

Abstract
The SARS-CoV-2 pandemic has entered an alarming new phase with the emergence of the variants of concern (VOC), P.1, B.1.351, and B.1.1.7, in late 2020, and B.1.427, B.1.429, and B.1.617, in 2021. Substitutions in the spike glycoprotein (S), such as Asn501Tyr and Glu484Lys, are likely key in several VOC. However, Asn501Tyr circulated for months in earlier strains and Glu484Lys is not found in B.1.1.7, indicating that they do not fully explain those fast-spreading variants. Here we use a computational systems biology approach to process more than 900,000 SARS-CoV-2 genomes, map their spatiotemporal relationships, and identify lineage-defining mutations followed by structural analyses that reveal their critical attributes. Comparisons to earlier dominant mutations and protein structural analyses indicate that increased transmission is promoted by epistasis, i.e., the combination of functionally complementary mutations in S and in other regions of the SARS-CoV-2 proteome. We report that the VOC have in common mutations in non-S proteins involved in immune-antagonism and replication performance, such as the nonstructural proteins 6 and 13, suggesting convergent evolution of the virus. Critically, we propose that recombination events among divergent coinfecting haplotypes greatly accelerates the emergence of VOC by bringing together cooperative mutations and explaining the remarkably high mutation load of B.1.1.7. Therefore, extensive community distribution of SARS-CoV-2 increases the probability of future recombination events, further accelerating the evolution of the virus. This study reinforces the need for a global response to stop COVID-19 and future pandemics.
Notice: This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
”Nothing in Biology Makes Sense Except in the Light of Evolution” -Theodosius Dobzhansky
1. Introduction
In late 2020, three SARS-CoV-2 variants of concern, VOC; B.1.1.7, B.1.351, and P.1 (also called alpha, beta, and gamma respectively) rapidly became the predominant source of infections due to enhanced transmission rates and have since been linked to increased hospitalizations and mortalities (Alpert et al. 2021; Challen et al. 2021; Davies et al. n.d.; Faria et al. 2021; Funk et al. 2021; Sabino et al. 2021; Volz et al. n.d.; Washington et al. 2021). In early 2021, several new VOC appeared including, B.1.427 (epsilon), B.1.526 (iota), and B.1.617 (delta). B.1.617 is of immediate concern because it is responsible for the COVID-19 crisis that recently began in India (J. Singh et al. 2021), is causing the majority of new infections in the United Kingdom (UK), and the United States (USA), and has now been observed in more than 70 countries worldwide. Notably, several of these VOC have rapidly spread even in regions such as the UK that depend on robust sampling efforts for early detection. There is therefore a critical need to identify accurate predictors and biological causes for the increased transmission of the next VOC, which will inevitably emerge if the viral spread is not globally restrained.
Although extensive efforts are underway to achieve these ends, integrating new findings is critical to unravel the multiple biomolecular and environmental factors influencing viral evolution. Toward a holistic understanding of VOC emergence, two major weaknesses need to be addressed: (1) currently, the mutations used to identify VOC and potentially explain the altered biology of the virus are predominantly focused on the changes observed in the spike glycoprotein (S) whereas those in other genomic regions are largely ignored, and (2) the molecular models used to reconstruct the evolutionary history of the virus employ phylogenetic trees that are useful for species-level but not population-based analyses and are unable to incorporate information and molecular events that are critical to understanding SARS-CoV-2 (Huson & Bryant 2006; Velasco 2013). Indeed, a recent study introduced novel methods to detect recombination in coronaviruses and found that their evolution shows very little resemblance to a tree structure (Müller et al. 2021).
For example, the Asn501Tyr substitution in S is likely key because it increases affinity for the host receptor, angiotensin-converting enzyme 2 (ACE2) (Liu et al. 2021), and is often used to identify the late 2020 VOC (Fratev n.d.; Luan et al. n.d.), but this mutation has been circulating widely at low frequency and only expanded seven months after being first detected. Similarly, the Glu484Lys substitution in S is often discussed in the context of P.1 and B.1.351 VOC and may allow escape from neutralizing antibodies (Greaney et al. n.d.; Starr et al. 2021), but is not found in B.1.1.7 and therefore does not explain the increased transmission of all three late 2020 VOC. These characteristics suggest that several mutations including those in S are being transmitted as a linked set, i.e. a haplotype and their combined effects (i.e., epistasis) may be contributing to the rapid viral spread.
Widely used molecular evolutionary models based on phylogenetic trees are also problematic because the algorithms that are applied assume that mutations appearing in different SARS-CoV-2 haplotypes are due to repeated, independent mutations and the scientific community is interpreting this as evidence for the same; i.e., the logic is circular. Alternatively, these apparent repeated mutations may represent recombination, which is a common mechanism to accelerate evolution compared to single site mutations in positive strand RNA viruses such as SARS-CoV-2 (Bentley & Evans 2018; Simon-Loriere & Holmes 2011). Furthermore, phylogenetic trees are unable to incorporate important molecular events and metadata such as geospatial and temporal data that would be highly informative for detecting current and future VOC.
In contrast, median-joining networks (MJN) are an efficient and accurate means to analyze haploid genomic data at the population level (Bandelt et al. 1999) such as SARS-CoV-2, (Garvin, Prates, et al. 2020). Unlike independently segregating sites represented by phylogenetic trees, the unit of interest in an MJN is the haplotype, which more accurately reflects the biology of coronaviruses and enables the detection of important evolutionary events such as recombination. Furthermore, a network can be annotated with information including frequency, geospatial location, demographic, or clinical outcomes associated with a unique haplotype to create interpretable patterns of genome variation.
Here, we processed more than 900,000 SARS-CoV-2 genomes using a computational workflow that combines MJN and protein structural analysis (Garvin, Prates, et al. 2020; E. T. Prates et al. 2020) to identify critical attributes of the mutations that define these VOC and provide substantial evidence that the genome-wide mutation load of the late 2020 VOC results from recombination between divergent strains. Using structural analysis and molecular dynamics simulations, we explore the individual effects of key mutations in S and other proteins of SARS-CoV-2 that are shared among different VOC. Additionally, we identify a signature of co-evolution between the residue 501 in S and ACE2, and propose the molecular basis for that. Overall, our results indicate that linked mutations in VOC generated from recombination act in an epistatic manner to enhance viral spread. This work emphasizes the important role of community spread in generating future VOC (Sheikh et al. 2021).
2. Results and Discussion
Network-based views of molecular evolution provide critical information that phylogenetic trees do not
The COVID-19 pandemic is both an unprecedented tragedy and an opportunity to study molecular evolution given the abundant and global sampling of the mutational space of SARS-CoV-2. The MJN is a valuable method of integrating these data to understand viral evolution because the model assumes single mutational steps in which each node represents a haplotype and the edge between nodes is a mutation leading to a new one. Typically, a subsample of extant haplotypes for a taxon is obtained and unsampled, or extinct lineages are inferred. In contrast, SARS-CoV-2 sequence data repositories provide extensive sampling of haplotypes and collection dates (the calendar date of the sample). Given that in an MJN, the temporal distribution of haplotypes is inherent (the model assumes time-ordered sets of mutations), the mutational history of the virus can be traced as a genealogy that can incorporate both the relative and absolute times of emergence of SARS-CoV-2 variants. Importantly, when the single-mutational step MJN model fails, it produces features such as loops or clusters of inferred haplotypes that can indicate biologically important processes such as recombination events, back mutations, or repeat mutations at a site that may be under positive selection.
In order to make a direct comparison, we generated a network and a phylogenetic tree of SARS-CoV-2 haplotypes that were identified from sequences sampled during the first four months of the pandemic and deposited into GISAID (A Global Initiative on Sharing Avian Flu Data, gisaid.org) (Figure 1). Clearly, important metadata such as haplotype frequency, date of emergence, and mutations of interest are easily displayed on the network but are not on the phylogenetic tree. Likewise, at day 96, reticulations (i.e., homoplasy loops) begin to appear in the MJN, indicating reverse mutations to the ancestral states at specific sites or possibly recombination events that can be explored further. Another important feature identified when using networks, but is lost when using phylogenetic trees, is the presence of polytomies. So-called soft polytomies often indicate unsampled genomic information at the species level and hard polytomies are molecular events often found in rapidly expanding populations. For example, haplotype H04 in the MJN (Figure 1) represents a hard polytomy and indicates that a frequent variant is further undergoing multiple independent mutational events, but the phylogenetic tree is unable to convey this information. This example demonstrates the significant gain in information an MJN provides compared to a tree-based view of coronavirus evolution.

a. MJN of SARS-CoV-2 haplotypes, 96, and 120 days. Node sizes in the MJN correspond to sample sizes for a given haplotype and node colors indicate the time of its first report relative to the putative origin of the pandemic in Wuhan. The most abundant haplotypes are named H02 - H05 and numerals 1 - 6 identify important mutations (Garvin, Prates, et al. 2020). Diamond shape nodes denote haplotypes that harbor a 3-bp mutation in the nucleocapsid gene (N) that is highly conserved and directly affects viral replication in vitro (Thorne et al. 2021; Tylor et al. 2009). b. The phylogenetic tree is unable to convey the same information. For example, rapidly expanding populations often display polytomies, i.e., single mutations from a common central haplotype. Those events are readily identified on the MJN but difficult to interpret on a tree because they are usually visualized as a multi-pronged fork (outlined in the dashed-line box) rather than a star pattern (compare H04 in (a) and (b)). These true biological processes also cause tree algorithms to perform poorly because they violate their assumptions, slowing convergence. Additionally, MJN are able to indicate reticulations (i.e., loops) that could denote recombination, reverse mutations, or other biologically important events whereas the forced bifurcation of phylogenetic tree algorithms is unable to display these. Reference sequence: NC_045512, Wuhan, December 24, 2019.
