

ABSTRACT
There is a high demand for the production of recombinant proteins in Escherichia coli for biotechnological applications but their production is still limited by their insolubility. Fusion tags have been successfully used to enhance the solubility of aggregation-prone proteins; however, smaller and more powerful tags are desired for increasing the yield and quality of target proteins. Herein, NEXT tag, a 53 amino acid-length solubility enhancer, is described. The NEXT tag showed outstanding ability to improve both in vivo and in vitro solubilities with minimal effect on passenger proteins. The C-terminal region of the tag was mostly responsible for in vitro solubility, while the N-terminal region was essential for in vivo soluble expression. The NEXT tag appeared to be intrinsically disordered and seemed to exclude neighboring molecules and prevent protein aggregation by acting as an entropic bristle. This novel peptide tag should have general use as a fusion partner to increase the yield and quality of difficult-to-express proteins.
IMPORTANCE Production of recombinant protein in Escherichia coli still suffers from the insolubility problem. Conventional solubility enhancers with large sizes represented by maltose-binding protein (MBP) have remained as the first-choice tags, however, the success in the soluble expression of tagged protein is largely unpredictable. In addition, the large tags can negatively affect the function of target proteins. In this work, NEXT tag, an intrinsically disordered peptide, was introduced as a small but powerful alternative to MBP. The NEXT tag could significantly improve both expression level and solubility of target proteins including a thermostable carbonic anhydrase and a polyethylene terephthalate (PET)-degrading enzyme that are remarkable enzymes for environmental bioremediation.
INTRODUCTION
Recombinant protein expression underpins protein engineering and metabolic pathway engineering for the production of biotherapeutics, bioreporters, biocatalysts, and various industrial chemicals as well as biochemical studies of protein. Bacterial hosts such as Escherichia coli have remained the preferred hosts for recombinant protein expression due to their fast growth rate to high cell densities, ease of genetic manipulation, and scale-up simplicity (1). However, the high rate of protein synthesis/folding and high-level accumulation of heterologous protein in E. coli often lead to the formation of inclusion bodies that are intracellular aggregates of misfolded, partially folded, or even fully folded proteins, limiting the yield of recombinant protein (1-3).
Fusion protein tags have been widely used as effective solubility enhancers for aggregation-prone proteins. When fused to the N termini of target proteins, these solubility tags not only improve the solubility of passenger proteins but also increase their expression level by providing sequence contexts for more efficient translation initiation (4). Despite numerous examples of their applications, the successful selection of effective tags for a given target protein still relies heavily on a trial-and-error approach. Although maltose-binding protein (MBP) and N-utilization substance A (NusA) are the best working tags (4, 5), they are relatively large proteins (> 40 kDa) that can impart a higher metabolic burden than smaller tags and increase the chance of full-length proteins undergoing incomplete synthesis (6), potentially leading to a lower yield of target protein. In addition, while fusion tags are generally removed by chemical or enzymatic cleavage after soluble expression and purification, proteins that have intrinsically poor solubilities need to be used in their tagged forms since they precipitate after the solubility tags are removed (1, 5, 7). In this case, smaller tags are more desirable to minimize the effect of fusion tags on the inherent properties of passenger proteins (7, 8). Collectively, there is still considerable demand to expand the repertoire of solubility tags that are more powerful but smaller than conventional tags.
The carbonic anhydrase of the marine bacterium Hydrogenovibrio marinus (hmCA) is a highly soluble protein (9). hmCA contains an unusual N-terminal extension that is not essential for its catalytic function. When the N-terminal sequence was truncated, the solubility of recombinant hmCA was drastically reduced, and it was expressed mostly in an insoluble form. Inspired by this observation, it was hypothesized that the 53 amino acid-length N-terminal extension sequence (designated as NEXT) could be used as a fusion tag to improve solubility. In this study, it was demonstrated that the small NEXT tag is an intrinsically disordered protein that is exceptionally powerful for improving the solubility and expression level of passenger proteins with minimal influence on the target protein.
