appears that the LDLa serves as an anchor that structures the linker region and this interaction is critical for receptor activation induced by the binding to H2 relaxin11, 17.
The LDLa-LRR linkers of RXFP1 and RXFP2 adopt distinct conformational ensembles
To understand the structural relationship between the LDLa and the linker, we performed SAXS on RXFP1(1-72) and RXFP2(1-65) (Fig. 2a). For RXFP1(1-72), the radius of gyration (Rg) was 21.06 ± 0.19 Å and the molecular weight as 7.68 kDa (expected M.W. ∼8.29 kDa) (Fig. 2b,c and Supplementary Table 2). For RXFP2(1-65), Rg and molecular weight were measured to be 15.98 ± 0.14 Å and 6.35 kDa (expected M.W. ∼7.09 kDa) respectively. The shapes of RXFP1(1-72) and RXFP2(1-65) were determined ab initio using DAMMIN (Supplementary Fig. 2). To further model different possible conformations of RXFP1(1-72) and RXFP2(1-65) in solution, we used the Ensemble Optimization Method (EOM)18, 19 to generate a pool of different conformations (Fig. 2d). The range of conformations for RXFP1(1-72) is smaller compared to RXFP2(1-65), which shows two distinct pools. Collectively, the SAXS models of RXFP1(1-72) suggest that the linker folds back onto the LDLa, providing a more globular shape, whereas for RXFP2(1-65), the models suggest an elongated linker extending away from the LDLa.
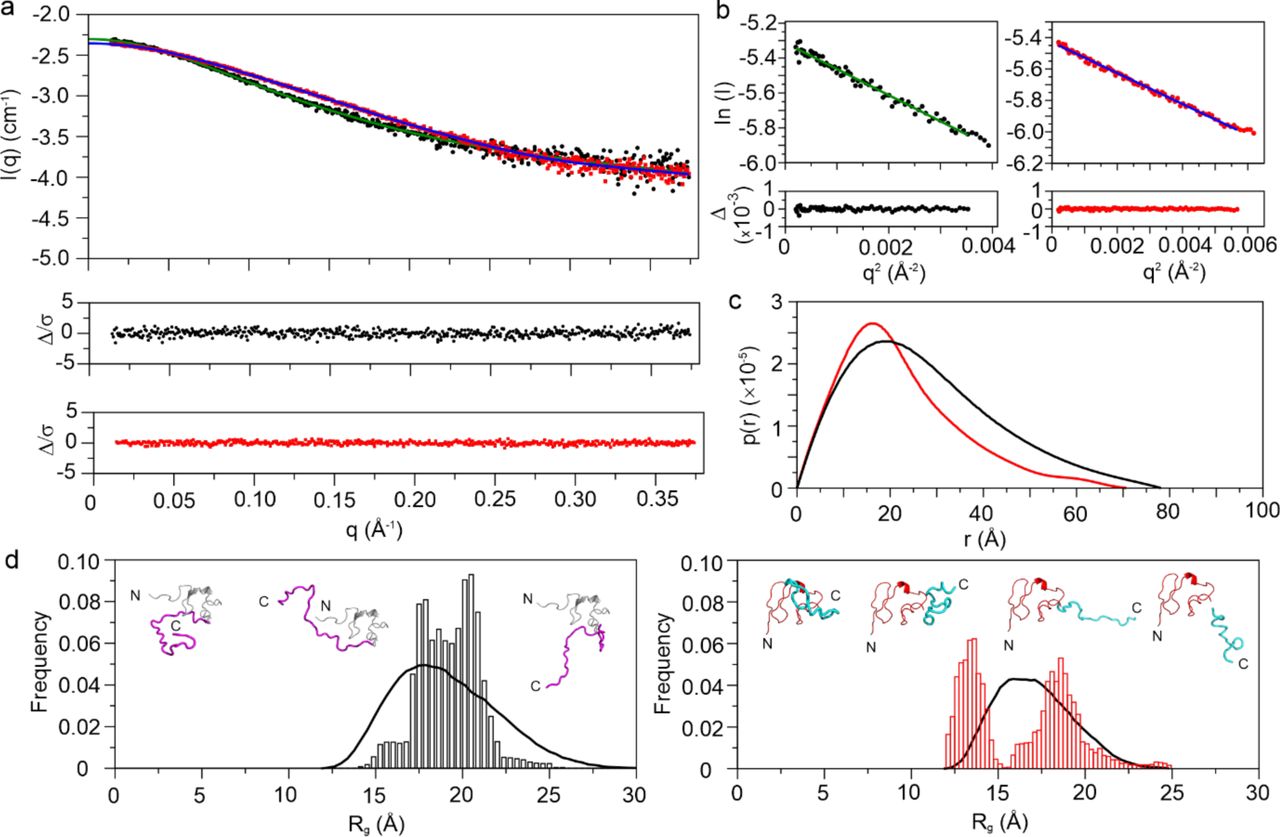
(a) Small Angle X-Ray Scattering data for RXFP1(1-72) (black) and RXFP2(1-65) (red). (b) Guinier Approximation curve for RXFP1(1-72) (black) and RXFP2(1-65) (red). (c) p(r) curve for RXFP1(1-72) (black) and RXFP2(1-65) (red) highlighting the difference in their radius of gyration. (d) EOM modeling distribution along with the models proposed for RXFP1(1-72) (left, LDLa module in grey, linker in magenta) and RXFP2(1-65) (right, LDLa module in red, linker in cyan) showing a bimodal distribution for RXFP2(1-65).