Networks identify lineage-defining mutations of Variants of Concern
We processed more than 900,000 SARS-CoV-2 genomes from human and mink, built a MJN network using the 640,211 genomes that survived our quality control workflow, and annotated with PANGO lineages defined in the GISAID database (Figure 2, see Methods). This genealogy-based approach to molecular evolution identifies the mutations that define a given haplotype based on the edge between nodes. Here, they define the variants of concern and variants of interest (VOI) based on the edge that initiates their corresponding clusters of nodes (Table 1).

a. MJN of haplotypes found in more than 30 individuals (N=640,211 sequences) using 2,128 variable sites. Colors identify PANGO lineages from GISAID. Diamond-shaped nodes correspond to haplotypes carrying a three base pair deletion in the nucleocapsid gene (N) at sites 28881-28883 (Arg203Lys, Gly203Arg). Black square nodes are inferred haplotypes, dashed-line box defines a subgroup of haplotypes within a lineage with a disjoint mutation that is also found in B.1.1.7. Several lineages show introgression from others (e.g., cyan nodes, B.1.160, into brick red, B.1.177). b. Several important mutations in S and non-S proteins appear in multiple lineages. For example, the B.1.1.7 variant carries four mutations that are in disjoint nodes: S Asn501Tyr, S Pro681His, a silent mutation in the codon for amino acid 1089 in nsp3, and theS His69_Val70del that is also found in a clade of haplotypes from mink (blue dashed-line box in (a)). c. Accumulation rate for common GISAID lineages including VOC represented by the ratio between the accumulated number of reported sequences of a given lineage per day since the appearance of that haplotype (Nacc) divided by the corresponding total number (Ntot) at the final sample date for this study. Colors of curves correspond to node colors in (a). All VOC display accumulation rates of at least 1% of the total for that variant per day. The remaining are less than 1% except for the VOI B.1.526 (not displayed in MJN) that is the highest with 3% per day, indicating further scrutiny of this variant is warranted. We also plotted the accumulation rate for lineages that carry the widely reported S Asp614Gly mutation but without the nsp12 Pro323Leu commonly found with it, supporting our previous hypothesis (Garvin, Prates, et al. 2020) that mutations in S alone are not responsible for the rapid transmission of these VOC/VOI but is a function of epistasis among S and non-S mutations. Reference sequence: NC_045512, Wuhan, December 24, 2019.
Center for disease control (CDC)-defined variants and their timing are listed under Lineages and discussed in the text. L-VOC denotes likely variants of concern, that is, those that we propose to have strong potential to become VOC. Non-VOC (N-VOC) are not identified by CDC as VOC. Potential epistatic non-S mutations lineage-defining mutations are listed for the VOC, L-VOC, and VOI. Sites in red font are discussed in the text.
This approach also enables the identification of all the common features acquired in different VOC/VOI, which can elucidate the set of molecular features underlying their rapid spread. For example, the B.1.1.7, B.1.351, and P.1 variants, here referred to as late_2020_VOC, can all be defined by a triple amino acid deletion in the nonstructural protein 6 (nsp6; Ser106_Phe108del) as well as Asn501Tyr in S. Notably this latter mutation has received considerable attention compared to the former (Figure 2a, Table 1), but both are likely key to the biology of these VOC. The MJN also reveals what appears to be a previous dominant but now rare variant (D.2) in Australia; an Ile120Phe mutation in nsp2 was followed immediately by S Ser477Asn, which seems to have led to its rapid expansion in April 2020, indicated by increased node size. In contrast, B.1.160 carries the S Ser477Asn mutation without nsp2 Ile120Phe, and did not demonstrate the same rapid expansion as D.2. This underscores the usefulness of the MJN approach as it is able to convey the sequence of timing of mutational events and the number of individuals carrying those haplotypes simultaneously.
In order to account for sampling bias (there are a disproportionate number of sequences contributed to GISAID by the UK and Australia as well as the high frequency of B.1.1.7 and D.2 in those two geographic locations), we plotted the number of daily samples of selected variants relative to their respective total number of cases to date (April, 2021) and compared the resulting slopes of the linear range of the curves (Figure 2c). The late_2020_VOC, early_2021_VOC, and early_2021_VOI (Table 1) display higher daily accumulation rates, between 1% and 3% of total observed cases per day, compared to other variants (e.g., B.1.177), which show less than 1% accumulation per day. Notably, the rapid increase in D.2 (2%), but not B.1.160 (0.7%) supports the MJN view of this as a likely VOC and the importance of epistasis (here S Ser477Asn and nsp2 Ile120Phe). This analysis and the MJN confirm the importance of monitoring these variants closely and identify both S and non-S mutations that define the current and potential SARS-CoV-2 VOC (Table 1).
Recombination is the likely source for the rapidly expanding variants
Haploid, clonally replicating organisms such as SARS-CoV-2 are predicted to eventually become extinct due to the accumulation of numerous slightly deleterious mutations over time, i.e., Muller’s ratchet (Muller 1964). Recombination is not only a rescue from Muller’s ratchet, it can also accelerate evolution by allowing for the union of advantageous mutations from divergent haplotypes (Bentley & Evans 2018). In SARS-CoV-2, recombination manifests as a template switch during replication when more than one haplotype is present in the host cell, i.e. the virus replisome stops processing a first RNA strand and switches to a second one from a different haplotype, producing a hybrid virus (Simon-Loriere & Holmes 2011). In fact, template switching is a necessary step during the negative-strand synthesis of SARS-CoV-2 when the replisomes functions as an RNA-dependent RNA polymerase and pauses at transcription-regulatory motifs of the sub-genomic template to add the leader sequence from the 5’ end of the genome (this “recombination” is not detected if only a single strain is present, i.e. there is no variation) (Kim et al. 2020),). Given this and the fact that recombination is a major mechanism of coronavirus evolution (Boni et al. 2020) it would be improbable for this process not to occur in the case of multiple strains infecting a cell (Gribble et al. 2021).
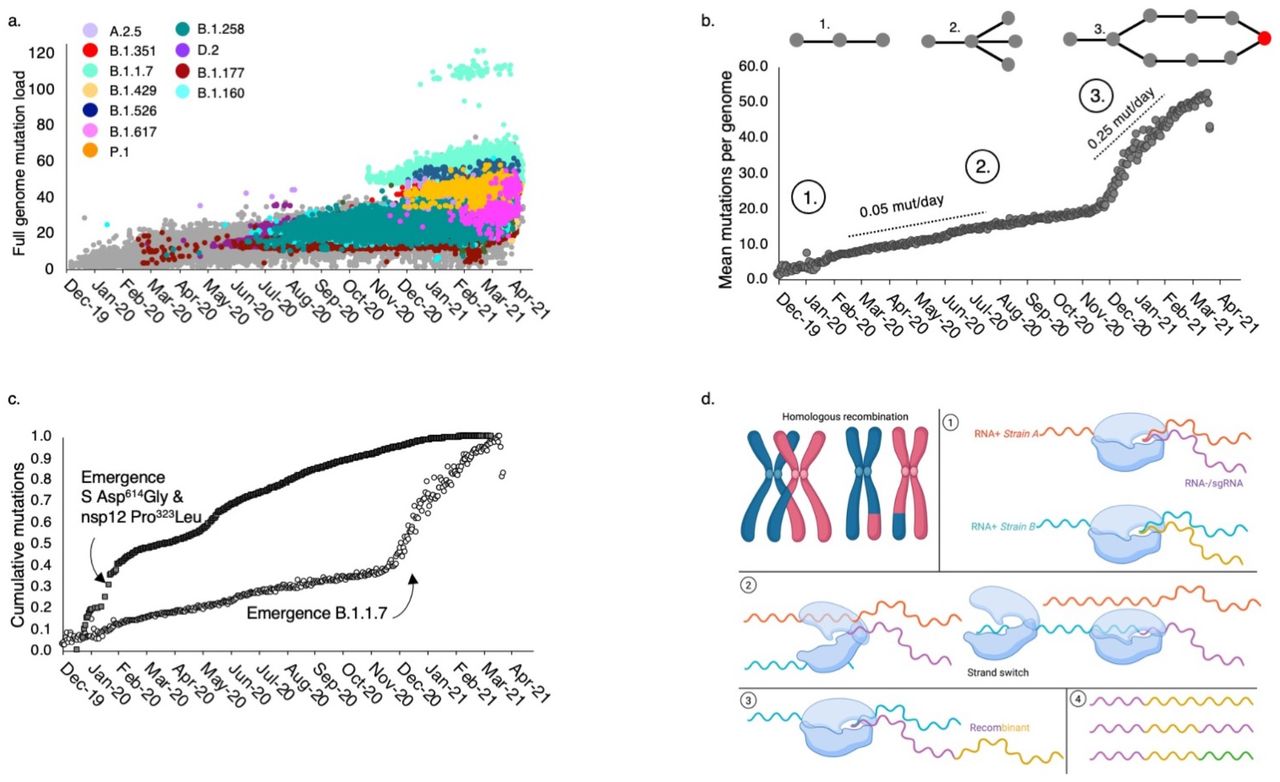
The late_2020_VOC exhibits large numbers of new mutations relative to any closely related sequence indicating rapid evolution of SARS-CoV-2 (Figure 3a). For example, the original node of B.1.1.7 differs from the most closely related node by 28 mutations. However, the majority of this total (15) corresponds to deletions that could be considered two single mutational events, as does a 3-bp change in N (28280-22883) since they occur in factors of three (a codon), maintaining the coding frame. By summing the two deletions and the full codon 3-bp change in N with the 10 remaining single site mutations, a conservative estimate would be 13 distinct mutational events leading to B.1.1.7. The plot of the accumulating mutations in the 640,211 haplotypes sampled to date reveals a linear growth of roughly 0.05 mutations per day (Figure 3b) and therefore, given this pace, it would be expected to take 260 days for these 13 mutational events to accumulate in a haplotype. For the B.1.1.7 to appear in October 2020 as reported, the genealogy would have to have been initiated in January 2020 and yet the nearest node harboring the S Asn501Tyr mutation was not sampled until June 2020 and no intermediate haplotypes have been identified to date.