RESULTS AND DISCUSSION
Effect of the fusion tags on the in vivo solubility of recombinant proteins
By convention, there are two types of protein solubilities: in vivo solubility and in vitro solubility (10). Low in vivo protein solubility is often observed when a recombinant protein is overexpressed in a bacterial host, generally resulting in the formation of inclusion bodies (1, 2). When a protein has low in vitro solubility, aggregates can be formed even with a folded, isolated protein (10, 11). In this case, the protein can remain soluble only at a low concentration.
To evaluate the efficiency of the NEXT tag in improving in vivo solubility, the tag was fused to the N termini of several difficult-to-express proteins via a flexible linker (Fig. 1a). The passenger proteins were selected based on their potential applications: human epidermal growth factor (hEGF, 6.4 kDa) as a therapeutic protein, green fluorescent protein (GFP, 26.9 kDa) as a bioreporter, and carbonic anhydrase from Thermovibrio ammonificans (taCA, 25.9 kDa) and polyethylene terephthalate (PET)-hydrolyzing enzyme from Ideonella sakaiensis (isPETase, 27.7 kDa) as biocatalysts for bioremediation. Other commonly used solubility tags, including MBP (12), glutathione S-transferase (GST) (13), and an 8-kDa protein from Fasciola hepatica (Fh8) (4), were also tested for comparison (Table 1). The fusion proteins were constructed, and their expression patterns were analyzed after soluble/insoluble fractionation.
Properties of solubility tags used in this study

In vivo solubility of recombinant proteins fused with different solubility tags. (a) Expression cassettes of fusion proteins. Protein expression is under the control of the IPTG-inducible T7lac promoter. The passenger proteins are fused with the N-terminal solubility tags via the flexible (GGGGS)2 linker and with the C-terminal His6-tag. Coomassie blue-stained SDS-PAGE gel for the analysis of the expression of (b) hEGF, (c) GFP, (d) taCA, and (e) isPETase fused with the different solubility tags at 37 °C or 25 °C (only for GFP). The fluorescence image in (c) is for GFP at 25 °C. The arrow indicates the band position of each recombinant protein. Lane: MW, molecular mass marker; S, soluble fraction; IS, insoluble fraction.
hEGF is a protein hormone that can be used as a wound healing agent by stimulating epidermal regeneration (14). Although the production of hEGF in bacterial cells has been reported, high-level soluble expression of hEGF in E. coli is still challenging. The expression level of soluble NEXT-hEGF was the highest among the tested constructs, and the percentage of the soluble fraction was 87 ± 9%, even at 37 °C (Fig. 1b), which was also one of the highest values ever reported (15-17). GFP is the most popular fluorescent protein for bioimaging and sensing applications (18). When GFP was fused to the solubility tags, a notable amount of soluble expression was not observed at 37 °C with all of the tested fusion proteins except for NEXT-GFP (Fig. 1c). At 25 °C, however, all of the constructs showed high in vivo solubility, and again, the most remarkable soluble expression was that of NEXT-GFP, as its fluorescence was the brightest (Fig. 1c). taCA is one of the most thermostable carbonic anhydrases and has potential applications in bioinspired CO2 capture and utilization (19-21). Despite its soluble expression, the relatively low protein yield and the low in vitro solubility (see below) have hampered intensive engineering and application of taCA. Although taCA was expressed mostly in soluble forms regardless of the fusion tags, the highest expression level was attained when the NEXT tag was used (Fig. 1d). Recently, isPETase has been extensively studied as a green biocatalyst that can degrade PET plastic under moderate temperature conditions (22-24). However, isPETase exhibits a low level of soluble expression in E. coli even at low temperature conditions, and it has not been demonstrated to have high-level soluble expression. The untagged isPETase was expressed almost exclusively in an insoluble form at 37 °C, as was the case for MBP and Fh8 fusion proteins. In contrast, surprisingly, NEXT-isPETase was expressed entirely in a soluble form (Fig. 1e). Collectively, these results demonstrate that the NEXT tag is an exceptionally powerful enhancer not only for in vivo solubility but also for the production yield of passenger protein.
