

Abstract
We proposed previously that aqueous non-covalent barriers arise from solute-induced perturbation of the H-bond network of solvating water (“the solvation field”) relative to bulk solvent, where the association barrier equates to enthalpic losses incurred from incomplete replacement of the H- bonds of expelled H-bond enriched solvation by inter-partner H-bonds, and the dissociation barrier equates to enthalpic + entropic losses incurred during dissociation-induced resolvation of H-bond depleted positions of the free partners (where dynamic occupancy is powered largely by the expulsion of such solvation to bulk solvent during association). We analyzed blockade of the ether-a-go-go-related gene potassium channel (hERG) based on these principles, the results of which suggest that blockers: 1) project a single rod-shaped R-group (denoted as “BP”) into the pore at a rate proportional to the desolvation cost of BP, with the largely solvated remainder (denoted as “BC”) occupying the cytoplasmic “antechamber” of hERG; and 2) undergo second-order entry to the antechamber, followed by first-order association of BP to the pore. In this work, we used WATMD to qualitatively survey the solvation fields of the pore and a representative set of 16 blockers sampled from the Redfern dataset of marketed drugs spanning a range of pro-arrhythmicity. We show that the highly non-polar pore is solvated principally by H-bond depleted and bulk-like water (incurring zero desolvation cost), whereas blocker BP moieties are solvated by variable combinations of H-bond enriched and depleted water. With a few explainable exceptions, the blocker solvation fields (and implied desolvation/resolvation costs) are qualitatively well-correlated with blocker potency and Redfern safety classification.
Introduction
Despite many years of intensive investigation into the possible causes of, and remedies for, inadvertent blockade of the hERG potassium channel by chemically diverse low molecular weight (LMW) hits, leads, preclinical/clinical candidates, and drugs, this liability remains one of the many unsolved problems in pharmaceutical R&D that are typically addressed via black box trial-and- error optimization. However, this approach is challenged by the convoluted nature of target/off-target potency (including hERG), solubility, permeability, and pharmacokinetics (PK), in which modulation of one property or behavior can positively or negatively affect one or more of the others. Lead optimization often culminates in residual hERG activity at the clinical candidate stage, resulting in potential no-go decisions or mandated clinical thorough QT (TQT) studies, depending on the benefit/risk ratio. The lack of significant progress toward the development of reliable hERG avoidance and mitigation strategies may be attributed to:
Poor general understanding of aqueous non-covalent binding between cognate partners, including drugs and targets/off-targets (described below).
Consideration of ion channel blockade as a typical binding process, when in fact, it is highly atypical due to:
The absence of native binding function of the ion conduction pathway, which serves as the binding site for all known blockers. We attribute native binding function, in general, to:
Complementarity between cognate binding partners in both steric size/shape.
The positions/H-bond propensities of polar groups vis-à-vis H-bond enriched solvation and non-polar groups vis-à-vis H-bond depleted solvation [1–3].
We postulate that blocker promiscuity results from the lack of H-bond enriched “gatekeeper” solvation within the pore, thereby relegating the association free energy barrier to steric size/shape complementarity and induced-fit costs, together with pore-mediated blocker desolvation cost (noting that binding is largely non-specific in the absence of H-bond enriched “gatekeeper” solvation).
Two-step binding, consisting of:
The capture of a single solvated blocker copy within the large cytoplasmic cavity of the channel adjoining the pore entrance (denoted as the “antechamber”), which is lined by the C-linker (denoted as “C”) and cyclic nucleotide binding homology (CNBH) domains. The on-rate is described by kc · [free antechamber] · [free blocker] (typical second-order binding, in which kon is capped at the 109 M-1 s-1 diffusion limit), where kc denotes the blocker-antechamber rate constant. The blocker-bound antechamber concentration builds and decays with free cytoplasmic concentration, which in turn, builds and decays with cardiac tissue uptake and plasma clearance, respectively.
Projection of a single quasi-rod-shaped blocker moiety (denoted as “BP”) into the open pore (denoted as “P”) [4]. The on-rate is described by kb · [antechamber-bound blocker] (atypical first-order binding), where kb denotes the BP-P association rate constant. The blocker off-rate is described by k-b · [bound blocker], where k-b is the dissociation rate constant of BP.
The lack of differentiation between trappable and non-trappable blockers, together with poor understanding of structure-trappability relationships. Non-trappable blocker occupancy builds and decays during each channel gating cycle (the peak magnitude of which occurs at the intracellular Cmax, where Cmax is the peak exposure during a given dosing cycle), whereas trappable blockers accumulate to their maximum fractional occupancy (given by the Hill equation: free Cmax/(free Cmax + IC50)), which decays during the clearance phase of the pharmacokinetic (PK) curve (noting that k-b is not usurped by channel closing). The highest maximum occupancy of non-trappable blockers is achieved when kb ≈ the channel opening rate and k-b ≈ the channel closing rate (i.e., noting that k-b is usurped by the rate of channel closing), whereas trappable blockers accumulate to their maximum occupancy over multiple channel gating cycles as the intracellular Cmax builds to n · IC50, where n is occupancy multiplier (n = 1 equates to 50% occupancy, n = 19 equates to 95% occupancy, etc.).
Measurement of hERG blockade under equilibrium conditions in status quo hERG assays, and the use of such data for generating hERG structure-activity relationship (SAR) models when native binding between hERG and non-trappable blockers is, in practice, highly non-equilibrium in nature. Typical native drug-target systems operate on far longer timescales than the ∼350 ms open channel time window of hERG, commensurate with significantly slower requirements for kon and koff. In our previous works, we simulated hERG blockade in the context of the cardiac AP using a modified version of the O’Hara-Rudy model of the undiseased human ventricular cardiomyocyte [5, 6]. Occupancy of hERG by certain blockers necessarily depends far more on fast kb than slow k-b (where k-b is the dissociation rate constant) due to fast buildup of the open state of the pore [5, 6].
Key deficiencies in the “Redfern hERG safety index” (SI), which was developed based on reported PK, IC50, and adverse clinical event data analyzed for 52 marketed drugs. In our previous work, we showed that the Redfern SI (hERG IC50/free Cmax > 30) equates to near zero safe fractional hERG occupancy at the therapeutic free Cmax, allowing for unintended exposure escalation due to overdose or drug-drug interactions (noting that all reported hERG blockade-induced arrhythmia cases in humans involved aberrant blood levels in excess of the therapeutic free Cmax). Improved mitigation depends on achieving a deeper, more in vivo-relevant understanding of hERG blockade and the safety implications thereof (versus black box trial-and-error approaches).