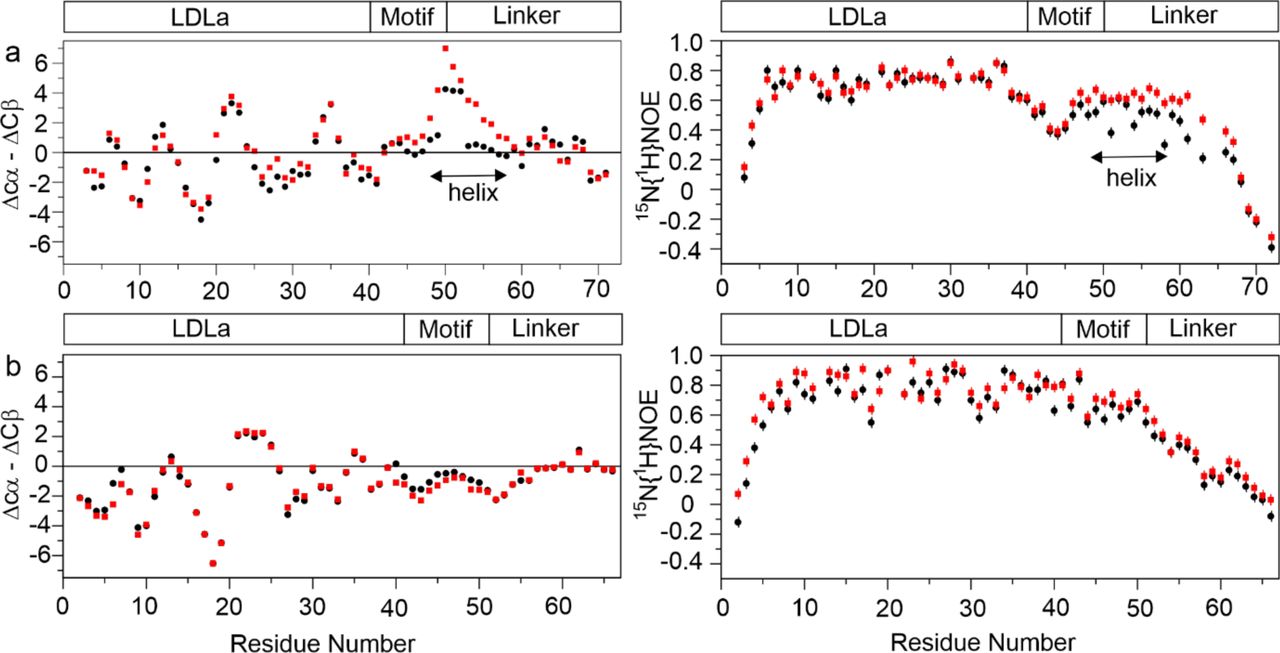
Due to lack of 1H-1H NOEs between the linker and LDLa, we could not solve the structure of the LDLa-linker by NMR. We collected 13Cαβ chemical shift and 15N{1H}-NOE data for RXFP1(1-72) in the presence of relaxin-NH2 and compared to data in the absence of relaxin-NH2 (11 and Fig. 3a). As expected, the ΔCα-ΔCβ and 15N{1H}-NOE data suggest a slight increase in the extent and stability of helix for the region Leu48-Tyr58 than previously observed for H2 relaxin11 due to stronger affinity and the monomeric nature of relaxin-NH2. For RXFP2(1-65) (Fig. 3b), we observed ΔCα-ΔCβ values consistent with the secondary structure of the RXFP2 LDLa10. In the absence of relaxin-NH2, the ΔCα-ΔCβ values suggest that the RXFP2 linker region is largely unstructured, except for the Gly42-His55 that showed negative ΔCα-ΔCβ values indicating the presence of transient β-structure. To investigate this structure, we also recorded 15N{1H}-NOEs on 15N-labelled RXFP2(1-65). The average 15N{1H}-NOE values (0.76 ± 0.10) (Supplementary Table 1) for the LDLa (Ser5-Cys41) agree with a folded structure (PDB 2m96). Notably, residues in the RXFP2-linker from Gly42-Phe51 showed 15N{1H}-NOE values ranging from 0.49 to 0.84 (average 0.62 ± 0.10), consistent with transient structure whereas the corresponding region in RXFP1(1-72) (Gly41-Phe50) is more flexible showing a range of 15N{1H}-NOE values of 0.35 to 0.70 (average 0.53 ± 0.09) (Fig. 3a; Supplementary Table 1). For RXFP2(1-65) following Phe51, the 15N{1H}-NOE value progressively decreases, implying a flexible C-terminal region extending away from the LDLa. These 15N{1H}-NOE data complement the SAXS EOM analysis for both RXFP1(1-72) and RXFP2(1-65), and further suggest that the linker region immediately C-terminal to the LDLa in RXFP2(1-65) is more ordered than in RXFP1(1-72). In contrast to RXFP1(1-72), on addition of relaxin-NH2 to 15N-labelled RXFP2(1-65), little change is observed in the ΔCα-ΔCβ and 15N{1H}-NOE profiles, indicating no significant structural change (Fig. 3).

Plots of 13Cαβ secondary chemical shifts (left panels) and 15N29-NOEs (right panels) for apo- (black circle) and in the presence of 20 equivalents of amidated relaxin (red square) for (a) RXFP1(1-72) and (b) RXFP2(1-65). The regions encompassing the LDLa module the GDxxGWxxxF motif and the remainder of the linker for both receptors are indicated as a block diagram. A distinct increase in stabilization of helical structure is observed for RXFP1(1-72) for residues within the linker (Leu48-Tyr58) whereas addition of relaxin to RXFP2(1-65) shows little change to structure despite clear binding (Figure 1).
Structural effect of mutations in the GDxxGWxxxF motif of RXFP1 and RXFP2
The conserved residues within the GDxxGWxxxF motif are critical for H2 relaxin binding and activation of RXFP1 and RXFP211, 12. Mutation of Asp42, Gly45, Trp46 and Phe50 from this motif to Ala in RXFP1 reduced H2 relaxin-induced cAMP production and weakened the affinity of H2 relaxin to the mutant RXFP111. Equivalent mutations in RXFP2 (Asp43, Gly46, Trp47 and Phe51) caused an even more profound result, completely preventing binding of H2 relaxin and abolishing cAMP production12. These mutations did not affect the binding of the cognate-ligand INSL3 to RXFP2, but reduced or abolished INSL3 activation of the receptor12. Here we translated these single residue mutations of the motif into our recombinant constructs RXFP1(1-72) and RXFP2(1-65) and characterized the impact of these mutations on structure by monitoring 1H,15N chemical shifts, 15N{1H}-NOE and relaxin-NH2 binding (Fig. 4, Table 1).

For (a-h) the first column is the average chemical shift changes of the 1HN and 15N (Δδ ppm) for mutant compared to wild-type; the second column shows the average chemical shift differences the 1HN and 15N (Δδ ppm) after titration with 20 equivalents of relaxin for mutant (red squares) and wild-type (black circles); the third column is the difference (wild-type less mutant) of the 15N{1H}-NOE where a positive difference indicates a lower NOE in the mutant. The following mutations were made for RXFP1(1-72) (a) (D42A) (b) G45A (c) W46A (d) F50A and for RXFP2(1-65) (e) D43A (f) G46A (g) W47A (h) F51A. A block diagram above (a) and (e) show the sequence location of the LDLa module, the GDxxGWxxxF motif and the remainder of the linker in RXFP1 and RXFP2.