Effect of the fusion tags on the in vitro solubility of purified proteins
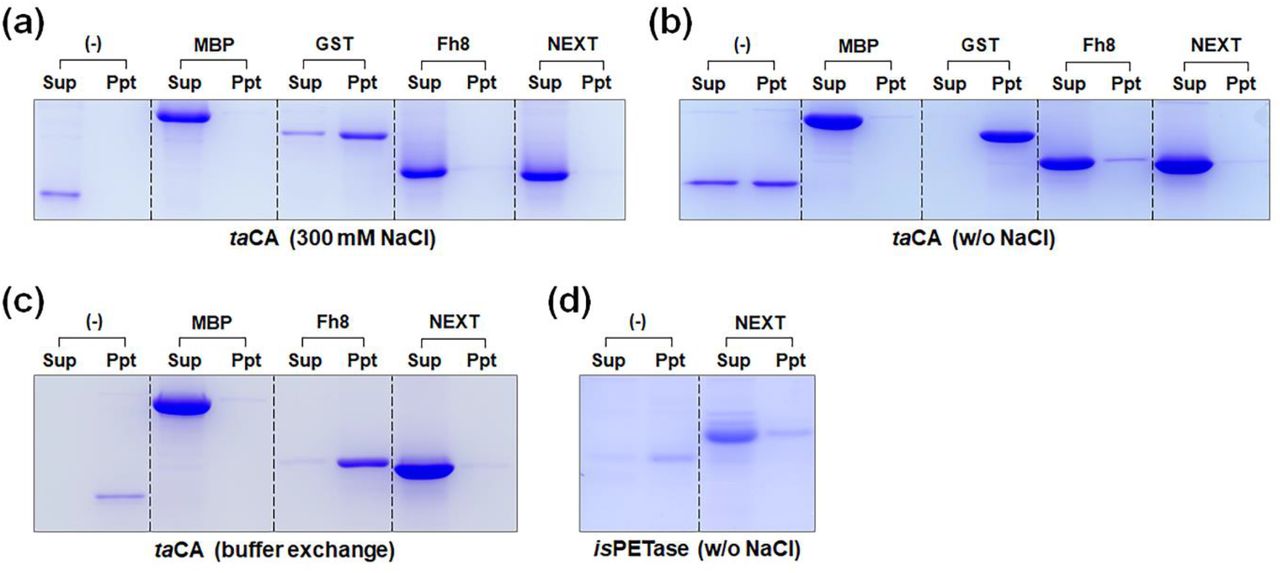
To test the ability of solubility tags to promote the in vitro solubility of passenger proteins, taCA and isPETase were further utilized for the test because these enzymes have low in vitro solubility and are susceptible to aggregation under low salt conditions (19, 24). The purified taCA enzymes with different solubility tags were exposed to buffer solutions with or without salt supplementation, and any protein precipitates were separated from the supernatant by centrifugation and analyzed by SDS-PAGE. When the buffer was supplemented with 300 mM NaCl, only GST-taCA showed a significant amount of precipitates, and the other proteins, including the untagged counterpart, remained soluble (Fig. 2a). After the enzymes were placed in a low salt condition, GST-taCA became completely insoluble, which was more severe than in the case of untagged taCA (Fig. 2b). Combined with the in vivo solubility results (Fig. 1), this confirms that GST is not an effective tag for improving protein solubility (1). On the other hand, almost all taCA enzymes were still in soluble forms when the other tags (MBP, Fh8, and NEXT) were used, demonstrating their effectiveness in improving the in vitro solubility of passenger proteins (Fig. 2b). Interestingly, after undergoing changes in the composition and pH of buffer under low salt conditions, only MBP- or NEXT-tagged taCA showed resistance to aggregation induced by changes in environmental conditions, indicating that both MBP and NEXT tags are superior to the Fh8 tag for enhancing in vitro solubility even under dynamic chemical environments (Fig. 2c). Similar to taCA, the poor in vitro solubility of isPETase under low salt conditions was successfully circumvented by fusion with the NEXT tag (Fig. 2d).