Mutual desolvation of P and BP depends on the existence of a water-accessible pathway between P and bulk-solvent. Since no obvious pathway exists at the extracellular end of P, displaced water is necessarily expelled from the free volume of the blocker-bound pore through the pore entrance, thereby precluding total occlusion of P by BP in fully buried blocker-bound states proposed elsewhere (e.g., [5,7,8]).
Taken together, the aforementioned atypical binding characteristics of hERG blockade engender a highly complex paradigm that is poorly described by conventional pharmacophore, quantitative structure-activity relationship (QSAR), and structure-based models compared with those of native cognate binding sites and ligands.
Binding free energy is contributed principally by solvation, and the implications thereof for hERG blockade
In several of our previous works, we postulated that non-covalent binding under aqueous conditions is governed principally by the solvation free energy contribution, and in particular, the desolvation and resolvation costs incurred during association and dissociation, respectively. Although the total magnitude of the solvation free energy is reflected in logP, free energy is distributed inhomogeneously around solute surfaces, such that desolvation and resolvation do not typically occur in an all-or-none fashion. As such logP (a scalar quantity) is a poor metric of these contributions. Most recently, we proposed that blockers are only partially desolvated during hERG association [4]. Therefore, knowing specifically where to increase the desolvation cost is essential for “surgical” hERG mitigation. We postulate that the H-bond network of solvating water behaves as a field that responds to solute-mediated perturbations at each position of solute surfaces, depending on local surface composition and topology, resulting in:
H-bond disruption/depletion, as reflected in slow and fast water exchanges to/from the solvation field and bulk solvent, respectively (denoted as “solvation dynamics”).
H-bond enhancement/enrichment, as reflected in fast and slow water exchanges to/from the solvation field and bulk solvent, respectively.
Bulk-like H-bond propensity, as reflected in equivalent exchange rates to/from bulk solvent and the solvation field.
We further postulate that:
Non-covalent free energy is released from binding interfaces during association, principally via mutual desolvation of H-bond depleted solvation (i.e., where such water is replaced by non-polar or weakly polar solute groups).
The canonical association barrier consists principally of the total desolvation cost of the binding interface (contributed by H-bond enriched solvation), and canonical dissociation barrier consists principally of the cost of resolvating H-bond depleted solvation positions expelled during association [1–3,9–12]. kon is proportional to the degree of replacement of the H-bonds of H-bond enriched solvation expelled during association (noting that the free energy gains from such replacements are typically ≤ the free energy lost from desolvation, resulting at best in a zero sum energy game). As such, binding specificity is limited to partners capable of mutually replacing the H-bonds of H-bond enriched water solvating the nascent binding interface.
Permeability and solubility (neglecting the dissolution component), both of which indirectly affect hERG blocker occupancy, likewise depend on desolvation and resolvation costs, as follows:
The kin for membrane surface penetration depends on membrane desolvation within the footprint of the permeant.
Solubility depends on the balance between H-bond enriched and depleted solvation. High solubility potentially equates to high desolvation cost (depending on the specific nature of the expelled solvation), resulting in slower kon to binding sites and slow kin to phospholipid membranes. Low solubility equates to high resolvation costs, resulting in slow binding site koff and slow kout from the hydrocarbon membrane phase.
We predicted from our earlier work with WaterMap that P is solvated predominantly by bulk-like and H-bond depleted solvation localized around the side chains of Phe656 and Tyr652 [5], which is consistent with the:
Largely non-polar composition of the lumen.
Screening effect of ordered, H-bond enriched solvation on the negative electrostatic potential within the ion conduction pathway [5].
High promiscuity of the channel in the absence of H-bond enriched “gatekeeper” solvation, relegating the association barrier principally to steric size/shape complementarity between P and BP and the desolvation cost of BP [1].
More recently, we proposed that the canonical hERG blocker motif consists of chemically diverse Y-shaped scaffolds similar to the motif proposed by Cavalli et al. [13]:
The stem of which (BP) projects into P. Basic amines that are prevalent among hERG blockers always reside somewhere with this moiety.
The V-shaped cap of which (BC), or a subset thereof resides within C, with the stem and cap straddled between P and C, respectively (the activation gate necessarily closes on the BP-BC interface of trappable blockers, whereas non-trappable blockers are expelled).
In this work, we used WATMD to characterize the solvation dynamics of P, C, BP, and BC among a representative subset of hERG-blocking drugs exhibiting a wide range of potencies and pro-arrhythmicities. We show that potency/pro-arrhythmicity is qualitatively proportional to the amount of high versus ultra-low occupancy voxels (denoted as HOVs and ULOVs, respectively) surrounding BP, corresponding to H-bond enriched versus depleted solvation, respectively. Replacement of the H-bonds of expelled H-bond enriched blocker solvation is precluded in the absence of accessible polar side chains lining P, and as such, kb is proportional to the full blocker desolvation cost incurred during association.
Materials and methods
Molecular dynamics (MD) simulations are used extensively for predicting intra- and intermolecular structural rearrangements of proteins and other biomolecules [14–17]. However, our simulations are focused on water exchanges between solvation and bulk solvent (which we refer to as “solvation dynamics (SD) simulations”), for which we believe that MD approaches are best suited. We used WATMD (fully described in reference [2]) to calculate the solvation fields of a set of marketed/withdrawn drugs exhibiting known hERG activity and pro-arrhythmicities selected from the dataset compiled by Redfern et al. (Table 1) [18]. Briefly, WATMD counts the number of visits of water oxygen (O) and hydrogen (H) atoms to each of a set of 1 Å3 cells (voxels) within a lattice fully surrounding a LMW or HMW solute of interest during the last 10 ns of a 100 ns MD simulation. The counts per voxel are always distributed in a Gaussian-like fashion around the mean H and O counts, which correspond to bulk-like solvation (normalized for the 2 H/O ratio). Voxels exhibiting ultra-low counts and ultra-high counts fall within the left and right tails of the distribution [2]. ULOVs and HOVs are represented as spheres, the radii of which are proportional to the counts, and the colors of which are assigned as follows:
Bright red = 100% O visits.
Varying shades of pink = mixture of O and H visits, tipped toward O.