Upon titration of D42A, G45A and W46A-RXFP1(1-72) with relaxin-NH2, both D42A and W46A showed a ∼4-fold decrease in affinity of relaxin-NH2 to the linker (Table 1). In comparison to wild-type RXFP1(1-72), D42A showed extensive amide chemical shift perturbation and a decrease in the 15N{1H}-NOE values for the linker region, Gly41-Phe50 and to a lesser degree for amide resonances up to Met60 (Fig. 4a). W46A had a significant impact on the amide chemical shifts and 15N{1H}-NOE values for the residues that form the helical structure (Leu48-Thr61) and hence affected relaxin-NH2 binding (Fig. 4c). Titration of F50A with relaxin-NH2 showed >6-fold loss of ligand binding (Table 1). F50A showed large chemical shift perturbations and a decrease in the 15N{1H}-NOE values for Asp51-Thr61, a region which is essential for ligand binding (11 and Fig. 4d). In contrast, G45A had minimal effect on relaxin-NH2 binding (Table 1) but showed similar chemical shift and 15N{1H}-NOE perturbations to W46A (Fig. 4b), although not as profound. Importantly, for all mutants we observed subtle changes in the chemical shifts and the 15N{1H}-NOE values of resonances of the LDLa. For D42A and W46A consistent changes were seen in the N-terminal region of the LDLa, Leu7-Gly10 and those immediately after the 310-helix, His24-Asn26, which supports the linker folding around the LDLa, as suggested by the SAXS EOM analysis (Fig. 2d).
For RXFP2(1-65), the equivalent mutations, D43A, W47A and F51A, showed near complete loss of binding to relaxin-NH2 and for G46A, significantly reduced binding (Fig. 4e-h, Table 1). Importantly, the mutations had widespread effect both on the amide chemical shifts and 15N{1H}-NOE values. Similar to RXFP1, we observed chemical shift perturbations and a decrease in the 15N{1H}-NOE values for the residues from the N-terminus of the LDLa, Ile2-Tyr10, but also to residues encompassing Cys26-Asp30, but not the 310-helix prior to Cys26. Notably, Cys26-Asp30 is the same region shown to participate in binding to relaxin-NH2 (Fig. 1b). Together, the data suggest that mutations in the motif GDxxGWxxxF structurally impact specific regions of the LDLa modules supporting interactions between the modules and linkers of the two receptors.
Functional effect of swapping the GDxxGWxxxF motif between receptors
The LDLa modules of RXFP1 and RXFP2 can be swapped with limited effect on H2 relaxin binding and ligand-induced activation of these chimeras20. For example, the chimera RXFP211, comprising the LDLa (Met1-Gly42) of RXFP2 with the linker, LRR domain and TMD of RXFP1, shows compared to RXFP1, similar affinity to H2 relaxin but with slightly lower efficacy in ligand-stimulated cAMP activity compared to RXFP1 (20 and Fig. 5a). To compare the role of the conserved GDxxGWxxxF motif in both receptors, we designed a chimera (RXFP2F11) made up of the LDLa and GDxxGWxxxF motif (Met1-Phe51) of RXFP2 and the remainder of RXFP1. This chimera expressed at the cell surface at a slightly higher level than RXFP1 and similar to the level of RXFP211 (Fig. 5c). However, compared to these receptors, RXFP2F11 had a greatly reduced signalling capacity in response to H2 relaxin (Fig. 5a,d), and showed minimal binding (KD >20 nM) of Europium-labelled H2 relaxin at concentrations up to 10 nM (Fig. 5b). These data suggest that swapping the RXFP2 LDLa including the GDxxGWxxxF motif markedly weakens ligand affinity pointing to critical structural differences in how this motif relates to the receptor to modulate H2 relaxin binding and activation.

(a) H2 relaxin-induced cAMP responses expressed as % 5 µM Forskolin response (b) Eu-H2 relaxin saturation binding where RXFP1 shows an affinity (KD) of 0.6 nM and RXFP2F11 an affinity of > 20 nM (the binding affinities reflect the sensitivity of the assay, maximum H2 concentration 15 nM). (c) Cell surface expression of chimeric receptors compared to RXFP1. Data are expressed as mean ± S.E.M of triplicate determinations from at least 3-7 independent experiments. (d) Pooled cAMP activity (pEC50 and Emax) and cell surface expression (%RXFP1) data for RXFP211 and RXFP2F11 in comparison to RXFP1. #p<0.01 compared to RXFP1.
We have previously shown H2 relaxin binding to RXFP1 is biphasic and to RXFP2 is monophasic, highlighting mechanistic differences17. Sequence alignments show there is a 7-residue insertion in the linker of RXFP1 that defines a part of its relaxin-binding site. By inserting these residues (Lys52-Tyr58) into the linker of RXFP2 we recapitulated the biphasic binding and dissociation of H2 relaxin17. This chimera, in response to H2 relaxin activation, showed greater potency and efficacy than RXFP2 and less potency, but similar efficacy to RXFP1. We inserted these seven residues into RXFP2(1-65) (RXFP2(1-65)-ExLink2), 15N-labelled it and showed in the presence of calcium the affinity to relaxin-NH2 was 2-fold weaker than RXFP1(1-72), but 3.5-fold stronger than RXFP2(1-65) (171 µM, Table 1). In the absence of calcium, the affinity weakened (482 µM) to be similar to RXFP1(1-72) (442 µM). As expected, these titrations showed a change in the relaxin-NH2 binding site to the extended linker region, similar to that of RXFP1(1-72) (Supplementary Fig. 2a, Fig. 1). Further, titration of 15N-labelled RXFP2(1-65)-ExLink2 with the TMD exoloop mimetic ssRXFP2, showed this mimetic also interacted within the extended linker, similar to RXFP1(1-72) and with comparable affinity (Table 1, Supplementary Fig. 1c).
To test whether the 7-residue insertion into RXFP2(1-65) influence or is influenced by the GDxxGWxxxF motif, we investigated the structural properties of the RXFP2(1-65)-ExLink2. 13Cαβ secondary shifts and 15N{1H}-NOE data showed that extending the linker region surprisingly did not induce helical propensity in either apo- or relaxin-bound states (Supplementary Fig. 2b,c). The average 15N{1H}-NOE value for the residues Lys53-Glu73 that are C-terminal to the GDxxGWxxxF motif was 0.17 ± 0.08 which supports a lack of structural ordering. The average 15N{1H}-NOE value for the GDxxGWxxxF motif in RXFP2(1-65)-ExLink2 (0.63 ± 0.15) was comparable to that recorded for RXFP2(1-65) (0.64 ± 0.10) suggesting no change in the dynamic properties of the motif. These data suggest that transferring the RXFP1 linker-relaxin binding site into the RXFP2 linker changes the ligand binding interface and affinity but does not allow the formation of helical structure and/or stabilization post-ligand binding, as observed for RXFP1. These data collectively demonstrate that the fold of the motif GDxxGWxxxF in RXFP1 and RXFP2 modulates the structuring of the linker and its association with the LDLa, explaining why swapping the motif between receptors (such as RXFP2F11) led to significant loss of H2 relaxin binding and potency.