In vitro solubility of purified enzymes fused with different solubility tags. Coomassie blue-stained SDS-PAGE gel for the analysis of precipitation of (a) taCA dialyzed against 20 mM phosphate buffer (pH 7.5) supplemented with 300 mM NaCl, (b) taCA dialyzed against 20 mM phosphate buffer (pH 7.5), (c) taCA after buffer exchange from 20 mM phosphate buffer (pH 7.5) to 20 mM Tris buffer (pH 8.3), and (d) isPETase dialyzed against 20 mM phosphate buffer (pH 7.5). Lane: Sup, supernatant after centrifugation; Ppt, precipitated protein pellet.
Effect of the fusion tags on protein quality
Using the purified taCA enzyme fused with the MBP, Fh8, or NEXT tag, the effect of the fusion tag on protein quality was investigated by examining the enzyme activity and stability, the two most important enzyme properties. The activity changes of taCA caused by the fusion of Fh8 (27%) and NEXT (14%) were relatively marginal when compared with that of the MBP-taCA, which showed an abnormally large increase in activity (115%) (Fig. 3a). The bulky MBP tag might interfere with the function of taCA more than the other small tags can. Similarly, the solubility tags affected the thermal stability of taCA corresponding with their size, and no apparent decrease in stability was seen when the NEXT tag was used (Fig. 3b). These results show that the smallest NEXT tag can be used as a noncleavable solubility tag that exerts only minimal influences on passenger proteins.

Activity and stability of purified taCA fused with different tags. (a) Relative CO2 hydration activity of solubility tag-fused taCA compared to the activity of taCA without a tag. (b) Thermal stability of taCA with or without a fusion tag. The enzyme activity was measured after incubation for 1 h at 90 °C, and the relative residual activity was obtained compared to the enzyme activity without heat treatment. Enzymes were prepared in 20 mM phosphate buffer (pH 7.5). Error bars represent standard deviations from two or three independent experiments. Asterisks indicate statistical significance of the tagged taCA compared with the untagged enzyme determined by t-test (*p < .05, **p < .01).
From a different standpoint, biochemical studies of proteins can also benefit from the small size of the NEXT tag. Larger solubility tags are expected to have more sites for posttranslational modification, such as phosphorylation (Table 1). For example, a candidate substrate for a kinase expressed with the fusion of MBP might be phosphorylated by the kinase within the MBP portion, not within the candidate substrate, leading to an incorrect conclusion that the candidate protein is actually a substrate for the kinase. In this situation, the NEXT tag with no potential phosphorylation site can be alternatively used.
N-terminal truncation of the NEXT tag
To identify the part of the NEXT tag that is most responsible for solubility enhancement, the tag was roughly divided into three regions with similar sizes, and the sequential N-terminal truncation of the regions was studied (Fig. 4a). First, hmCA, from which the NEXT tag originated, was expressed with the partial or full deletion of the N-terminal extension (Fig. 4b). Full-length hmCA and the two partial deletion variants (ΔN19 and ΔN36) were expressed in soluble forms despite the different expression levels. However, when the N-terminal extension of hmCA was fully truncated, the protein was expressed in a form that was almost insoluble, as previously reported (9). This result clearly indicates that the C-terminal part of the NEXT tag (NEXTC16 peptide) is the part most responsible for the soluble expression of hmCA.