White = no preference for O or H. All ULOVs are colored white due to the typically non-polar environment of these voxels. Alternatively, white HOVs may be indicative of mixed donor/acceptor voxel environments or water molecules that are trapped within non-polar cavities (depending on the context).
Varying shades of purple = mixture of O and H visits, tipped toward H.
Bright blue = 100% H visits.
A subset of the hERG blockers and data compiled by Redfern et al. selected for our study. The data consists of the adverse event Class assigned by those authors (1 = antiarrhythmic drugs that were also to be pro-arrhythmic; 2 = drugs that were withdrawn due to high arrhythmic risk/benefit ratio; 3 = drugs with numerous reported cases of arrhythmia; 4 = drugs with isolated reports of arrhythmia; 5 = drugs with no reported cases of arrhythmia), minimum and maximum reported hERG IC50, and minimum and maximum reported plasma Cmax. Trappable and non-trappable blockers reported by Stork et al. [19] and Windisch et al. [20] are denoted by * and #, respectively.
The LMW SD protocol differs from the HMW protocol described in reference [2] in that LMW structures are fully restrained during the simulations (which would otherwise distribute over a large number of conformations in proportion to their force-field-calculated energies), whereas HMW structures are fully unrestrained (self-limited to high frequency rearrangements among the side chains and loops). All blocker structures were generated using the Build Tool of Maestro release 2021-2 (Schrodinger, LLC), and minimized using the default minimization protocol. The structures were overlaid manually on our previously published qualitative blocker overlay model [4] using the manual superposition tool of Maestro. Each structure was then simulated using AMBER 20 PMEMD CUDA (GAFF and ff99sb force-fields) for 100 ns in a box of explicit TIP3P water molecules, and the last 10 ns of each trajectory (40,000 frames) was processed into voxel counts via WATMD, and visualized as spheres using PyMol 2.4.1 (Schrodinger, LLC). We tested the conformational sensitivity of our results by calculating the solvation fields for mildly modified terfenadine and fexofenadine conformations relative to those in the overlay model (noting that significant variation of the solvation fields among highly dissimilar conformations is expected).
The open hERG structure (PDB code = 5VA1 [21]) was prepared using the default settings of the PPrep tool in Maestro. The starting POPC phospholipid membrane-bound structure was taken from reference [8] and subjected to a fully unrestrained 100 ns SD simulation in a box of explicit TIP3P water molecules using AMBER 20 PMEMD CUDA (ff14sb and Lipid14 force-fields). The resulting trajectory was processed into voxel counts as described above and in reference [2]). Astemizole was docked in the astemizole-bound structure (PDB code = 7CN1 [7]) using the Glide tool in Maestro (noting that astemizole was omitted from this structure for unexplained reasons). We qualitatively compared the binding modes of quinidine (PDB code = 6LQA [22]) and flecainide (PDB code = 6UZ0 [23]) in Nav1.5 with that of astemizole and our binding model.
Results
We postulated previously that pore occupancy by non-trappable blockers builds and decays transiently during each action potential (AP) cycle, whereas that of trappable blockers accumulates across APs [5, 6]. Blocker occupancy is governed by solubility and permeability (underlying free intracellular blocker concentration), pKa, and the desovlation/resolvation costs of the BP and BC moieties (noting that k-b < the rate of channel closing has no impact on potency under native conditions). Surgical hERG mitigation is often constrained by the typically narrow separation between these properties among the therapeutic target versus hERG and other off-target occupancies, all of which stem from (or are modulated by in the case of pKa) the solvation field. Here, we have used WATMD to qualitatively explore the desolvation costs of the blockers listed in Table 1, as described in Materials and methods. We postulate that blockers project their BP moieties into P at a rate governed by the full and partial desolvation costs of BP and BC, respectively, as reflected qualitatively in the sizes of the HOVs surrounding those moieties. We overlaid the blockers as described in Materials and methods (Figure 1) and reference [4], which we then used to predict the approximate BP-BC demarcation line and compare the calculated solvation fields across the dataset. The objectives of this work consisted of:
Testing our straddled BP-in/BC-out blocker binding hypothesis. If correct, the larger HOVs should be concentrated around the BC moiety of each blocker, which is predicted to remain bound within the well-solvated antechamber (possibly undergoing partial desolvation in the peri-entrance region [4]).
Assessing the relationship between HOV position/size/number and blocker potency. Fewer or smaller HOVs should occur on the BP moieties of Classes 1-3 compared with Class 4-5 drugs.

Qualitative manual overlay of the blockers listed in Table 1, as described in Materials and methods (keeping in mind that the blocker-bound state distribution cannot be captured in a single overlay model). The BP and BC moieties are labeled. The approximate positions of the pore entrance and selectivity filter relative to the overlaid blockers are shown for reference (red and blue horizontal lines, respectively).