Fast timescale dynamics of apo- and relaxin-bound RXFP1(1-72) and RXFP2(1-65)
The differences in conformation and relaxin-binding properties of the LDLa and linker of RXFP1(1-72) and RXFP2(1-65) suggested differences in dynamics. To investigate this possibility, we acquired 15N spin relaxation experiments, 15N-R1, 15N-R2 and 15N{1H}-NOE at 81.1 MHz for RXFP1(1-72) and RXFP2(1-65) in the presence or absence of relaxin-NH2 and at 25 or 15 °C (Fig. 6). In addition, the data were subjected to a reduced spectral density analysis (supplementary Fig. 3). For RXFP1(1-72) the spin-relaxation values (Fig. 6) show that the first six and last ten residues are disordered and insensitive to relaxin-NH2, so we focused our analysis on Ser6-Cys40 of the LDLa, and Gly41-Met60 of the linker. Similarly, the first four residues of the LDLa of RXFP2(1-65) and residues after Gly52 are essentially disordered. Therefore, for RXFP2(1-65) we focused on Ser5-Cys41 of the LDLa and Gly42-Gly52 of the linker. The average values for 15N-R1, 15N-R2 and 15N{1H}-NOE, and the reduced spectral parameters J(0), J(0.87ωH) and J(ωN) for these regions are provided in Supplementary Table 1.

Backbone amide 15N relaxation parameters of 100 µM RXFP1(1-72) (a-d) and RXFP2 (1-65) (e-h) in apo-state at 25 °C (black circles), and at 15 °C (green triangle) and bound with amidated relaxin at 25 °C (red circle) and at 15 °C (blue triangle) measured at 15N frequency of 81.1 MHz and pH 6.8. Sample concentrations represent 83% saturation of RXFP1(1-72) and RXFP2(1-65) with ligand. Error bars are calculated using Monte Carlo Simulations for 15N-R1, 15N-R2 measurements and based on average estimated noise level for 15N{1H}-NOE. The sequence locations of the LDLa module, the GDxxGWxxxF motif and the remainder of the linker in the receptors are shown above (a) and (e).
For RXFP1(1-72) the 15N{1H}-NOE values change little for the LDLa under all conditions, consistent with a well-ordered structure. In contrast, 15N{1H}-NOE values for the linker of RXFP1(1-72) increase at 15 °C or on addition of relaxin-NH2 suggesting a gain in order. As expected, at 25 °C and in the presence of relaxin-NH2, the 15N-R2 for residues of the linker increase, especially Gln49-Met60, which is clearly observed in the R2/R1 ratios (Fig. 6), reflecting the fast-timescale interaction with relaxin-NH2. However, at 15 °C and in the absence of relaxin-NH2, there are marked increases in R2/R1 (>10) for residues in the relaxin-binding site, Phe50-Ala55, Lys59-Thr61, supporting the presence of transient structure. Notably, there are similar increases in R2/R1 (>10) for residues within the LDLa, Gly8, Phe10, Cys12, and Asn14-Cys18 (Fig. 6b). These pronounced changes pose the question: are the dynamics for these residues of the LDLa connected to those of the linker? To partly address this question, we collected 15N spin relaxation data for the LDLa alone at 15 and 25 °C (Supplementary Fig. 4). The only residue to show a significant R2/R1 (>10) was Lys17 at 15 °C. We investigated the effect of decrease in temperature (15 °C) on the dynamics of RXFP1(1-72) bound to relaxin-NH2. With the exception of the GDxxGWxxxF motif, the LDLa and linker show similar average 15N{1H}-NOE and 15N-R2 values (Fig. 6d, Supplementary Table 1). However, significant R2/R1 (>10) are observed for the entire linker from Gly45 to Gln63 and for extensive regions of the LDLa, Ser6, Cys12, Asn14, Ile15, Lys17-His24, Gly27, Val28, Asp30, Gly32, Gln34, Ala35 and Asp38. Importantly, the reduced spectral density parameters J(0) and J(0.87ωH) for the LDLa and linker were now similar, reflecting similar ordering despite the distinct chemical exchange. Furthermore, on addition of relaxin-NH2, J(0) increased and J(0.87ωH) decreased for the linker (Supplementary Table 1), consistent with an increase in ordering within the linker.
For RXFP2(1-65) under all conditions, the 15N{1H}-NOE over Gly5-Gly52, covering the LDLa and a substantial part of the linker, are similar, reflecting an overall similar tumbling and ordering of these residues with little conformational change, consistent with the SAXS analysis (Fig. 2), and distinctly different to RXFP1(1-72). Addition of relaxin-NH2 at 15 °C shows increased 15N-R2 and decreased 15N-R1, clearly observed in the increased R2/R1, although notably these differences are substantially less than those of RXFP1(1-72). Three regions show distinct changes in R2/R1 (>8): the N-terminal of the LDLa, Phe4-Lys8; a region within the LDLa, His25-Asp31; and the C-terminal of the LDLa that includes Cys41-Gly46, and Ile50 of the linker. At 15 °C in the absence of relaxin-NH2 (Fig. 6c), we observed small but distinct increases in R2/R1 for residues Ile2-Tyr10 and Glu38-Ile50. These changes are consistent with the reported spatial proximity of N- and C-terminal regions12 and so the change in R2/R1 in the presence of relaxin-NH2 for Ile2-Tyr10 may not reflect a direct interaction with relaxin-NH2. The remaining two regions (His25-Asp31 and Cys41-Gly46), however, correlate well with the chemical shift differences observed upon relaxin-NH2 binding (Fig. 1), and therefore we propose that these increases in R2/R1 reflect chemical exchange from an interaction with relaxin-NH2 and in contrast to RXFP1(1-72), not a conformational change. The reduced spectral density parameters J(0) and J(0.87ωH) for the LDLa (Ser5-Cys41) and linker (Gly42-Gly52) of RXFP2(1-65) reflect similar ordering and structure (Supplementary Table 1). Furthermore, on addition of relaxin-NH2 at 15 °C, the more modest increases in J(0) and decreases in J(0.87ωH) for the LDLa and linker are consistent with the notion of little conformational change in RXFP(1-65).
15N-relaxation dispersion on RXFP1(1-72) and RXFP2(1-65)
To further understand the influence of relaxin binding on the dynamics of RXFP1(1-72) we acquired 15N-CPMG relaxation dispersion experiments in the absence and presence of relaxin-NH2 at 15 °C at 70.9 and 81.1 MHz (Table 2; Supplementary Fig. 5a,b). In the absence of relaxin-NH2 four residues, Cys12 and Thr16 from the LDLa and Tyr53 and Ser56 from the linker, showed distinct dispersion fitting to a slow-limit exchange, two-state model. As Thr53 and Ser56 are within the transient helix they likely reflect its fluctuations; whereas Cys12 and Thr16 are located in the loop between the β-strands of the LDLa. Both pairs of residues show a population of 2-3% for the minor state. The kinetics vary for the two sites where the linker residues show a kex of 800 s-1 whereas the LDLa residues, a kex of 250 s-1. The kinetics of the linker residues may reflect both the formation of transient structure and interaction with the LDLa, whereas the kinetics of the LDLa residues may only reflect an interaction with a structured linker.