Effect of truncating the NEXT tag on the solubility of the target protein. (a) Design of truncation constructs. The sequence of the NEXT tag is presented along with the selected residue numbers. The first methionine was excluded for residue numbering. (b) In vivo solubility of N-terminal truncated hmCA variants. (c) In vivo solubility of the recombinant proteins (GFP, hEGF, and taCA) fused with the NEXT or NEXTC16 tag. Proteins were expressed at 37 °C with 1 mM IPTG. (d) In vitro solubility of purified taCA with the NEXT or NEXTC16 tag prepared in 20 mM phosphate buffer with or without 300 mM NaCl. Lane: MW, molecular mass marker; S, soluble fraction; IS, insoluble fraction; Sup, supernatant after centrifugation; Ppt, precipitated protein pellet. (e) Activity and stability of taCA fused with the NEXT or NEXTC16 tag. taCA enzymes were prepared in 20 mM phosphate buffer (pH 7.5). For the stability test, enzymes were incubated for 1 h at 90 °C. Error bars represent standard deviations from two independent experiments.
To test whether the 16 amino acid-length NEXTC16 can substitute the full-length NEXT tag, the expression patterns of hEGF, GFP, and taCA fused to NEXTC16 were evaluated. Unfortunately, the soluble expressions of both hEGF and GFP were significantly hampered by the replacement of the NEXT tag with the NEXTC16 tag, suggesting that the N-terminal region of the NEXT tag is crucial for improving the in vivo solubility of the passenger protein (Fig. 4c). In the case of taCA, soluble expression of NEXTC16-taCA was observed as expected, although the production yield appeared to be reduced compared to that of NEXT-taCA (Fig. 4c). Intriguingly, when the in vitro solubility of purified NEXTC16-taCA was tested, no apparent protein precipitation was observed regardless of NaCl supplementation (Fig. 4d). Additionally, the activity and stability of NEXTC16-taCA were identical to those of NEXT-taCA (Fig. 4e). These results show that in contrast to the in vivo solubility result, the high in vitro solubility of passenger proteins can be retained by using the NEXTC16 region alone instead of the full-length NEXT tag. The use of a very short NEXTC16 tag might be beneficial, e.g., for the immobilization of the target enzyme onto a solid matrix with a limited surface area to maximize the immobilization yield and overall catalytic efficiency of biocatalysts (25).
Intrinsic disorder of the NEXT tag
The mechanisms of solubility enhancement by various solubility tags are still not clear, and there seems to be multiple routes for the promoted solubility (4). The in vivo solubility results (Fig. 1) did not fit into the solubility prediction by the modified Wilkinson−Harrison model (Table S1) (26). Although machine learning-based SOLpro (27) predicted the solubility patterns of fusion proteins more accurately, it could still not discriminate the NEXT tag from the others, especially for isPETase fusions (Table S1, Fig. 1e). Protein acidity is known to be one of the determinants of solubility (28, 29), which cannot explain the remarkable enhancement of solubility by the NEXT tag, which possesses a net positive charge (Table 1). The classical structure-function paradigm of proteins has been challenged by the concept of intrinsically disordered proteins (IDPs). IDPs exist as highly dynamic structural ensembles with undefined three-dimensional structures (30). It has been proposed that an IDP region within a protein can act as an intramolecular entropic bristle (EB) (31, 32). The EB domain is expected to have an extended conformation, and by thermally driven random motion, it can occupy a significantly large space around the protein molecule (33). By entropically excluding neighboring molecules, EB can prevent protein aggregation, thus leading to improved protein solubility.
Sequence-based prediction showed that the NEXT tag is highly disordered, whereas the MBP, GST, and Fh8 tags possess low disorder propensities (Fig. 5a). Its low hydrophobicity, a measure that is correlated with protein disorder (34), also distinguishes the NEXT tag from the other tags (Table 1). The C-terminal region of the NEXT tag was predicted to be the most disordered (Fig. 5a), which corresponds to the NEXTC16 region that was crucial for improving solubility (Fig. 4). The NEXT tag was separately expressed and purified (Fig. 5b). The circular dichroism (CD) spectrum of the purified NEXT tag coincided with that of a random coil without any secondary structural element (Fig. 5c) (35). These results strongly suggest that the NEXT tag is an IDP that can improve the solubility of passenger proteins as a function of EB.