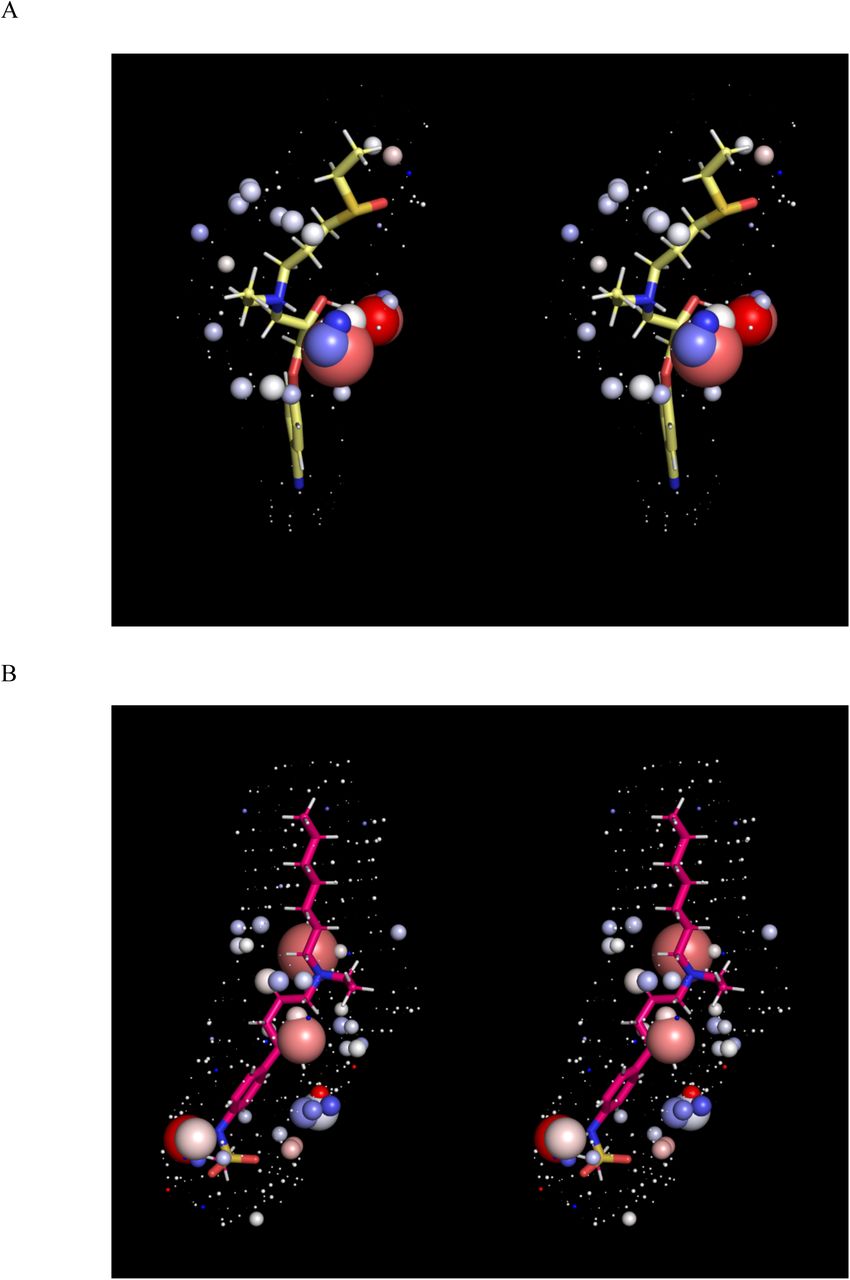
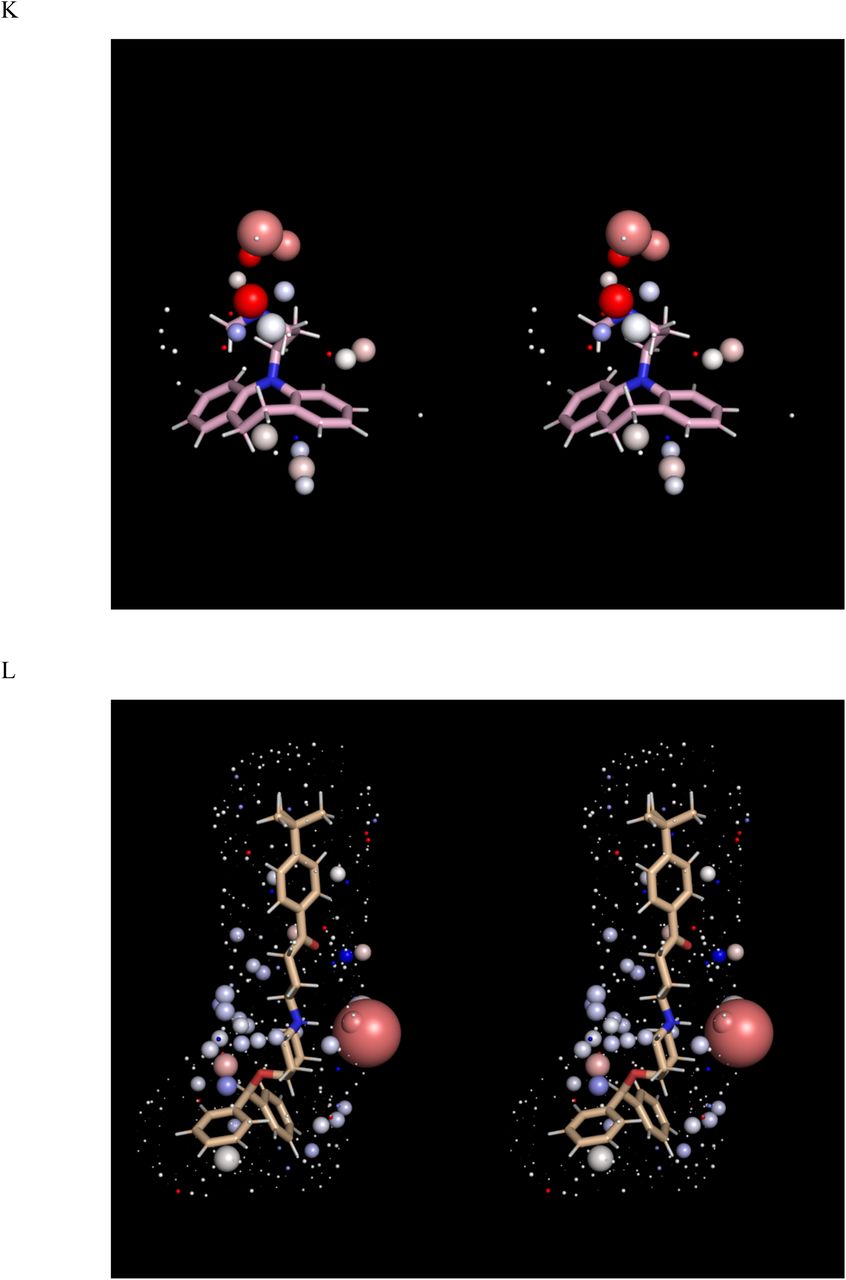
The calculated solvation field within the blocker-accessible region of P is predicted to consist principally of H-bond depleted solvation (Figure 2), in agreement with our previously reported WaterMap results [5] (consistent with the largely non-polar lining of P). We note, however, that this conclusion is subject to the considerable rearrangement of the pore during the unrestrained simulation (possibly resulting from truncation of the cytoplasmic domain, a question that will be addressed in future work). The solvation fields of the blocker conformations and charge state assumed in our overlay model (Figure 1) are shown in Figure 3, ordered by Redfern Class. In addition, we tested the sensitivity of our results to the choice of conformation and charge state (Figure 5). All blockers are solvated by H-bond depleted water (reflected in numerous diffusely distributed ULOVs) that govern koff from therapeutic targets and off-targets and k-b from P. The relatively non-polar BP groups of the Class 1 and 2 blockers almokalant, ibutilide, and terfenadine (Figures 3A-D) are largely devoid of HOVs, consistent with the low expected desolvation cost of the BP moieties (noting that the HOVs proximal to the aromatic groups in terfenadine and other blockers likely result from slight electrostatic attraction between water molecules and planar aromatic groups that are unobstructed by tetrahedral hydrogens, the free energy contribution of which is likely negligible). Conversely, the BP moiety of the Class 2 blocker terodiline (Figure 3E) is dominated by large HOVs surrounding the basic amine, the desolvation cost of which seems more comparable to that of the Class 3 blocker flecainide (Figure 3F) and the Class 4 blocker desipramine (Figure 3J)) than other Class 2 blockers. This inconsistency may be partially explained by the additive bradycardic and hERG blocking effects of this drug [6, 24]. The t-butyl acid and hydroxymethyl groups of the BP moiety in fexofenadine (a metabolite of terfenadine) are spanned by numerous HOVs (Figures 3G and H), consistent with higher BP desolvation cost and the Class 4 designation of this drug. The basic amine and hydroxymethyl groups positioned midway along the BP moiety of propafenone (Figure 3I) are likewise consistent with higher desolvation cost and the Class 4 designation of this drug. The predicted solvation field of verapamil (Figure 3K) is consistent with that of Class 2, rather than Class 5 blockers, which may be explained by the comparatively low maximum reported Cmax, together with compensatory blockade of the inwardly conducting Cav1.2 channel (the therapeutic target of this drug). The highly similar solvation fields of ebastine and terfenadine are inconsistent with their respective Class 5 and 2 designations, which is attributable to the large conformational difference between the t-butylphenylketone of ebastine (which is sterically incompatible with P) and the t-butylphenylmethane of terfenadine (Figure 4). Overall, our WATMD results are consistent with our proposed straddled BP-in/BC-out binding paradigm, and in general agreement with the Redfern classification (with certain explainable exceptions).