a. A rapid increase in the number of mutations per individual genome is evident in the late_2020_VOC. The outliers of the B.1.1.7 lineages (mint green) are a subset of that lineage due to a single, 57 base pair deletion in ORF7a (amino acids 5-23). b. Mean mutation load per individual, based on 2,128 high-confidence sites by date. The SARS-CoV-2 virus accumulated an estimated 0.05 mutations per day until the appearance of B.1.1.7, when it increased five-fold. Circles with numbers denote three processes occurring at different timepoints: (1) emergence, (2) haplotype expansion, and (3) recombination of divergent lineages. c. A population-level analysis of new mutations per day over the same time period (dark squares) displays a declining rate of mutations with a slight increase around the emergence of D.2 in Australia but not an increase with the emergence of B.1.1.7 that could explain the rapid accumulation of mutations shown in (b) plotted as percent accumulation (unfilled circles). d. Recombination in a diploid organism results from the crossover of homologous chromosomes during meiosis. In RNA+ betacoronaviruses, recombination occurs when two or more strains (haplotypes) infect a single cell (1). The replisome dissociates (2) from one strand and switches to another, (3) generating a hybrid recombinant. The resulting chimera (4) can be as simple as a section of strain A fused to a section of strain B or more complex recombinants if strand switching occurs more than once or there are multiple strains per cell (green section). Reference sequence: NC_045512, Wuhan, December 24, 2019.
Alternatively, it could be that the 13 mutational events occurred between June and October (122 days), but the probability of this is about one in 1015 (Supplemental Methods). Furthermore, all 28 differences between the Wuhan reference sequence and B.1.1.7 appeared in earlier haplotypes (Table S1) and therefore, if rapid evolution were the cause, it would require the extremely unlikely process of 28 independent, repeat mutations to the same nucleotide state. In order to test this, we plotted the population-level mutations per day (including repeat mutations at variable sites), which did not reveal any increase in mutation rate at the time of the B.1.1.7 and in fact displayed a decrease with the emergence of the late_2020_VOC (Figure 3c, Figure S1). Possible explanations are either a large increase in mutations in a small number of individuals over a short time period (that would have to occur on multiple continents to explain B.1.1.7, B.1.351, and P.1), or recombination between two or more divergent haplotypes carrying the VOC mutations (Figure 3d).
Recombination is the most parsimonious explanation given (1) the absence of a substantial increase in mutation rate at any time prior to the appearance of the VOC along with their increased mutation load, which can easily be explained by template-switching during replication (Simon-Loriere & Holmes 2011), (2) the widespread and early circulation of the majority of the mutations associated with them in other haplotypes and, (3) that several mutations appear disjointly across the MJN (Figure 2a). The first notable disjoint mutation is Ser477Asn in S that defines D.2 along with nsp2 Ile120Phe (Figure 2a), which then appears in B.1.160. Likewise, Asn501Tyr and Pro681His in S appear in divergent haplotypes, including one mink subgroup from Denmark and a basal node to B.1.351 (without the nsp6 deletion). It could be argued that those in S (Asn501Tyr, Ser477Asn, and Pro681His) are the result of multiple independent mutation events because they are under positive selection (Martin et al. 2021), but we also identified a mutation in nsp3 that is one of the lineage-defining mutations for B.1.1.7 and appears in disjoint nodes, but is unlikely to be under selection because it is synonymous. Furthermore, mutation and selection are separate events, i.e., even if sites appearing in multiple lineages are under positive selection, their appearance in disjoint nodes still requires repeated mutations in the absence of recombination.
It should also be noted that recombination can generate a high number of false-positives when testing for signs of positive selection (Anisimova et al. 2003), and the complexity of coronavirus recombinants compared to those generated in diploid organisms through homologous chromosome crossovers (Figure 3d) makes that process difficult to detect. Therefore, analyses that test for positive selection based on multiple independent mutations at a site may, in fact, be false positives that result from recombination events. The majority of the mutations found in B.1.1.7 could be explained by the admixture and recombination among lineages and a random scan of 100 FASTQ files from B.1.1.7 available in the NCBI SRA database identified two co-infected individuals in further support of this hypothesis (Table S2). Large-scale analyses of these data with newer methods may enable the detection of recombinants (Müller et al. 2021).
A recent analysis of sequences in the U.K. that leveraged the geographical and temporal presence of specific sequences identified recombinants whose parents appeared to be from B.1.1.7 and B.1.1.177-derived strains (Jackson et al. n.d.). Although this demonstrates that recombination is occurring, it does not answer if B.1.1.7 itself arose through recombination. As noted above, the majority of B.1.1.7-defining mutations appeared much earlier in the pandemic outside the U.K. and therefore approaches that focus on a single geographic location will miss parental variants. The probability of proving B.1.1.7 originated from recombination using this method is further reduced if the parental lineages were less fit or poorly sampled and therefore rare. The present study demonstrates that detecting recombination events requires the use of data on a global scale and the inclusion of rare variants.
The potential functional impact of key mutations in S and non-S proteins
Given the results from the MJN analysis and our previous hypothesis (Garvin, Prates, et al. 2020) that the cooperative effects of mutations in S and non-S proteins (i.e., epistasis) define and are responsible for the increased transmission of prevalent SARS-CoV-2 variants (Lauring & Hodcroft 2021), we performed protein structural analyses and discuss below the functional effects of these individual and combined mutations in SARS-CoV-2 VOC. We analyze ten likely key mutation sites (red font, Table 1) in S and non-S proteins.
S Asn501Tyr
Located in the receptor-binding domain (RBD) of SARS-CoV-2 S, immunoprecipitation assays reveal that site 501 plays a major role in the affinity of the virus to the host receptor, ACE2 (Shang et al. 2020). Via structural analysis and extensive molecular dynamics simulations, Ali et al. highlighted the importance of the interactions with human ACE2 (hACE2) near the site 501 of the receptor-binding domain of S, particularly via a sustained hydrogen bond between RBD Asn498 and hACE2 Lys353 (Ali & Vijayan 2020). Deep mutational scanning of SARS-CoV-2 RBD reveals that the naturally occurring mutations at site 501, Asn501Tyr and Asn501Thr, lead to an increased affinity to hACE2 (Starr et al. n.d.). Additionally, this site is located near a linear B cell immunodominant site (B.-Z. Zhang et al. 2020), and therefore the mutation may allow SARS-CoV-2 variants to escape neutralizing antibodies (Figure 4, Figure 5). Indeed, neutralizing antibodies derived from vaccinations and natural infection have significantly reduced activity against pseudotyped viruses carrying this mutation (Wang et al. 2021).