15N CPMG relaxation dispersion kinetics parameters for 15N RXFP1(1-72) and 15N RXFP2(1-65)
Upon addition of relaxin-NH2, Tyr53 and Ser56 from the linker continue to show chemical exchange, but also Trp46 and Met60. These residues show similar kinetics as viewed in the apo-state with a minor-state population of 2-5% and a kex of 700 s-1. The similar kinetics is surprising, but exchange rates now will reflect a combination of stabilizing transient structure, interactions with both the LDLa and non-saturating relaxin-NH2. Within the LDLa, Cys12 and Thr16, but also Tyr9, Phe10, Leu22 and Leu23 show dispersion. These residues all show a significantly larger minor-state population of 6-9% and a 4-fold lower kex of 65 s-1. As previous experiments11 and the data presented here show that the LDLa does not interact with relaxin, we interpret these kinetics for the LDLa residues as reflecting only an interaction with the linker, presumably when structured, which is more populated in the presence of relaxin-NH2 compared to the apo-state.
Previously we showed that mutation of several residues within the LDLa, mainly L7K and K17A, decreased ligand-binding induced cAMP production which was interpreted to be important for interacting with the TMD and therefore activation of the receptor9. Our work here presents an alternative view, in that they interact with the linker to induce a conformation within the GDxxGWxxxF motif that forms the tethered agonist. To test the role of these residues in stabilizing the LDLa-linker/relaxin complex, we prepared the double-mutant RXFP1(1-72)-L7K/K17A. 15N-CPMG relaxation dispersion experiments of RXFP1(1-72)-L7K/K17A in the apo-state show no residues within the LDLa have dispersion, but the linker residue Tyr53 retained dispersion with similar kinetics (kex 940 s-1, 2% minor state) to wild-type (Table 2). On titration of RXFP1(1-72)-L7K/K17A with relaxin-NH2, the majority of residues of the linker, with the notable exception of Trp46, that showed dispersion in wild-type also show dispersion with similar kinetics (kex 700 s-1, 3% minor state), however no residues within the LDLa show dispersion. As the derived apparent dissociation constants of the linker residues of both wild-type (25 μM) and RXFP1(1-72)-L7K/K17A (30 μM) are similar we conclude that this apparent KD predominantly reflects both the stabilization of helical structure and the association with relaxin-NH2. Furthermore, the loss of all dispersion in the mutant LDLa supports that the dispersion observed in wild-type is indicative of stabilization and association of the linker, supporting that the major functional role of the LDLa is to stabilize structure within the linker for receptor activation.
Similar 15N-CPMG relaxation dispersion experiments were performed for RXFP2(1-65) (Supplementary Fig. 5c). In the absence of relaxin-NH2, two residues show dispersion, Ser5 (kex 112 s-1, 8% minor state) in the disordered N-terminal of the LDLa and Gly42 (kex 71 s-1, 11% minor state) in the linker C-terminal to the LDLa module (Table 2). This dispersion likely reflects an interaction between these regions as previously described20. On addition of relaxin-NH2, these residues continue to show dispersion, but with the addition of His25 of the LDLa module and Trp47 of the linker. These residues show minor-state populations of 25-36% (Table 2), however, as all residues except Ser5 show chemical shift changes in the presence of relaxin-NH2 (Fig. 1), these data are consistent with the direct weak interaction of non-saturating relaxin-NH2 and not a conformational change.
Discussion
Most class A GPCRs are activated by the binding of an agonist into an orthosteric site of the TMD shifting the conformational equilibrium to favour the active state. While this mechanism appears simple, variations of the nature of the agonist are extensive, especially for peptide- and protein-activated GPCRs. The LGR family, that includes the GHPRs and RXFP1/2, presents complex activation mechanisms that appear to utilize internal agonists, but each receptor shows significant differences. The current mechanism for the GPHRs21 proposes that binding of the hormone to the LRR domain induces a conformational change in a C-terminal hinge, structurally rearranging a 10-residue peptide, considered the agonist, to modulate the ECL region of the TMD leading to receptor activation. In contrast, for RXFP1 and RXFP2, binding of the hormone induces conformational changes in the N-terminal region of the receptor. Truncation of the N-terminal LDLa and analysis of N-terminal splice variants suggested that this module is the internal or tethered agonist for these receptors14. Mutagenesis experiments and receptor signalling assays suggested that key residues for RXFP1 were in the N-terminal region of the LDLa8, 9 while those for RXFP2 were in the C-terminal10, proposing that the role the LDLa played in activation differed for the two receptors. Importantly, despite extensive mutagenesis studies no essential residues of the LDLa were identified to implicate a direct role of the module in activation. However, mutation of the conserved residues within the GDxxGWxxxF motif, immediately C-terminal of the LDLa in both RXFP111 and RXFP212, did have significant effects on activation suggesting that these residues along with the LDLa may constitute the internal agonist. In our present study, we demonstrate that the function of the LDLa is not to directly activate the receptors, but to interact with the GDxxGWxxxF motif to modulate its shape and interaction with the TMD, and thus trigger receptor activation.