Cutaway stereo view of the solvation field within the pore of the time-averaged structure of the hERG channel (5VA1 with the cytoplasmic domain truncated), noting the significant rearrangement of the hERG structure during the unrestrained 100 ns SD simulation due in whole or part to the absence of the cytoplasmic domain. The solvation field consists primarily of ULOVs (consistent with the highly non-polar environment of P), accompanied by a small number of HOVs corresponding to weakly H-bond enriched solvation (the latter of which may be exaggerated by the loss of 4-fold structural symmetry of P during the simulation).


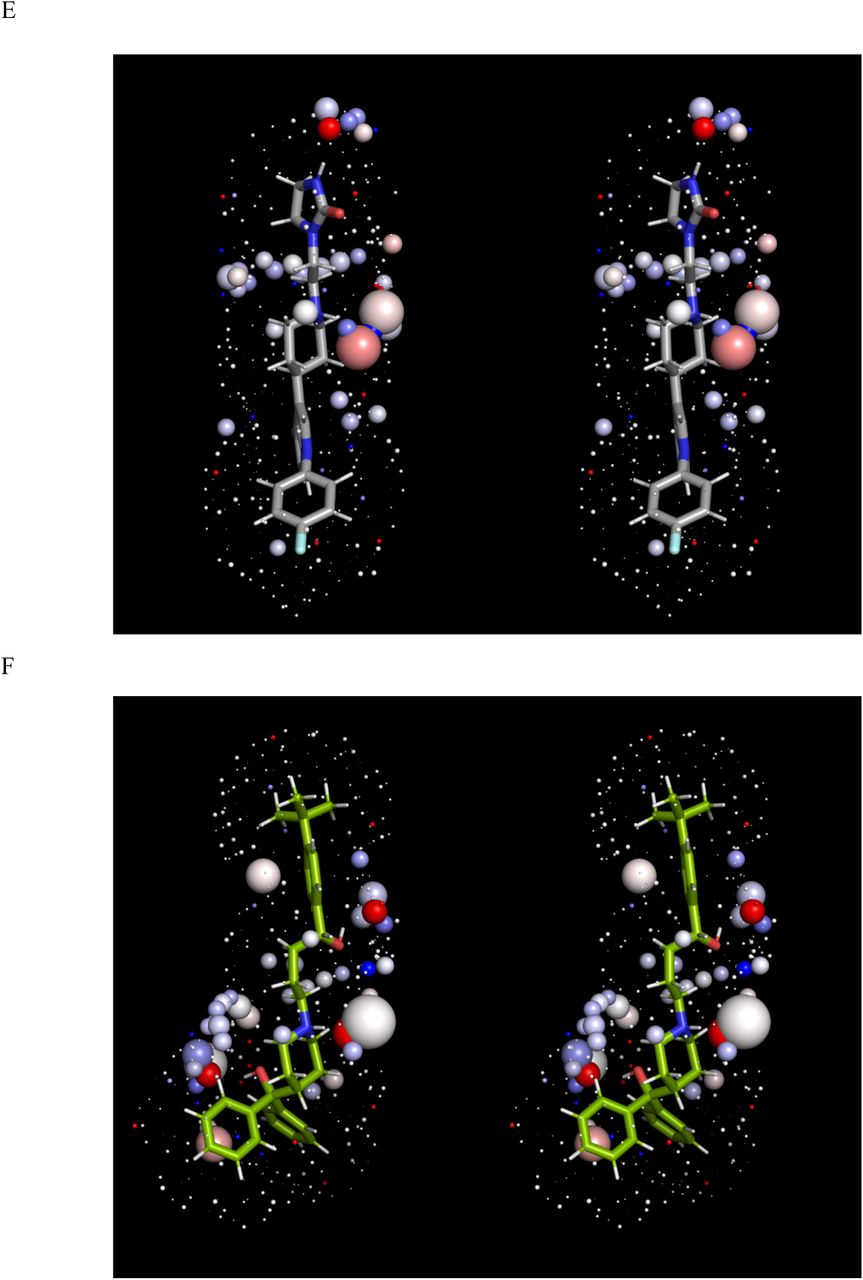




Stereo views of the solvation fields (represented by spheres as described in Materials and methods) and structures of the hERG blockers listed in Table 1 (sticks), the conformations of which correspond to those in Figure 1. The solvation fields of Class 1-2 blockers are highly similar and equate to low desolvation costs, with the exception of basic groups, which contribute heavily to solubility (as reflected in their larger HOV sizes). (A) Almokalant (Class 1). (B) Ibutilide (Class 1). (C) Quinidine (Class 1). (D) Cisapride (Class 2). (E) Sertindole (Class 2). (F) Terfenadine (Class 2). (G) Terodiline (Class 2). (H) Flecainide (Class 3). (I) Fexofenadine (Class 4). (J) Propafenone (Class 4). (K) Desipramine (Class 4). (L) Ebastine (Class 5). (M) Verapamil (Class 5).