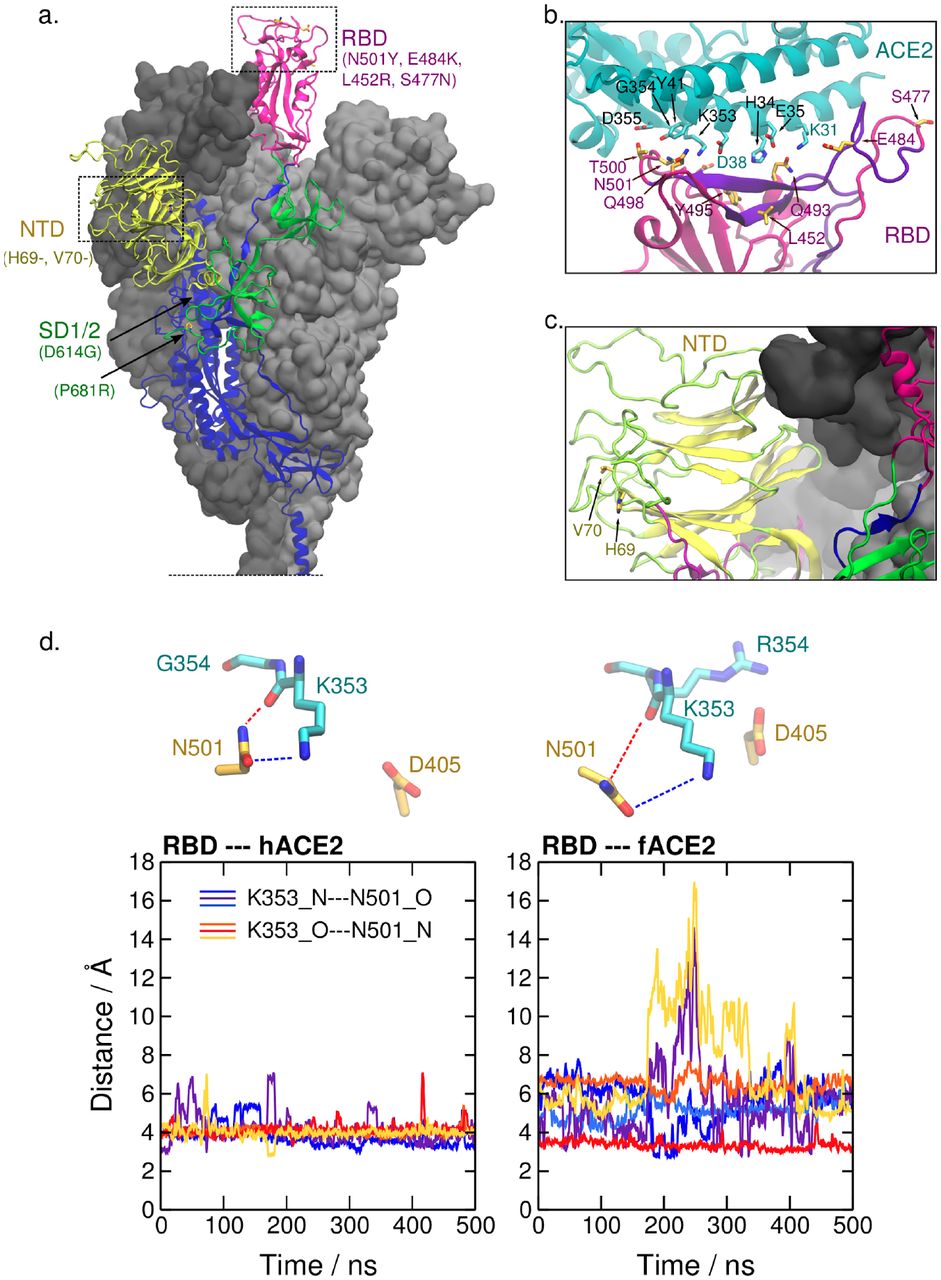
a. Several mutations associated with dominant haplotypes are located in the receptor-binding domain (RBD, aa. 331-506), N-terminal domain (NTD, aa. 13-305), and subdomains 1 and 2 (SD1/2, aa. 528-685) of S. The structure of S in the prefusion conformation derived from PDB ID 6VSB (Wrapp et al. 2020) and completed in silico (Casalino et al. 2020) is shown. Glycosyl chains are not depicted and the S trimer is truncated at the connecting domain for visual clarity. The secondary structure framework of one protomer is represented and the neighboring protomers are shown as a gray surface. b. Mutation sites in the S RBD of SARS-CoV-2 VOC, such as 484, 452, 477, and 501 are located at or near the interface with ACE2. Notably, site 452 and 484 reside in an epitope that is a target of the adaptive immune response in humans (aa. 480-499, in violet) and site 501 is also located near it (B.-Z. Zhang et al. 2020). Dashed lines represent relevant polar interactions discussed here. PDB ID 6M17 was used (R. Yan et al. 2020). c. The sites 69 and 70 on the NTD, which are deleted in the VOC B.1.1.7, are also found near an epitope (aa. 21-45, in violet) (B.-Z. Zhang et al. 2020). d. Time progression of N---O distances between atoms of Asn501 in RBD and Lys353 in human and ferret ACE2 (hACE2 and fACE2, respectively) from the last 500 ns of the simulation runs. Colors in the plots correspond to the distances Lys353_N---Asn501_O (cold colors) and Lys353_O---Asn501_N (warm colors) in three independent simulations of each system. These distances are represented in the upper part of the figure.

a. As part of the innate immune response, (Step 1) the SARS-CoV-2 virus is internalized into endosomes and degraded. (Step 2) viral RNA activates the mitochondrial antiviral innate immunity (MAVS) pathway and (Step 3) degraded proteins activate the toll receptor pathway (TLR3/TLR7), which result in the (Step 4) phosphorylation of TBK1 and translocation of NF-kB and IRF3 to the nucleus, where they regulate the transcription of immune genes including interferons (IFNs, Step 5). IFNs recruit CD8+ T cells that, (Step 6) recognize fragments of the virus on the cell surface via their class I major histocompatibility complex (MHC I) receptors and are activated by dendritic cells (antigen processing cells, or APC). If the virus bypasses innate immunity (orange arrows) nonstructural proteins (nsp6 and nsp13) block the IRF3 nuclear translocation. b. APCs recruit B lymphocytes and stimulate the production of antibodies that recognize SARS-CoV-2 S (whereas T cells recognize fragments of S bound to MHC I). c. The neutralizing antibodies block binding of the virus to the ACE2 receptor and can prevent re-infection but mutations in the receptor-binding domain (RBD), e.g., S Asn501Tyr, prevent binding of the antibodies and the virus is then able to bind the receptor again even if individuals experienced exposure to an earlier strain or were vaccinated. Created with BioRender.com.
Transmission between human and non-human hosts for SARS-CoV-2 provides further information on the evolutionary selectivity of site 501 in S. Repeated infection of mice with human SARS-CoV-2 resulted in the selection of a mouse-adapted strain carrying S Tyr501 (Gu et al. 2020). It is possible that Asn501Tyr results in an additional stabilization of the RBD-ACE2 interaction via π-stacking of Tyr501 with Tyr41 in ACE2 (Figure 4a-b). In contrast, several introductions into farmed mink (Neovison vison), which caused a substantial increase in their mortality (Oude Munnink et al. 2021), have not led to the same selection. To date, reported sequences in GISAID of SARS-CoV-2 in this host carry either S Asn501, which is prevalent, or S Thr501, which appeared independently in mink farms (Table S3) (Oude Munnink et al. 2021). In ACE2 of these taxa, Tyr41 is conserved, but near this site, a larger, positively charged amino acid, His354, replaces Gly354. Table 2 shows that the amino acids in the RBD 501-binding region of the ACE2 orthologues are conserved, except for Gly354, indicating that this site may play a key role in viral fitness.
Relative to the human sequence, almost all these residues are either conserved (“|”) or replaced by a nearly equivalent amino acid in mouse, American mink, European mink, ferret, and pangolin. Notably, there is a nonconservative substitution of Gly354 to a bulky positively charged amino acid in most species. Our structural analyses suggests that this substitution contributes to a putative host-dependent selective pressure at site 501 of SARS-CoV-2 S. Prevalent residues reported at this site are informed in order of frequency.
Similarly, ferrets (Mustela putorius furo) and pangolins (Manis pentadactyla), relevant potential reservoirs of SARS-CoV-2, carry a large basic residue at site 354 (an arginine and histidine, respectively). Sawatzki et al. reported that the constant exposure of ferrets to infected humans did not result in natural transmission in a domestic setting, suggesting that ferret infection may require improved viral fitness (Sawatzki et al. 2021). In agreement with that, Richard et al. (Richard et al. 2020) reported that the adaptive substitution Asn501Thr was detected in all experimentally infected ferrets in the laboratory. In order to further investigate the role of the Gly354 versus Arg354 in the adaptive mutation of site 501 in S RBD, we performed extensive molecular dynamics simulations of the truncated complexes of Asn501-carrying RBD of SARS-CoV-2 and ACE2, from human (hACE2) and ferret (fACE2). The simulations indicate that there is a remarkable difference in the interaction pattern between the two systems in the region surrounding site 501 of RBD. Firstly, we identified the main ACE2 contacts with Asn501, which were the same for both species, namely, Tyr41, Lys353, and Asp355, and we also show that the intensity of these contacts is lower in the simulations of fACE2 (Table S4).
To investigate further, we analyzed structural features in the interaction between ACE2 Lys353 and RBD Asn501. Distances between polar atoms computed from the simulations indicate a weaker electrostatic interaction between this pair of residues in ferret compared to human (Figure 4d). This effect is accompanied by a conformational change of fACE2 Lys353. Figure S2 shows that, in ferret, the side chain of Lys353 exhibits more stretched conformations, i.e., a higher population of the trans mode of the dihedral angle formed by the side chain carbon atoms. This conformational difference could be partially attributed to the electrostatic repulsion between the two consecutive bulky positively charged amino acids in ferrets, Lys353 and Arg354. Additionally, the simulations suggest a correlation, in a competitive manner, between other interactions that these residues display with the RBD. For example, Figure S3 shows that the salt bridge fACE2_Arg354---RBD_Asp405 and the HB interaction fACE2_Lys353---RBD_Tyr495 (backbone) alternate in the simulations. This also suggests that the salt bridge formed by fACE2 Arg354 drags Lys353 apart from RBD Asn501, weakening the interaction between this pair of residues in ferrets relative to humans.
Altogether, these analyses indicate that site 354 in ACE2 significantly influences the interactions with RBD in the region of site 501 and is likely playing a major role in the selectivity of the size and chemical properties of this residue in SARS-CoV-2. We propose that, in contrast to Tyr501, a smaller HB-interacting amino acid at site 501 of RBD, such as the threonine reported in farmed mink and ferrets, may ease the interactions on the region, e.g., the salt bridge between fACE2 Arg354 and RBD Asp405. The differences in the region of ACE2 in contact with site 501 seem to have a key role for host adaptation and are worth further investigation as it may also reveal details of the origin of this zoonotic pandemic.