To understand how this internal agonist presents itself in the receptor we compared the structural relationship of the GDxxGWxxxF motif with the LDLa in the two receptors and used the common ligand, relaxin-NH2, to probe differences in binding sites and interactions. We propose that the motif in RXFP1 undergoes significant conformational change upon binding and activation by H2 relaxin (Fig. 7). The motif forms a loosely structured loop due to the association of the helical region, Leu48-Thr61, of the linker with the LDLa. Residues of the linker weakly interact with residues in the N-terminal region of the module, for example, Leu7 or Lys17. Mutagenesis of Leu7 and/or Lys17 lead to a weakening of both the linker/LDLa interaction and binding of H2 relaxin, and consequently a reduction in efficacy of activation as previously observed9. Therefore, in the context of the whole receptor, this helical region is likely to become well-structured upon H2 relaxin binding, due to interaction with both the LDLa and H2 relaxin. The GDxxGW portion of the motif does not directly participate in H2 relaxin binding, however, mutagenesis of the conserved residues, Asp42, Gly45 and Trp46, affect how the helical region of the linker associates with the LDLa, thus indirectly impacting on H2 relaxin affinity. Nevertheless, mutation of these residues results in a profound loss of activation, pointing to critical roles in TMD interactions. In contrast, in RXFP2 the GDxxGWxxxF motif is intimately associated and structured with the LDLa (Fig. 7). Its interaction with the module forms a binding-surface for H2 relaxin, involving residues in the C-terminal of the module, with little conformational change upon binding. For this receptor, mutation of the conserved residues, Asp43, Gly46 and Trp47 have a greater impact on H2 relaxin binding and activity than observed for RXFP1. However, these residues are likely to have similar key roles in receptor activation for both H2 relaxin and the cognate ligand of RXFP2, INSL3. Importantly, INSL3 does not interact with the linker or LDLa and the exact mechanism of activation for this ligand remains to be elucidated. Previous studies have highlighted that the N-terminus of the INSL3 A-chain is essential for RXFP2 activation22 suggesting that it may directly contact the extracellular loops or TMD of RXFP2 to enable the interaction of the GDxxGWxxxF motif for receptor activation12.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) RXFP1: In the absence of relaxin, the linker of RXFP1 weakly associates with the LDLa and this complex weakly interacts with EL2 of the transmembrane domain (TMD). The association of the linker with the LDLa induces a helical turn within the linker (red), while the GDxxGWxxxF motif (blue) remains largely unstructured. Relaxin associates strongly with the LRR domain, and weakly with the linker and not the LDLa. In combination the LDLa and relaxin stabilize and extend the linker helix (red), enabling the motif to form a conformation that leads to receptor activation. (b) RXFP2: In the absence of relaxin, the GDxxGWxxxF motif (magenta) closely associates with the LDLa. The LDLa-motif interacts with both EL2 of the TMD and relaxin. While little conformational change to the motif occurs, the strong association of relaxin with the LRR domain leads to the essential interaction of the LDLa-motif for receptor activation. (c) RXFP2F11: Replacing the LDLa-motif in RXFP1 with the RXFP2 LDLa-motif results in a receptor that weakens the binding of relaxin (20-fold) and reduces potency by 3000-fold. We propose that the structure of the RXFP2 LDLa-motif cannot effectively engage with relaxin or EL2 in this chimera (d) RXFP2(1-65) ExLink2: Insertion of the 7-residues that encompass the relaxin binding site of RXFP1 into RXFP2 results in a chimera that behaves more like RXFP1, although helical structure is not induced in the linker. The chimeras highlight the different roles of the LDLa-motif in linker structure formation and stabilization in both receptors. Figure created with BioRender.com.
The difference in structure and interaction of the GDxxGWxxxF motif, as a requirement for H2 relaxin binding and activation, is reinforced by the behaviour of the construct RXFP2F11. Compared to the RXFP211 chimera, RXFP2F11 showed ∼3000-fold loss in H2 relaxin potency and >10-fold loss in H2 relaxin affinity. The only differences between these two chimeras are the non-conserved residues of the motif, which previously when mutated did not affect ligand-induced activation of RXFP2/H2 relaxin or RXFP2/INSL312. Sequence alignments (Supplementary Fig. 6) show these non-conserved residues are conserved in the context of RXFP1 or RXFP2. Importantly, for organisms where no H2 relaxin gene has been identified but an INSL3 gene has, for example cow and rabbit, the residues are poorly conserved in RXFP1 but well conserved in RXFP2 pointing to the importance of these residues in maintaining the shape of the motif within the receptor. While very little is known about the structures of these sister receptors, our present study provides critical structural evidence of the differences in their H2 relaxin binding and activation mechanism. By studying the linker, a region that potentially differentiates these two receptors, we can bring to light some of the ways in which these two highly similar receptors are able to behave differently to one another in the biological context and explain how H2 relaxin has evolved into a cognate/specific ligand for RXFP1.
Methods
Peptides
Native recombinant H2 relaxin (serelaxin) was kindly provided by Corthera Inc. Amidated H2 relaxin with both the A and B-chain C-termini amidated, was synthesized using Fmoc chemistry and regioselective disulfide bond formation as previously described for amidated H2 relaxin and other analogues13, 23. After synthesis, the peptide was purified and characterized by reverse phase RP-HPLC and MALDI-TOF MS.
Protein Expression and Purification
The DNA sequences of RXFP1(1-72), RXFP2(1-65), RXFP1(1-40) and RXFP2(1-41) were amplified by PCR and inserted into the vector pGEV2, an N-terminal thrombin-cleavable GB1 fusion expression vector using BamHI and XhoI restriction sites. For RXFP2(1-65), Pro4 was mutated to phenylalanine to remove cis-trans isomerism as previously reported12. All site-specific mutants were prepared using PrimeStar DNA Taq Polymerase (Takara Clonetech). Expression and purification of recombinant wild-type and mutant RXFP1(1-72), RXFP2(1-65), RXFP1(1-40) and RXFP2(1-41) followed our previous methods with minor modifications8–11. Briefly, proteins were expressed into BL21(DE3) trxB (Novagen) using autoinduction24, for producing unlabelled protein samples for SAXS experiments. For uniform 15N isotopic labelling, protein was expressed in N5052 minimal medium24 using 15NH4Cl (Sigma-Aldrich). For 13C,15N labelling, cells were grown in a 1-L Braun Biostat fermenter supplemented with 15NH4Cl and D-[13C] glucose (Sigma-Aldrich) as the sole source of nitrogen and carbon for the bacterial cells, as suggested by Cai et al.25 Cells were harvested, pelleted, and stored at −20 °C. Following resuspension in 50 mM Tris, 150 mM NaCl, 5mM EDTA-Na and 1mM phenylmethylsulfonyl fluoride (PMSF) at pH 7.4, cell pellets were lysed using an Avestin EmulsiFlex C3 cell crusher. Cell debris was removed by centrifugation (13,000 × g, 4 °C for 40 min), the soluble fraction was filtered using a 0.45 µm syringe filter. The GB1-LDLa linker fusion proteins were purified over IgG Sepharose 6 Fast Flow beads, eluted with 50mM acetic acid, pH 3.4. Further, the eluted protein was buffer exchanged (50 mM Tris-HCl, 150mM NaCl, pH 8.5) via dialysis, and refolded (100–300 mg.ml-1, 3 mM GSH, 0.3 mM GSSG, 50mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, pH 8.5) by incubating overnight at 4 °C with gentle stirring. The GB1 tag was cleaved from the oxidized proteins by incubating overnight with thrombin (10 U.mg−1 protein, Sigma-Aldrich). The cleaved protein was purified by reversed phase high-performance liquid chromatography (RP-HPLC) (buffer A: 0.1% trifluoro-acetic acid in water, buffer B 100% acetonitrile with 0.1% trifluoro-acetic acid; Agilent Zorbax 300SB-C18 column). Collected protein fractions were lyophilized and stored at −20 °C.