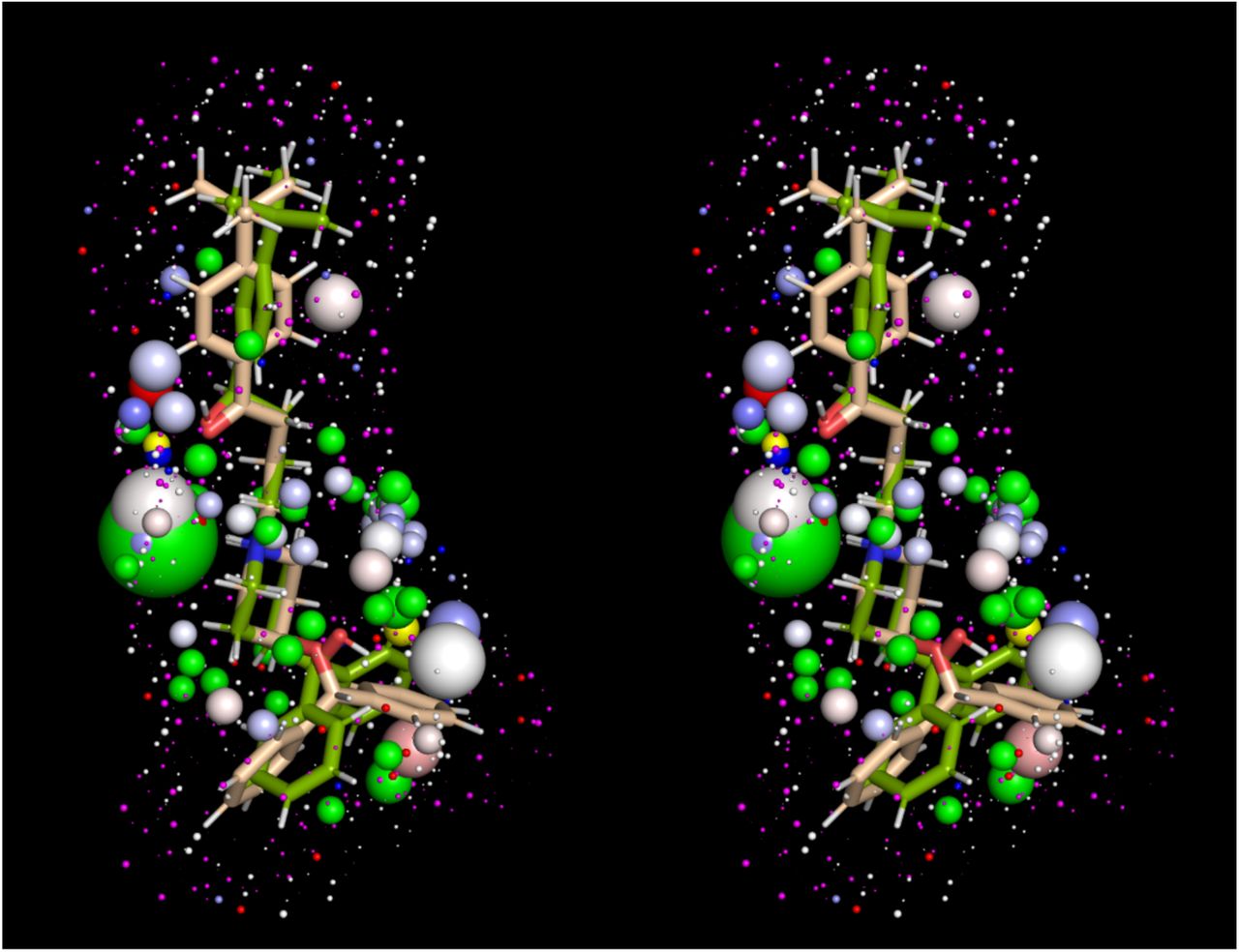
Stereo view of the solvation fields of ebastine (salmon sticks, green spheres = O preferred HOVs, yellow spheres = H preferred HOVs, magenta spheres = ULOVs) and terfenadine (green sticks, white spheres = ULOVs, standard red/white/blue spectrum = HOVs). The two blockers exhibit similar solvation fields despite their vastly different Redfern Class designations (5 and 2, respectively), leaving the significant conformational difference between the t-butylphenylketone of ebastine versus the t-butylphenylmethane of terfenadine to explain the large difference in hERG effects.


Stereo views of the solvation fields of alternate (though similar) conformations and neutral forms of selected blockers from Table 1 used to test the sensitivity of our results to conformational choice. (A) An arbitrary alternate terfenadine conformation overlaid on the conformation shown in Figure 1 (alternate form = salmon sticks, green spheres = O preferred HOVs, yellow spheres = H preferred HOVs, magenta spheres = ULOVs; reference form = green sticks, white spheres = ULOVs, standard red/white/blue spectrum = HOVs). (B) Same as A, except for fexofenadine (reference form = cyan sticks). (C) Neutral form of terfenadine overlaid on the charged form (neutral form = salmon sticks, green spheres = O preferred HOVs, yellow spheres = H preferred HOVs, magenta spheres = ULOVs; charged form = green sticks, white spheres = ULOVs, standard red/white/blue spectrum = HOVs). (D) Same as C, except for fexofenadine (neutral form = orange sticks, green spheres = O preferred HOVs, yellow spheres = H preferred HOVs, magenta spheres = ULOVs; charged form = cyan sticks, white spheres = ULOVs, standard red/white/blue spectrum = HOVs.
Discussion
hERG blockers undergo atypical binding
In our previous work [4], we proposed a canonical Y-shaped hERG blocker scaffold (or a subset thereof), in which the stem and cap of the Y straddle the pore entrance between the antechamber and pore, respectively, which is qualitatively analogous to the observed bound state of the detergent GDN in the cryo-EM structure of Nav1.4 (PDB code = 6AGF) [25]. However, this hypothesis is contradicted by subsequent experimental evidence and previous computational predictions that blockers are fully buried within the ion conduction pathway, including:
The MD simulations of Dickson et al. [8], in which the authors predicted that both blockers and activators are fully bound within the pore.