S His69_Val70 deletion
The His69_Val70 deletion (in B.1.1.7) is adjacent to a linear epitope at the N-terminal domain of S (Figure 4a,c) (B.-Z. Zhang et al. 2020), suggesting it too may improve fitness by reducing host antibody effectiveness.
S Leu452Arg
The Leu452Arg mutation in S is a core change in the early_2021_VOC (Table 1, Figure 4a-b). Although Leu452 does not interact directly with ACE2, this mutation was shown to moderately increase infectivity in cell cultures and lung organoids using Leu452Arg-carrying pseudovirus (Deng et al. 2021). It is possible that the substitution of the leucine, hydrophobic, to arginine, a positively charged residue, creates a direct binding site with ACE2 via the electrostatic interaction with Glu35. However, in Starr et al., experiments with the isolated RBD expressed on the cell surface of yeast show that this mutation is associated with enhanced structural stability of RBD, while it only slightly improves ACE2-binding (Starr et al. n.d.). An alternative but not mutually exclusive hypothesis is that it causes a local conformational change that impacts the complex dynamic interchange between interactions of RBD with the spike trimer itself and with the host receptor. Noteworthy, site 452 resides in a significant conformational epitope in RBD and Leu452Arg was shown to decrease binding to neutralizing antibodies (Figure 4b) (Deng et al. 2021; Li et al. 2021).
As noted in Deng et al., S Leu425Arg has been reported in rare variants starting in March 2020 from Denmark, i.e., several months before the surge of the VOC that carry this mutation (B.1.427, B.1.429, and B.1.617) (Deng et al. 2021). This indicates that the high transmissibility of the early_2021_VOC is not fully explained by the increased infectivity caused by Leu425Arg and combined mutations may be essential for the rapid spread. Besides the other mutations in the spike in these VOC, the substitution Asp260Tyr in the SARS-CoV-2 helicase (nsp13, below) is especially interesting, as it was identified in the MJN analysis as a defining mutation of both B.1.427 and B.1.429 variants.
S Ser477Asn
Variants carrying the S Ser477Asn mutation spread rapidly in Australia (Figure 1, Figure 3b). This site, located at the loop β4-5 of the RBD, is predicted not to establish persistent interactions with ACE2 (Ali & Vijayan 2020). However, deep scanning shows that this mutation is associated with a slight enhancement of ACE2-binding. Molecular dynamics simulations suggest that Ser477Asn affects the local flexibility of the RBD at the ACE2-binding interface, which could be underlying the highest binding affinity with ACE2 reported from potential mean force calculations (A. Singh et al. n.d.). Additionally, this site is located near an epitope and may alter antibody recognition and counteract the host immune response (Figure 4b).
S Glu484Lys
A recent computational study suggests that Glu484 exhibits only intermittent interactions with Lys31 in ACE2 (Ali & Vijayan 2020). Deep scanning shows that this mutation is associated with higher affinity to ACE2 (Starr et al. n.d.) and may be explained by its proximity to Glu75 in ACE2, which would form a salt bridge with Lys484. Aside from the potential impact of Glu484Lys between virus-host cell interaction, this site is part of a linear B cell immunodominant site (B.-Z. Zhang et al. 2020) and this mutation was shown to impair antibody neutralization (Wang et al. 2021).
S Pro681Arg and Pro681His
These mutations in the multibasic furin cleavage site are particularly relevant given the importance of this region for cell-cell fusion (Hoffmann et al. 2020; Papa et al. n.d.). The presence of the multibasic motif of SARS-CoV-2 has shown to be essential to the formation of syncytia (i.e., multinucleate fused cells), and thus it is thought to be a key factor underlying pathogenicity and virulence differences between SARS-CoV-2 and other related betacoronaviruses. Hoffmann et al. recognized the importance of the furin cleavage site in
SARS-CoV-2 and its biochemically basic signature and generated mutants to determine the effects of specific amino acids. Notably, they showed that pseudotyped virion particles bearing mutant SARS-CoV-2 S with additional basic residues in this region, including the substitution Pro681Arg (present in B.1.617), exhibits a remarkable increase in syncytium formation in lung cells in vitro (Hoffmann et al. 2020), which may explain the increased severity of the disease (Sheikh et al. 2021). Hoffman et al did not include a Pro681His change that is a defining mutation of B.1.1.7, and so it is not known if this too increases syncytium formation given that it is a basic amino acid, but should be the target of future studies.
Nucleocapsid Arg203Met
The main function of the nucleocapsid (N) protein in SARS-CoV-2 is to act as a scaffold for the viral genome and it is also the most antigenic protein produced by the virus (Dutta et al. 2020). In a previous study, we reported that the Ser-Arg-rich motif of this protein (a.a. 183-206), shown in vitro to be necessary for viral replication (Garvin, Prates, et al. 2020; Tylor et al. 2009), displays a high number of amino acid changes during the COVID-19 pandemic and is likely under positive selection. We propose that the RNA gene segment coding this particular subsequence may be linked to improved fitness of specific SARS-CoV-2 haplotypes including the rapidly spreading delta variant and is linked to epigenetic alterations (Figure 6a).

a. Location of 41 epigenetic sites reported in Kim et al. 2020 (red bars on SARS-CoV-2 genome). One of the sites in the nucleocapsid gene (nucleotides in red box of aligned sequences) is highly conserved across diverse host-defined coronaviruses. All bats and human coronavirus species from China are completely conserved at the epigenetic site 28881-28883, except for a 3-bp mutation in SARS-CoV-2 that occurred early in the pandemic and now corresponds to ∼50% of all sequences globally (diamond nodes in Figure 1). b. Kim et al. proposed that N6-methyladeonsine modification of the genome (purple hexagons), common in RNA viruses, caused the strand to pause while traversing the nanopore sequencing apparatus. We propose that loss of this site via mutations at site 203 in N may increase the replication rate of the RNA strand through the SARS-CoV-2 replisome. Aselliscus stoliczkanus - Stoliczka’s trident bat, Chaerephon plicata - wrinkle-lipped free-tailed bat, Rhinolophus pusillus - least horseshoe bat, R. pearsoni - Pearson’s horseshoe bat, R. macrotis - big-eared horseshoe bat, R. ferrumequinum - greater horseshoe bat, R. monoceros - Formosan lesser horseshoe bat, R. affinis intermediate horseshoe bat, R. sinicus Chinese rufous horseshoe bat, R. mayalanis - Mayalan horseshoe bat, SARS - Severe Acute Respiratory Syndrome, Manis javanica - Malayan pangolin. Created with BioRender.com.
A recent deep transcriptome sequencing study used Oxford NanoporeTM technology to detect epigenetic modifications at 41 sites in the RNA genome that are associated with leader sequence addition to sub-genomic RNA transcripts, a recombination-like process of SARS-CoV- 2 (Kim et al. 2020). Nanopore instruments can detect epigenetic modifications based on disruptions of the electrical current as the RNA molecule passes through the molecular pore (Rand et al. 2017; Simpson et al. 2017), which Kim et al propose is responsible for the pause that occurs before leader sequence addition. Twenty-five of the 41 modified sites reside in the N gene and the majority of the sites in this subset are found near the Ser-Arg-rich motif (Figure 6a).
Furthermore, one specific epigenetic site is linked to two highly successful SARS-CoV-2 haplotypes. The first is a triple mutation at sites 28881-28883 (GGG to AAC, Arg203Lys) that is now found in nearly half of all sequences sampled across the globe (diamond nodes, Figure 1) and the second is Arg203Met, which is a defining mutation for the rapidly spreading B.1.617. Notably, this region of the genome is highly conserved across several hundred years of coronavirus evolution (Boni et al. 2020) (Figure 6a). Given that these epigenetic sites were discovered because the RNA pauses as it passes across the pore of the molecular nanopore sequencer, one interesting hypothesis is that mutations at this region remove the epigenetic modification and speed the SARS-CoV-2 genome through the replisome (Figure 6b), increasing the production of virions, which is consistent with the more than 1000-fold higher virion count in those infected with B.1.617 (Lu et al. n.d.).
nsp2 Ile120Phe
The main role of the nonstructural protein 2 (nsp2) in viral performance is not yet defined. Instead, this protein appears to be part of multiple interactions with host proteins involved in a range of processes including the regulation of mitochondrial respiratory function, endosomal transport, and ribosome biogenesis (Verba et al. 2021). Very recently, deep learning-based methods of structure prediction and cryo-electron microscopy density were combined to provide the atomic model of nsp2 (PDB id 7MSW). In a preprint from Verba et al., structural information was used to localize the surfaces that are key for protein-protein interaction with nsp2 (Verba et al. 2021). From structural analysis and mass spectrometry experiments, the authors pose the interesting hypothesis that nsp2 interacts directly with ribosomal RNA via a highly conserved zinc ribbon motif to bring ribosomes close to the replication-transcription complexes.