The expression and purification of RXFP2(1-65) ExLink 2 was performed as previously described17. Protein was expressed as a His6-tag fusion in BL21(DE3) trxB (Novagen) using autoinduction24. 15N and 13C,15N labelling were performed as described above. Cells were harvested, lysed and purified using Talon Superflow resin (Takara Clontech). Further, the eluted protein was refolded using the reduced and oxidized glutathione and His6-GB1 tag was cleaved using thrombin, followed by RP-HLC, also described above.
The peptide mimetics of the exoloops, ssRXFP1 (EL1(475-486)/EL2-RXFP1) and ssRXFP2 (EL1(475-486)/EL2-RXFP2), were purified as previously described11, 15. Proteins were expressed from BL21(DE3) as His6-tag fusions. Expression was induced by isopropyl –D-1-thiogalactopyranoside (IPTG) induction in LB medium for 16 h at 16 °C. Cells were harvested, pelleted, lysed and purified over Talon Superflow resin (Takara Clontech). The His6-tag was removed by thrombin cleavage and the proteins further purified with a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare) in 20 mM Tris HCl (pH 7.4), 150 mM NaCl.
NMR spectroscopy
The 15N-labeled RXFP1(1-72) sample, which comprises the first 72 residues of the receptor, was identical to the material used for the assignment of the backbone chemical shifts as previously described26. For 15N RXFP2(1-65) and RXFP2(1-65) ExLink2, the assignments were adapted from previously reported assignments for C-terminal GB1 fused RXFP2(1-65) and RXFP2(1-65) ExLink2 construct12, 17. All protein samples were prepared in a buffer containing 50 mM Imidazole, 10 mM CaCl2 at pH 6.8 for all experiments. Protein samples used for titrations were dialyzed in the same buffer and same vessel overnight to ensure sample buffer conditions. NMR spectra were acquired on a Bruker Avance II 800 MHz spectrometer equipped with TXI cryoprobe and a single axis field gradient (Gz). NMR spectra were processed using NMRPipe 27 as described in 26 and data analyzed in NMRFAM-SPARKY 28. The 1H chemical shifts were referenced directly to DSS at 0 ppm and the 13C and 15N chemical shifts were subsequently referenced using the 13C/1H and 15N/1H ratios 29. The ΔCα – ΔβC smoothed values were calculated as previously11, 30. The 15N,1H chemical shift mapping was computed using 31:

SAXS data collection and analysis
Small-angle X-ray scattering (SAXS) measurements were conducted at the Australian Synchrotron SAXS/WAXS Beamline equipped with a co-flow system to avoid radiation damage and enable higher X-ray flux (11,500 eV) and with optimized in-line size exclusion chromatography (SEC) to limit protein sample dilution 32–34. Fifty microliters of the purified RXFP1(1-72) and RXFP2(1-65) at 5 mg/ml was injected over a Superose 6 5/150 increase column (GE Healthcare) equilibrated and eluted with TBS (pH 7.4) with a solution containing 10 mM calcium chloride and 0.2% sodium azide. The sample to detector length used was 1426 mm, providing a q range of 0.007 - 0.515 Å-1. Collected SAXS data was reduced using the Sactterbrain software, analysed by CHROMIXS35, and the ATSAS software package 3.0.2 (r12592)36. SAXS patterns, the radius of gyration (Rg), the maximal particle dimension (Dmax), and the pairwise distance distribution histogram [P(r) plot] were determined by using the ATSAS software36. Ab initio modelling was performed using DAMMIN37. Ensemble of conformations and models were generated using EOM 2.019 in the ATSAS software suite. A summary of the SAXS data acquisition parameters is provided in Supplementary Table 2.
Construction of RXFP2F11
The chimeric construct RXFP2F11 consists of the RXFP2 sequence up to and including Phe51, followed by the RXFP1 sequence from Asp51 to the end of the TM domain. It was made using overlap PCR by the methods described in 20, with primers Fwd: CATCATGGATCCGCCACCATGGACAGCAAAG and Rev: 5’ CATTTTGTAGTAACTGGCAAAATATTTGTCAAATATGGTCGCCCATCCACTAGTG 3’ on RXFP2 template, and Fwd: 5’ CACTAGTGGATGGGCGACCATATTTGACAAATATTTTGCCAGTTACTACAAAATG 3’ and Rev: GAGAGCTCGAGTCATGAATAGGAATTGAGTCTCGTTG on RXFP1 template to make the two DNA portions. The final annealed product was cut with BamHI and XhoI restriction enzymes for insertion into a freshly cut pcDNA3.1TM/Zeo+ AmpR mammalian expression vector (Invitrogen, Carlsbad, CA, USA) and then the entire insert sequenced to ensure accuracy.
Receptor expression in HEK293T cells
HEK293T cells (ATCC #CRL-1573; American Type Tissue Culture Collection) were used for receptor expression and were grown in Dulbecco’s modified eagle medium supplemented with 10% foetal bovine serum, 1% l-glutamine and 1% penicillin/streptomycin in incubators maintained at 37 °C with 5% CO2 and 85% humidity. Transient transfections were achieved using lipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instructions.
Binding and signaling assays
The affinity of relaxin for the mutant receptors in comparison to wild-type RXFP1 was assessed using Europium (Eu3+)-labelled H2 relaxin (Eu-H2) 38 at increasing concentrations in the presence or absence of 1 μM unlabeled ligand. Following a 1 h incubation, media was removed and 100 μl Delfia Enhancement solution (PerkinElmer) was added to each well. Time-resolved fluorescence with excitation at 340 nm and emission at 614 nm was read on an Omega POLARstar plate reader after incubation in low light for 20–30 mins with shaking. Data from at least three independent experiments, all performed in triplicate were pooled and presented as mean fluorescent specific binding ± SEM. Binding data was analysed by nonlinear regression one-site binding curves using GraphPad PRISM 6 to calculate KD values. cAMP activation in response to ligand stimulation was measured by co-transfection with a pCRE β-galactosidase reporter gene as previously described 14. Briefly, co-transfected cells were stimulated with increasing concentrations of H2 relaxin and 5 μM Forskolin or media only were used as a positive and negative controls, respectively. Following a 6 h incubation at 37 °C media was aspirated and plates frozen at −80 °C for ≥ 24 h. β-galactosidase activity in plates was achieved as previously described 14 with absorbance readings measured on a Benchmark Plus Microplate Reader (Bio-Rad) at 570 nm. All experiments were performed in triplicate a minimum of three times and data were pooled and presented as percentages of the 5% Forskolin response. GraphPad PRISM was used to fit a nonlinear regression sigmoidal dose-response curve and resulting pEC50 and maximum response (Emax) values were subjected to one-way ANOVA and uncorrected Fisher’s least square difference comparison test.