Trapping of MK-499 within closed hERG channels inferred by Mitcheson et al. based on the observed slow recovery from block [27].
The first cryo-EM hERG structures solved by Wang et al. [28], in which the authors proposed that blockers occupy some or all of the “hydrophobic pockets” residing inferior to the selectivity filter.
A recent cryo-EM structure of astemizole-bound hERG solved by Asai et al. [7] (7CN1).
Recent cryo-EM structures of flecainide and quinidine-bound Nav1.5, in which both blockers are fully buried within the pore (as claimed for astemizole-bound hERG and may be assumed for other hERG blockers as well).
It is nevertheless reasonable to question whether modeled and experimentally determined structures solved under equilibrium conditions at high blocker concentrations recapitulate the physiologically relevant blocker-bound states of hERG and Nav1.5 channels, in which blocker-driven equilibration is likely precluded by the dynamic forces on the S5 and S6 helices by the voltage-sensing domain and membrane dipole potential [29]. It is likewise reasonable to question whether the structural states of bound blockers can be differentiated electrophysiologically. Both fully and partially buried trappable blocker scenarios can exhibit extended recovery times since, unlike for non-trappable blockers, k-b is not usurped by channel closing (which governs dissociation exclusively from the open state). It is apparent that under fully native conditions, channel gating imposes severe time constraints on blocker association and dissociation that may be many fold lower under in vitro conditions. Blocker association and dissociation under physiological conditions are limited to:
The open/activated/inactivated states of hERG, which approximately span the ∼350 ms duration of the cardiac action potential (noting that the pore is likely accessible in the inactivated state).
The decaying open/activated sub-population of Nav1.5 channels that conducts the late Na+ current throughout the AP duration (noting that the peak current conducted by the fully populated open state of Nav1.5 is limited to the initial ∼2 ms of the AP and that the pore is likely inaccessible in the inactivated state).
The fully buried blocker-bound states observed in the cryo-EM and modeled structures are inconsistent with the aforementioned timescales for the following reasons:
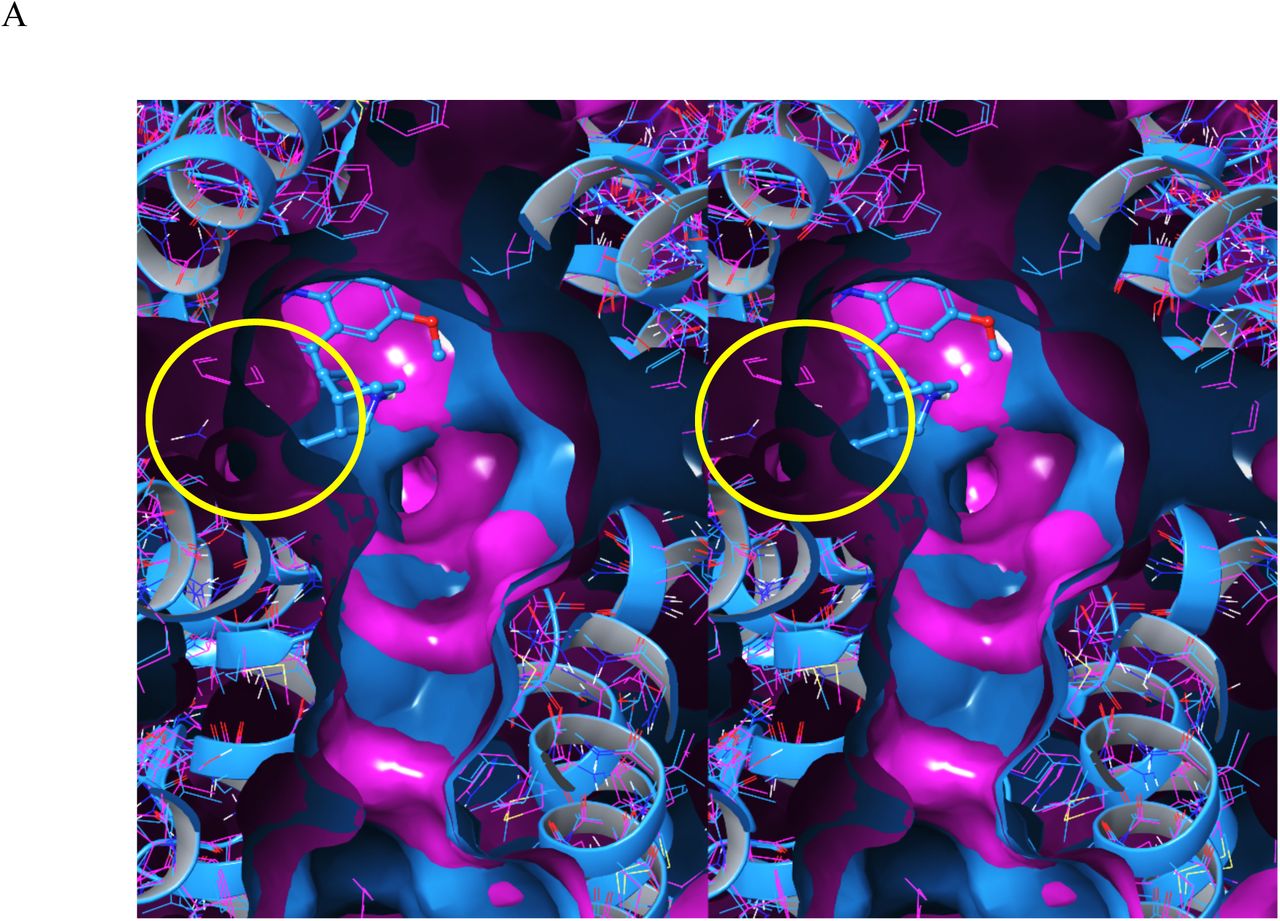
Steric constraints on the passage of the bulky, chemically diverse BC moiety of most blockers through the pore entrance, which can be reasonably assumed to depend on time-consuming induced-fit rearrangements. The pore entrance is partially closed in the apo Nav1.5 structure (Figure 6A), and quinidine and flecainide are trapped behind the closed pore in their respective structures (Figures 6B and C, respectively). The fact that blocker association is limited to the open state serves as further evidence for equilibrated channel populations in the cryo-EM preparations.