Intrinsic disorder propensities of solubility tags. (a) Position-dependent prediction of disordered regions by three different methods (IUPred2A, PONDR, and DISpro). The region corresponding to NEXTC16 is highlighted in a yellow box. (b) Purified NEXT tag with a C-terminal His6-tag (6.5 kDa) analyzed by SDS-PAGE. Lane: MW, molecular mass marker; P, purified protein. (c) Circular dichroism (CD) spectrum of purified NEXT tag in the far-UV region.
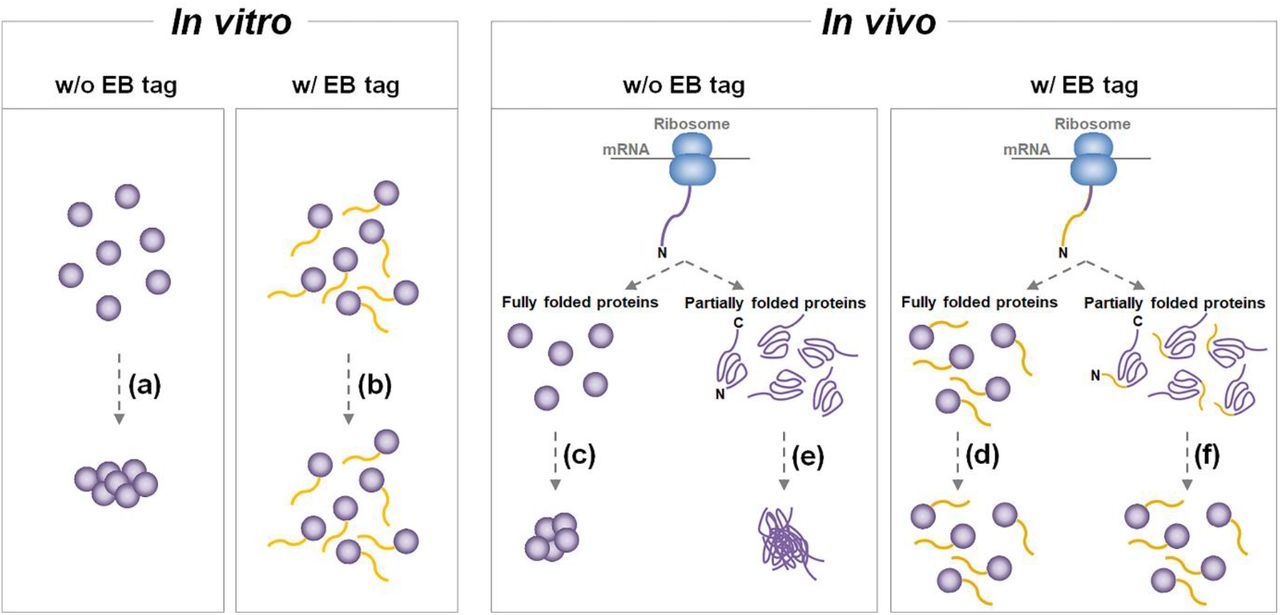
As previously demonstrated, the fusion of the NEXT tag can prevent protein aggregation both in vitro and in vivo (Fig. 6). A fully folded protein with low in vitro solubility is prone to aggregation (Fig. 6a), which can be circumvented by the fusion of EB (Fig. 6b). The accumulation of overexpressed, fully folded protein with low in vitro solubility can result in protein aggregation in the cytosol (Fig. 6c). This apparently low in vivo solubility can also be overcome by the utilization of EB (Fig. 6d). The accumulation and subsequent aggregation of partially folded proteins before the completion of folding is another cause of low in vivo solubility (Fig. 6e). The interaction between the folding intermediates can be reduced with N-terminal fusion of EB, facilitating correct protein folding (Fig. 6f).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Solubility enhancement by the fusion of entropic bristle (EB) (a, b) A protein with low in vitro solubility aggregates in its folded state and after the fusion of EB, the protein can remain soluble. (c, d) The accumulation of fully folded protein with low in vitro solubility can lead to protein aggregation in the cytosol, leading to protein expression that seems to be insoluble. This situation can also be circumvented by the fusion of EB. (e, f) Low in vivo solubility during recombinant protein overexpression can occur by the aggregation of partially folded proteins, which can be overcome by the N-terminal fusion of EB, preventing the interaction between the folding intermediates and thus allowing protein folding to complete.