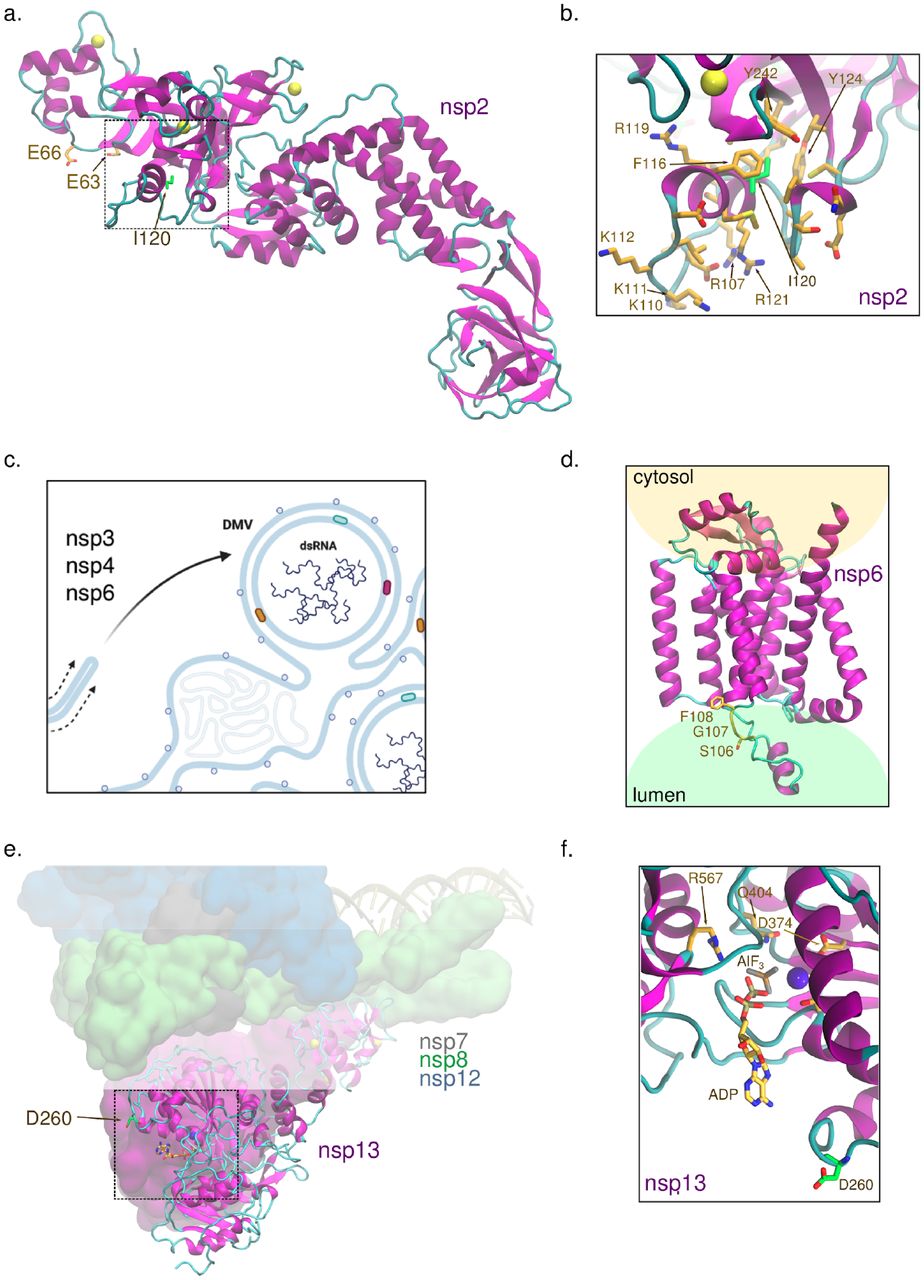
Here we are particularly interested in the functional impact of the mutation Ile120Phe in nsp2 present in the D.2 variant. Site 120, identified in the nsp2 structure on Figure 7a, is a point of hydrophobic contact between a small helix, rich in positively charged residues, and a zinc binding site. The positively charged surface of the helix may be especially relevant for a putative interaction with the phosphate groups from ribosomal RNA. Normal mode analysis from DynaMut2 predicts that the substitution has a destabilizing effect in the protein structure (estimated ΔΔGstability = -2 kcal/mol) (Rodrigues et al. 2021). Possibly, this could be caused by π-π stacking interactions of the tyrosine with aromatic residues in the same helix that would disrupt the contacts anchoring it to the protein core (Figure 7b). Additionally, site 120 is spatially close to Glu63 and Glu66, which were shown to be relevant for interactions with the endosomal/actin machinery via affinity purification mass spectrometry in HEK293T cells. Remarkably, upon mutation of these glutamates to lysines, there is increased interactions with proteins involved in ribosome biogenesis (Verba et al. 2021).

a. Site 120 in nsp2 is located in a small helix near a zinc-binding site and residues Glu63 and Glu66, which play a role in the interaction with proteins involved in ribosome biogenesis and in the endosomal/actin machinery (Verba et al. 2021). PDB id 7MSW was used. b. Ile120 forms some of the hydrophobic contacts that anchor the helix at the surface of nsp2, where this site resides, to the protein core. c. Nsp6 participates in generating double-membrane vesicles (DMV) for viral genome replication. Natural selection for the biological traits of viral entry and replication may explain the increased transmission of variants with adaptive mutations in both S and nsp6. DMVs isolate the viral genome from host cell attack to provide for efficient genome and sub-genome replication and generate virions. d. Sites 106-108 are predicted to be located at/near the protein region of nsp6 embedded in the endoplasmic reticulum lumen (structure generated by AlphaFold2 (Jumper et al. n.d.)). e. Nsp13 is the SARS-CoV-2 helicase and it is part of the replication complex. f. Asp260 in nsp13 is mutated to tyrosine in B.1.427 and B.1.429 and it is located at the entrance of the NTP-binding site. PDB id 6XEZ was used (Chen et al. 2020).
nsp6 Ser106_Phe108deletion
The nsp6 protein plays critical roles in viral replication and suppression of the host immune response (Figure 5a and Figure 7c) (Gupta et al. 2020). Along with nsp3 and nsp4, nsp6 is responsible for producing double-membrane vesicles from the endoplasmic reticulum (ER) to protect the viral RNA from host attack and increase replication efficiency (Figure 7c) (Santerre et al. 2020). The nsp6 Ser106_Phe108del is predicted to be located at a loop in the interface between a transmembrane helix and the ER lumen based on a preliminary structural analysis of the model generated by the AlphaFold2 system (Figure 7d), and we hypothesize that the deletion may affect functional interactions of nsp6 with other proteins. In addition, in agreement with the enhanced suppression of innate immune response reported for B.1.1.7 (Thorne et al. 2021), changes in immune-antagonists, such as nsp6 Ser106_Phe108del, may be key to prolonged viral shedding (Calistri et al. 2021).
nsp13 Asp260Tyr
The nonstructural protein 13 is a component of the viral replication-transcription complex, (nsp13; or SARS-CoV-2 helicase) and plays an essential role in unwinding the duplex oligonucleotides into single strands in a NTP-dependent manner (L. Yan et al. 2020). Hydrogen/deuterium exchange mass spectrometry demonstrates that the helicase and NTPase activities of SARS-CoV nsp13 are highly coordinated, and mutations at the NTPase active site impair both ATP hydrolysis and the unwinding process (Jia et al. 2019). Here we note that the substitution Asp260Tyr, present in B.1.427 and B.1.429, is located at the entrance of the NTPase active site and may favor π-π stacking interactions with nucleobases (Figure 7e-f). Given that, at high ATP concentrations, SARS-CoV nsp13 exhibits increased helicase activity on duplex RNA (Jang et al. 2020), it is possible that, similarly, the putative optimization on NPT uptake in nsp13 Asp260Tyr favors RNA unwinding.
Additionally, nsp13 was shown to play an important role as an innate immune antagonist (Figure 5a). It contributes to the inhibition of the type I interferon response by directly binding to TBK1 and, with that, it impedes IRF3 phosphorylation (Guo et al. 2021). The dual role of nsp6 and nsp13 in immune suppression and viral replication may suggest a convergent evolution of SARS-CoV-2 manifested in most of the VOC, which carries either nsp6 Ser106_Phe108del or nsp13 Asp260Tyr.
3. Concluding Remarks
From our thorough analysis of the spatiotemporal relationships of SARS-CoV-2 variants, we propose that the rapid increase of mutations in the late 2020 VOC is likely a consequence of the recombination of haplotypes carrying adaptive mutations in S and in non-S proteins that act cooperatively to enhance viral fitness. For example, as indicative of that, we call attention to five mutations that occur independently in disjoint clusters of our MJN, four of which (S Asn501Tyr, S His69_Val70del, S Phe681His/Arg, and nsp6 Ser106_Phe108del) are shared by different VOC, including B.1.1.7. Notably, S His69_Val70del appeared in human and mink populations simultaneously in August 2020, prior to the emergence of B.1.1.7, indicating that mink should be further investigated as a possible component of a recombination event. In turn, our molecular dynamics simulations indicate that the molecular forces at site 501 in S and how they are altered upon mutation (S Asn501Tyr in B.1.1.7) are a key component to describe the history of transmission among other putative zoonotic reservoirs, such as farmed minks, ferrets, and pangolins.