Cell surface expression assays
The presence of mutant receptors at the surface of cells was gauged by virtue of the FLAG epitope present on their N-termini using a plate-based ELISA assay. HEK293T cells were seeded at 2 × 105 cells per well in 24-well plates (Costar) pre-coated with poly(l-lysine) and transfected with 1 mg per well of plasmid DNA. After a further 24 h of growth, cells were washed in TBS/CaCl2 (50 mM Tris pH 7.4, 150 mM NaCl and 1 mM CaCl2) and fixed with 3.7% formaldehyde in TBS/CaCl2 for 20 min. Two washes followed and then incubation for 45 min at room temperature with 1% bovine serum albumin (BSA) in TBS/CaCl2 to block non-specific binding. The cells were then incubated with 10 mg.ml-1 of anti-FLAG M1 monoclonal antibody (Sigma-Aldrich) in TBS/CaCl2 for 2 h at room temperature. Cells were washed twice more before a 15-min reblock in 1% BSA/TBS/CaCl2; then incubated for 1 h with 2 mg.ml-1 goat anti-mouse Alexa Fluor 488 suspended in 1% BSA/TBS/CaCl2. Cells were then washed thrice and stored frozen at −80 °C overnight. Finally, cells were thawed and lysed by incubating for 30 min with 200 ml per well of lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.25% Triton X-100) at room temperature with shaking and scraped and transferred to black 96-well plates (180 ml in each well; Costar) to be read on an Omega POLARstar plate reader (BMG labtech) with excitation at 490 nm and emission at 520 nm. Non-specific background was determined using cells transfected with empty vector and mutant receptor expression was expressed as the percentage of the wild-type receptor expression. Data are mean ± s.e.m. from at least three independent experiments each performed in triplicate. Pooled data were analysed in GraphPad PRISM 6 using one-way analysis of variance and uncorrected Fisher’s least square difference multiple comparison test.
Backbone 15N relaxation parameters and reduced spectral density mapping
15N relaxation parameters R1, R2, and steady state 15N{1H}-NOE of RXFP1(1-72) were measured at 15N frequency of 81.1 MHz with 1024 and 150 complex data points over spectral widths of 13.0 and 26.0 ppm for 1H (F2) and 15N (F1), respectively at pH 6.8 and at 15 and 25 °C, using pulse sequences with sensitivity enhancement provided in the Bruker pulse sequence library, as described previously39–41. R1 and R2 experiments were collected with a recycle time of 2.6 s and 16 scans per FID and 15N{1H}-NOE experiments were collected with a saturation pulse of 4 s and an additional relaxation delay of 5 s and 32 scans per FID. Relaxation delays of 10 (×2), 50, 100, 300 (×2), 500, 700, 900 (×2) and 1500 ms for R1 experiments and for R2 relaxation delays of 16.96 (×2), 33.92, 67.84, 101.76 (×2), 135.68, 169.6 (×2), 203.52 and 237.44 ms were used. The repeated spectra were used to estimate instrumental error. 15N relaxation parameters were determined using the program relax 42–44 (version 3.3.4). For R1 and R2 rate constants, errors were estimated using 500 Monte Carlo Simulations. The steady state 15N{1H}-NOE values for RXFP1(1-72) were estimated from the ratios of peak intensities obtained from spectra acquired with and without proton saturation using relax. Errors for 15N{1H}-NOE experiment were calculated based on the noise level in the spectra.
Reduced Spectral Density Mapping
The relaxation data acquired at 15N frequency of 81.1 MHz on the 800 MHz spectrometer was also analyzed using spectral densities 41. The reduced spectral density parameters were calculated using Mathematica 45; based on the following equations:



 where J(0), J(ωN) and J(0.87ωH) are values of spectral density sampling frequencies ω = 0, ωN (81 MHz), and ωH (800 MHz), respectively. The other parameters are given by d = (μ0hγNγH/(8π2))/ < r3NH >, c = ωN(Δσ)/√3, μ0 is the permeability of the free space, h is Planck’s constant, γH and γN are the gyromagnetic ratio of 1H and 15N respectively, rNH is the average amide bond length (1.02 Å), and Δσ is the chemical shift anisotropy for 15N nuclei (−170 ppm).
where J(0), J(ωN) and J(0.87ωH) are values of spectral density sampling frequencies ω = 0, ωN (81 MHz), and ωH (800 MHz), respectively. The other parameters are given by d = (μ0hγNγH/(8π2))/ < r3NH >, c = ωN(Δσ)/√3, μ0 is the permeability of the free space, h is Planck’s constant, γH and γN are the gyromagnetic ratio of 1H and 15N respectively, rNH is the average amide bond length (1.02 Å), and Δσ is the chemical shift anisotropy for 15N nuclei (−170 ppm).
15N-relaxation dispersion on RXFP1(1-72)
Backbone amide 15N Carr-Purcell-Meiboom-Gill (CPMG) constant time relaxation dispersion experiments were acquired on apo 15N labeled RXFP1(1-72) and in complex with H2 relaxin at 18.8 and 16.5 T with 1024 and 128 complex data points over spectral widths of 13.0 and 26.0 ppm for 1H (F2) and 15N (F1), respectively at pH 6.8 and at 298 and 288 K. A series of interleaved two-dimensional spectra were collected at different υCPMG frequencies of 50, 75 (×2), 100, 150 (×2), 200, 300, 500, 700 (×2), 800, 900, 1000, 1500 MHz, with a total relaxation delay τCPMG of 80 ms. All NMR spectra were processed using NMRPipe 27 and analyzed in NMRFAM-SPARKY 28.
The Effective relaxation rates, R2,eff, were extracted by fitting the dispersion data to equation (6) using NESSY 46:
 where I( υCPMG) is the peak intensity in the spectrum recorded at various υCPMG values and I0 is the peak intensity in the reference spectrum with no relaxation delay (τCPMG = 0 ms). R2,eff(υCPMG) values obtained for RXFP1(1-72) in apo- and relaxin bound form at 15 °C were fitted to Carver-Richard equations for two-site exchange model undergoing slow exchange (kex << Δω) regime on the Sherekhan webserver 47.
where I( υCPMG) is the peak intensity in the spectrum recorded at various υCPMG values and I0 is the peak intensity in the reference spectrum with no relaxation delay (τCPMG = 0 ms). R2,eff(υCPMG) values obtained for RXFP1(1-72) in apo- and relaxin bound form at 15 °C were fitted to Carver-Richard equations for two-site exchange model undergoing slow exchange (kex << Δω) regime on the Sherekhan webserver 47.







Acknowledgements
This research was supported by National Health and Medical Research Council of Australia project grant 1100676 (R.A.D.B., D.J.S, M.D.W.G and P.R.G.), the Victorian Government Operational Infrastructure Support Program and equipment grants from the Australian Research Council (LE120100022). R.A.D.B. is supported by an NHMRC Research Fellowship.
References