In conclusion, the successful use of a small-sized NEXT tag was demonstrated to improve both the in vivo and in vitro solubilities of the selected recombinant proteins. Because the degree of solubility enhancement by EB fusion appeared to depend on the length of the tag (31), a more powerful IDP-based solubility tag should be artificially engineered by optimizing not only the amino acid sequence but also the length of the tag. Further experimental analyses using a variety of potential EB proteins, including the NEXT tag, will provide insight into the engineering principles for the de novo design of IDP-based solubility tags customized for each passenger protein.
MATERIALS AND METHODS
Construction of expression vectors
The strains, plasmids, and primers used in this study are listed in Table S2. The E. coli TOP10 strain (Thermo Fisher Scientific, USA) was used for gene cloning. E. coli was routinely cultured in Luria-Bertani (LB) medium supplemented with appropriate antibiotics (10 μg/mL streptomycin or 50 μg/mL ampicillin) at 37 °C in a shaking incubator (Jeiotech, Korea). The genes for MBP, GST, NEXT, GFP and taCA were cloned by polymerase chain reaction (PCR) using pMAL-c5X (New England Biolabs, USA), pGEX-4T-1 (GE Healthcare, USA), pET-hmCA (9), pTH-GFP (36), and pET-taCA (19) as the templates. The primers for the solubility tags contain the sequence for flexible linker (GGGGS)2 along with NdeI and NcoI restriction sites. The PCR fragments were cloned into the pGEM-T Easy vector (Promega, USA) and the amplified sequences were confirmed by direct sequencing. The genes for the Fh8 tag (GenBank accession number: AF213970) , hEGF (GenBank accession number: M15672) and isPETase (GenBank accession number: 6EQD_A) were chemically synthesized along with the linker sequence (only for Fh8) and the restriction sites (Genotech, Korea). The genes were subcloned into pET-22b(+) (Novagen, USA). All of the recombinant genes have a hexahistidine (His6)-tag sequence at their 3’ termini provided by the parent vector.
Expression of recombinant proteins
Recombinant E. coli BL21(DE3) strains transformed with the constructed vectors were incubated in LB medium with 50 μg/mL ampicillin at 37 °C in the shaking incubator. Protein expression was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG; Duchefa Biochemie, Netherlands) to a final concentration of 1 mM (at 37 °C) or 10 μM (at 25 °C) when the optical density at 600 nm (OD600) reached 0.6-0.8. For the expression of taCA variants, 0.1 mM ZnSO4 (Junsei, Japan) was also added to the culture medium. After cultivation for 10 h at 37 °C or for 20 h at 25 °C, the cells were collected by centrifugation at 4 °C and 4,000 g for 10 min. The cells were resuspended in lysis buffer (50 mM sodium phosphate, 300 mM NaCl, and 10 mM imidazole; pH 8.0) and disrupted by an ultrasonic dismembrator (Sonics and Materials, USA) in ice water. After centrifugation of the lysate at 4 °C and 10,000g for 10 min, the supernatant was removed, and the soluble fraction (S) was designated; the remaining debris was designated the insoluble fraction (IS).