The S Asp614Gly mutation has been shown to increase infectivity and is now predominant in the circulating virus (L. Zhang et al. 2020), and S Asn501Tyr is associated with higher virulence (Gu et al. 2020). We show that the expansion of the strains carrying these mutations only occurred upon the additional substitutions in nsp12 Leu323Pro (Figure 2b) (Garvin, Prates, et al. 2020) and nsp6 Ser106_Phe108del, respectively. A hypothesis consistent with these observations is that the changes in S enhance viral entry into the host cells but they do not easily transmit due to rapid suppression by a robust innate immune response. A secondary mutation is able to counteract the immune-driven suppression. In the case of S Asp614Gly, the nsp12 Leu323Pro may have increased the replication rate of the virus, which was supported by quantitative PCR from clinical samples with different viral strains in Korber et al. (Garvin, Prates, et al. 2020; Korber et al. 2020). However, the separate effects of S Asp614Gly and nsp12 Leu323Pro could not be described in the referred study because it did not include individuals infected with variants harboring only one of the mutations.
For the late 2020 VOC, nsp6 Ser106_Phe108del may affect viral replication in DMVs or suppress the interferon-driven antiviral response (Xia et al. 2020). It is likely that other mutations also enhance viral mechanisms that impair the host immune response. For example, Thorne et al. recently showed that the B.1.1.7 VOC suppresses the innate immune response by host cells in vitro and attributed it to the increased transcription of the orf9b gene, nested within the gene coding the nucleocapsid protein. (Thorne et al. 2021), although they could not rule out the possibility that this was due to nsp6 Ser106_Phe108del.
Via focused protein structural analysis, we identify other mutations shared among different VOC that reside in key locations of proteins involved in viral replication and/or in suppressing the innate immune suppression, such as nsp13, suggesting a convergent evolution of SARS-CoV-2. This emphasizes the importance of tracking mutations in a genome-wide manner as a strategy to avoid the emergence of future VOC. For example, an earlier dominant variant in Australia (D.2) that carried the mutations Ser477Asn in S and Ile120Phe in nsp6 was successfully restrained. However, we note that variants harboring only the S Ser477Asn substitution are currently circulating in several European countries (Figure 2, Table S5) and may only need to recombine with a variant carrying an advantageous complementary mutation to become the next VOC.
A second and equally significant outcome from recombination-driven haplotypes is the generation of variants that allow escape from neutralizing antibodies produced by an adaptive immune response (Garvin, T Prates, et al. 2020) (Figure 5c). As a case in point, the resurgence of COVID-19 in Manaus, Brazil, in January 2021, where seroprevalence was above 75% in October 2020, is due to immune escape of new SARS-CoV-2 lineages (Sabino et al. 2021). Broad disease prevalence and community spread of COVID-19 increase the probability that divergent haplotypes may come in contact, thereby dramatically accelerating the evolution and transmission of the virus. This emphasizes that regions with low sequence surveillance can be viral breeding grounds for the next SARS-CoV-2 VOC.
4. Methods
Sequence data pre-processing
We downloaded SARS-CoV-2 sequences in FASTA format and corresponding metadata from GISAID and processed as we have reported previously (Garvin, Prates, et al. 2020; E. Prates et al. 2021). To ensure that deletions were accounted for, full genome sequences were aligned with MAFFT (Katoh et al. 2002) to the established reference genome (accession NC_045512) , uploaded into CLC Genomics Workbench, and trimmed to the start and stop codons (nsp1 start site and ORF10 stop codon). Aligned sequences in tab-delimited format were imported into R to count the number of variable accessions at each of the 29,409 sites.
Variable sites were determined with all sequences downloaded up through the end of January, 2021. In order to reduce false-positive mutation sites (those that were due to technical error), we selected sites that were variable in 25 or more individuals (0.01%) compared to the reference (all 25 were required to be the same state: A, G, T, C, or -). We further pruned these by removing sites in which 20% or more of the accessions harbored an unknown character state (“N”), leaving 2,128 variable sites for downstream analyses. After removing sequences with an “N” at any of these sites, we retained 280,409 individuals. Prior to submission, we updated the number of sequences through April 19, 2021, keeping the same 2128 variable sites, which allowed us to capture the most up-to date metadata and produced 640,211 for analysis. We kept haplotypes that occurred in more than 35 individuals to remove rare or artifact-derived haplotypes. For the comparison of median-joining networks and phylogenetic trees, we used sequences from the pandemic sampled through the end of April, 2020. We used variable sites found in more than ten individuals and haplotypes found in five or more individuals as we had in previous work (Garvin, Prates, et al. 2020). This produced 410 unique haplotypes based on 467 variable sites.
Median-joining network (MJN)
Haplotypes were coded in NEXUS format and uploaded to PopArt (Leigh & Bryant 2015). An MJN was produced with the epsilon parameter set to 0. The networks were exported as a table and visualized in Cytoscape (Shannon et al. 2003) with corresponding metadata. The date of emergence of each haplotype was defined by the sample date subtracted from the report date for the Wuhan reference sequence (December 24, 2019) and then one day was added to remove zeros. For samples that only reported the month but no day, we recorded the day as the 15th of that month. We excluded samples with no sampling date.
Phylogenetic tree
We used the program MrBayes to generate a phylogenetic tree (Ronquist & Huelsenbeck 2003). Parameters were set to Nucmodel=4by4, Nst=6, Code=Universal, and Rates=Invgamma. We performed 5,000,000 mcmc generations, which produced a stable standard deviation of split frequencies of 0.014. A consensus tree was generated using the 50% majority rule and visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Estimation of genome mutation load
We estimated the mutation load using two data sets. First, we used the 640,211 sequences based on 2,128 variable sites used for the MJN because these represent high-confidence mutations. For each of the 640,211 accessions, we counted the number of differences of the 2,128 variable sites compared to the reference genome (accession NC_045512) and recorded the day of emergence. The mutational load for all accessions for a given day was then averaged and this was plotted across time. For the second estimate of mutation rate, we used all variable sites across the full genome (29,409 sites) to include rare variants and removed all sequences with at least one ambiguous site, leaving 584,119 accessions.
For the population-level estimate of mutation accumulation, we applied the filters used to identify the 2,128 variable sites that were used for the MJN for all sequences up through April 19, 2021. We did not include new mutations because the B.1.1.7 VOC and its downstream haplotypes had become the predominant variants globally at that time and, consequently, much early information of the molecular evolution is lost when applying frequency filters on the entire GISAID database. This is exacerbated with the MJN approach because the software algorithm used to generate the network is computationally intractable with greater than 1,000 haplotypes and therefore future efforts will either need to ignore early molecular events or use new methods that can handle the large datasets and any recombination events that occur (an alternative approach would be to now use the alpha or delta variant as the reference sequence because they are now the predominant strains globally).
For calculations of population-level mutation accumulation, it is possible (and necessary) to include all sequences to determine if mutation or recombination are the cause of the high mutation load seen in the late 2020 VOC. After applying the frequency and haplotype filters, we retained 5,011 variable sites that define 12,282 unique haplotypes for further analysis. Mutations to five possible states (A, G, T, C, and -) were counted at each site on the first date that they appeared and their appearance at later dates were excluded. Multiple mutations at a site to different states were counted with this method.
For lineage-specific mutation curves, we extracted all sequences based on their PANGO lineage listed in the metadata from GISAID that also had a sample data and plotted the cumulative number over time, where time is represented by days from first appearance. To estimate the rate of accumulation, we calculated the slope for the linear portion of each of the curves.
Probability of mutation accumulation
To calculate the chance of accumulating several mutations in a certain period, the probability density function for a normal distribution is used:
 where μ is the expected number of mutations for that date, x is the measured value, and σ is the standard deviation of error calculated from the data shown in Fig. 1b, considering the difference between the actual and predicted number of mutations. The expected value of mutations μ for a given time period is computed from the estimated rate of mutations per day (Figure 3, 0.05). c. The period of interest to our discussion (June-October 2020) corresponds to 122 days, for which, the integral of PDF(x=13) gives the probability of 1*10-15 to accumulate 13 mutational events.
where μ is the expected number of mutations for that date, x is the measured value, and σ is the standard deviation of error calculated from the data shown in Fig. 1b, considering the difference between the actual and predicted number of mutations. The expected value of mutations μ for a given time period is computed from the estimated rate of mutations per day (Figure 3, 0.05). c. The period of interest to our discussion (June-October 2020) corresponds to 122 days, for which, the integral of PDF(x=13) gives the probability of 1*10-15 to accumulate 13 mutational events.
Screen for coinfected individuals with UK B.1.1.7
We extracted 25 samples from the Sequence Read Archive at NCBI for each of the months of October, November, December, and January listed as variant B.1.1.7 from the UK (Table S2) for a total of 100 samples to check for coinfection. The reads were mapped to the NC_045512 Wuhan reference using CLC Genomics Workbench using the default parameters except for length fraction and similarity fraction were set to 0.9. Three sites specific to UK B.1.1.7 were analyzed for possible heterozygosity. Of the 100 we sampled, two appeared to be cases of coinfection. This supports the hypothesis that the large expansion in overall mutations seen in UK B.1.1.7 are likely due to recombination. In addition, it also supports the case that coinfection is occurring at a baseline sufficient to allow for occasional recombination.
Protein structure analysis
VMD was used to visualize the protein structures and analyze the potential functional effects of mutations (Humphrey et al. 1996). Figure 3 was created using Inkscape (https://inskape.org/) and Gimp 2.8 (https://www.gimp.org) (‘The GIMP Development Team. (2019). GIMP. Retrieved from https://www.gimp.org’ n.d.).
Molecular dynamics simulations
Molecular dynamics (MD) simulations were used to study interactions between SARS-CoV-2 RBD and ACE2 from ferret and human. Three independent extensive MD simulations were performed for each species using GROMACS 2020 package (Lindahl et al. 2020) and the CHARMM36 force field for protein and glycans (Guvench et al. 2011; J. Huang & MacKerell 2013). Each simulation ran up to 800 ns, being the last 500 ns used for analysis. PDB id 6M17 was used to build the ACE2-RBD complexes. Given the high sequence identity between human and ferret ACE2 (83%), we performed local modeling of the non-conserved amino acid residues in ferret ACE2 using the human homolog as the template, via RosettaRemodel (P.-S. Huang et al. 2011).
The inputs for simulations were generated using CHARMM-GUI (Jo et al. 2008). Counterions were added for electroneutrality (0.1 M NaCl). The complexes were surrounded by TIP3P water molecules to form a layer of at least 10 Å relative to the box borders (Jorgensen et al. 1983). Simulations were performed using the NPT ensemble. The temperature was maintained at 310 K with the Nosé–Hoover thermostat using a time constant of 1.0 ps (Evans & Holian 1985). The pressure was maintained at 1 bar with the isotropic Parrinello–Rahman barostat using a compressibility of 4.5 × 10−5 bar−1 and a time constant of 1.0 ps in a rectangular simulation box (Parrinello & Rahman 1981). The particle mesh Ewald method was used for the treatment of periodic electrostatic interactions with a cutoff distance of 1.2 nm (Darden et al. 1993). The Lennard–Jones potential was smoothed over the cutoff range of 1.0–1.2 nm by using the force-based switching function. Only atoms in the Verlet pair list with a cutoff range reassigned every 20 steps were considered. The LINCS algorithm was used to constrain all bonds involving hydrogen atoms to allow the use of a 2 fs time step (Hess et al. 1997). The suggested protocol for nonbonded interactions with the CHARMM36 force field when used in the GROMACS suite was followed.
The Hbonds plugin in VMD was used to identify hydrogen bond interactions along the simulations (Humphrey et al. 1996). The geometric criteria adopted are a cutoff of 3.5 Å for donor-acceptor distance and 30° for acceptor-donor-H angle. The Timeline plugin was used to count contacts formed by a given amino acid residue. We defined the distance of 4 Å between any atom pairs as the cutoff for contact.
5. Data Access
All SARS-CoV-2 sequences used in this study are available from the public repositories Genome Initiative on Sharing Avian Influenza Data (GISAID, gisaid.org), the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/sars-cov-2/) and the COVID-19 Genomics UK Consortium (COG, https://www.sanger.ac.uk/collaboration/covid-19-genomics-uk-cog-uk-consortium/
Author Contribution
MR Garvin: Conceptualization, Data curation, Funding acquisition, Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing.
ET Prates: Formal Analysis, Investigation, Visualization, Writing-original draft, Writing - review & editing
J Romero: Conceptualization, Formal Analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing.
A Cliff: Methodology, Software, Writing – review & editing
JGFM Gazolla: Software, Formal Analysis, Investigation, Data Curation, Visualization, Writing - Review and Editing.
M Pickholz: Investigation, Visualization, Writing – original draft, Writing – review & editing
M Pavicic: Investigation, Writing – original draft, Writing – review & editing
DA Jacobson: Conceptualization, Funding acquisition, Formal Analysis, Investigation, Project administration, Supervision, Resources,Writing – original draft, Writing – review & editing
Supplementary Material
1. Supplementary Figures

Number of novel mutations sampled across the globe for each day are plotted against time (days from the Wuhan outbreak). Emergence of major VOC are provided for context and show small increases in the number of new mutations but there is an overall decrease across time, even accounting for multiple mutations at a site to different nucleotide states and deletions.
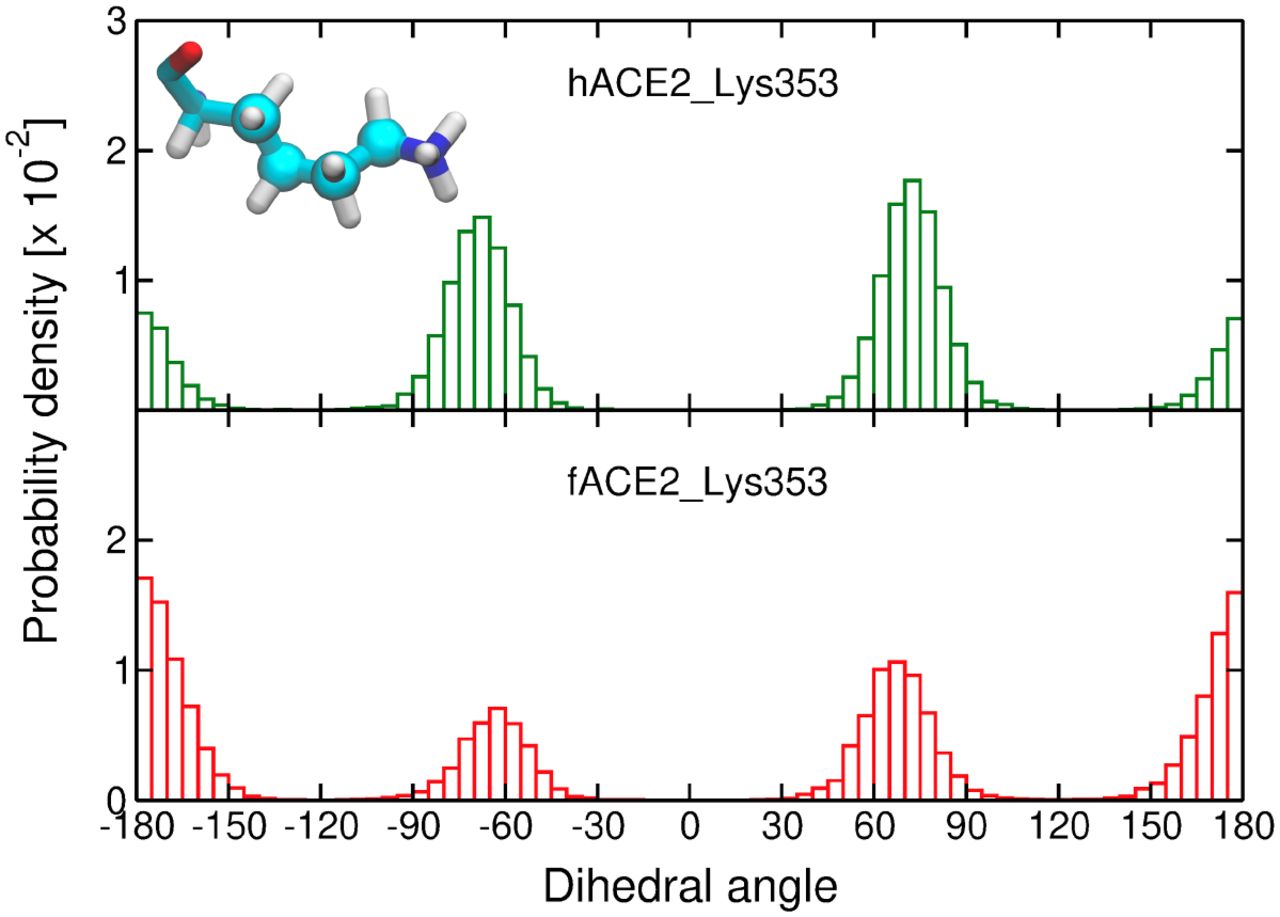
Histograms of the distribution of a dihedral angle of the Lys353 side chain carbon atoms in human ACE2 (hACE2, upper figure) and ferret ACE2 (fACE2, lower figure) in complex with the SARS-CoV-2 S receptor-binding domain. The atoms forming the selected dihedral are depicted as spheres in the molecular representation of Lys353. Three independent simulations are considered for the calculation of the histograms. Dihedral angles near ±180° correspond to a more stretched conformation (i.e., trans).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Time evolution of the number of hydrogen bonds (HB) that fACE2 Arg354 and Lys353 form with Asp405 and Tyr495 from the SARS-CoV-2 S receptor-binding domain. The columns correspond to the three simulation replicas. The geometric criteria adopted for hydrogen bonds are a cutoff of 3.0 Å for donor-acceptor distance and 20° for acceptor-donor-H angle.
2. Supplementary Tables
A distance of 4 Å between any atom pairs was defined as the cut-off for contact statistics.
6. Acknowledgments
The viral evolution research was funded by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC for the US Department of Energy (LOIS:10074) and the structural implication work was funded via the DOE Office of Science through the National Virtual Biotechnology Laboratory (NVBL), a consortium of DOE national laboratories focused on the response to COVID-19, with funding provided by the Coronavirus CARES Act. This work was also funded by the United States Government. This research used resources of the Oak Ridge Leadership Computing Facility (OLCF) and the Compute and Data Environment for Science (CADES) at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725. Figures generated with Biorender and VMD. We gratefully acknowledge the Originating laboratories responsible for obtaining the viral specimens and the Submitting laboratories where genetic sequence data were generated and shared via the GISAID Initiative, on which this research is based.
Footnotes
We incorporated information from a recent analysis that identified recombination in the UK and also further emphasized the importance of epistasis as the reason for the improved fitness of the VOC.
7. References