Purification of recombinant proteins
The soluble fraction of the cell lysate was mixed with Ni2+-nitrilotriacetic acid agarose beads (Qiagen, USA), and the His6-tagged recombinant proteins were purified by immobilized metal affinity chromatography (IMAC) according to the manufacturer’s instructions. The enzymes were eluted using elution buffer (50 mM sodium phosphate, 300 mM NaCl, and 250 mM imidazole; pH 8.0). The eluates were thoroughly dialyzed against 20 mM sodium phosphate buffer (pH 7.5) with or without 300 mM NaCl. After dialysis was completed, the protein precipitates (Ppt) were removed by centrifugation at 4 °C and 10,000 g for 10 min. The supernatants (Sup) were used for subsequent activity and stability tests. In some cases, the enzyme buffer was further exchanged with 20 mM Tris-sulfate buffer (pH 8.3).
Protein analyses
For protein quantification, the purified enzyme was denatured in denaturing buffer (6 M guanidine hydrochloride GuHCl/20 mM sodium phosphate buffer, pH 7.5), and the absorbance of the denatured protein was measured at 280 nm. The protein concentration was determined using the measured absorbance and the molar extinction coefficient at 280 nm for each protein calculated by ProtParam (http://web.expasy.org/protparam/) (37). Proteins were separated and visualized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Brilliant Blue R-250 (Bio-Rad, USA) staining. The percentage of soluble expression was estimated by densitometric analysis of the band intensities of soluble and insoluble fractions on the protein gel performed using ImageJ.
Activity and stability test for taCA variants
CA activity was measured by a colorimetric CO2 hydration assay (38, 39). The purified enzyme in 20 mM phosphate buffer (pH 7.5) was diluted to a concentration of 1 μM. Ten microliters of sample was added to a disposable cuvette containing 600 μL of 20 mM Tris buffer (pH 8.3) supplemented with 100 μM phenol red. The reaction was performed at 4 °C inside the spectrometer by adding 400 μL of CO2-saturated deionized water prepared in ice-cold water. The absorbance change was monitored at 570 nm. The time (t) required for the absorbance to drop from 1.2 (corresponding to pH 7.5) to 0.18 (corresponding to pH 6.5) was obtained. The time (t0) for the uncatalyzed reaction was also measured by adding a corresponding blank buffer instead of an enzyme sample. The Wilbur-Anderson unit was calculated as (t0− t)/t. For the stability test, the enzyme sample was incubated for 1 h at 90 °C and the residual enzyme activity was measured. Relative residual activity was calculated based on the activity of the untreated sample.
Circular dichroism spectroscopy
Circular dichroism (CD) spectrum was recorded on a CD spectropolarimeter (Jasco, Japan). The purified solution of the NEXT tag in 20 mM phosphate buffer (pH 7.5) was scanned in a quartz crystal cuvette with a 2 mm path length (Hellma Analytics, Germany) for the far-UV region (190-250 nm) at 25 °C. Based on the CD spectrum, secondary structural elements were analyzed using BeStSel (40).
In silico calculations
Protein parameters including amino acid length, molecular weight, net charge and pI, were calculated by ProtParam (37). Phosphorylation sites were predicted by NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos/) (41). Kyte-Doolittle hydropathicity was calculated by ProtScale (https://web.expasy.org/protscale/) using a window size of 5, and the values were averaged to obtain a mean hydropathy (37, 42). Sequence-based prediction of protein solubility was performed by the modified Wilkinson−Harrison method (26) and SOLpro (http://scratch.proteomics.ics.uci.edu/) (27). Disordered protein regions were predicted by IUPred2A (https://iupred2a.elte.hu/) (43), PONDR (http://www.pondr.com/) (44) and DISpro (http://scratch.proteomics.ics.uci.edu/) (45).
CONFLICT OF INTEREST
The author declares no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant (20182010600430) funded by the Ministry of Trade, Industry, and Energy, Korea, and by the National Research Foundation grants (NRF-2020M3A9I5037642, NRF-2021R1F1A1057310 and NRF-2021R1A5A8029490) funded by the Ministry of Science and ICT, Korea.
REFERENCES