Time-consuming induced-fit rearrangements between quinidine, flecainide, and the backbone/side chains of the Nav1.5 blocker binding site (assessed by comparison of 6LQA and 6UZ0 with the apo 6UZ3 (Figures 7A and B, respectively)). Relatively minor astemizole-driven induced-fit is apparent from a comparison of apo (5VA1) and astemizole-bound hERG (7CN1) structures (Figure 7C). However, the blocker was omitted from the PDB file for unknown reasons, and the bound state described by the authors was not reproducible via docking into the original 7CN1 structure using Glide XP (see Materials and methods).


Stereo views comparing the pores of apo hERG with apo and bound Nav1.5 cryo-EM structures, looking from the cytoplasmic to extracellular direction. (A) The open state of apo hERG (5VA1, red) overlaid on the partially closed state of apo Nav1.5 (6UZ3, magenta). (B) Same as A, except for quinidine-bound Nav1.5 (6LQA). (C). Same as A, except for flecainide-bound Nav1.5 (6UZ0). The pore entrance in apo Nav1.5 is a small fraction of that in hERG, and nearly fully closed in the bound forms (consistent with equilibration).


Stereo views of the pore in Nav1.5 (6UZ3/apo, 6LQA/quinidine, and 6UZ0/flecainide) and hERG (5VA1/apo and 7CN1/astemizole), looking from the cytoplasmic to extracellular direction (induced-fit rearrangements are circled in red). (A) Quinidine-bound Nav1.5 overlaid on apo Nav1.5. (B) Flecainide-bound Nav1.5 overlaid on apo Nav1.5. (C) Astemizole-bound hERG overlaid on apo hERG (absent astemizole). The primary difference is limited largely to the lower right region, which may or may not result from induced-fit.
The fully buried blocker-bound states observed in the cryo-EM structures are additionally questioned by the following discrepancies:
The lack of an obvious explanation for trappable versus non-trappable blocker subtypes based on the observed binding modes in 7CN1, 6LQA, and 6UZ0 compared with our proposed straddled BP-in/BC-out paradigm [4]. Time-consuming induced-fit dependent association and dissociation steps seem particularly implausible for non-trappable blockers, the occupancy of which necessarily builds and decays within each AP cycle (noting that trappable blockers likewise associate and dissociate dynamically during the open channel time window).
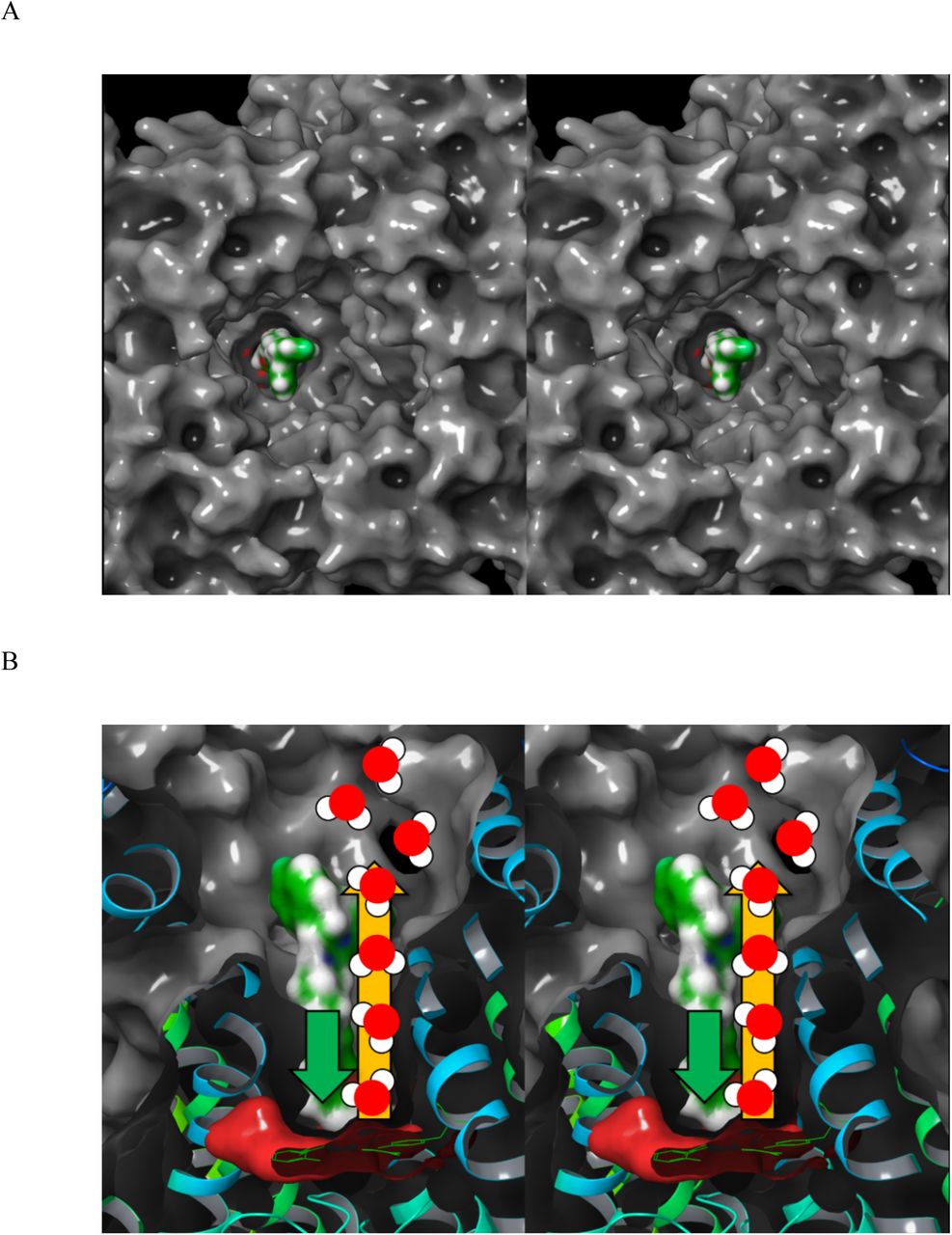
The lack of an obvious desolvation path from the fully occupied pore and one or more of the hydrophobic pockets in hERG. The pore volume is only partially filled by blocker BP moieties in our model (analogous to a syringe in which the plunger diameter < barrel diameter) (Figure 8A). We postulate that water expelled during blocker association flows through the unoccupied pore volume between the blocker surface and pore lumen anti-parallel to the BP association direction, exiting into the antechamber via the pore entrance (Figure 8B).
Overweighted interatomic contacts between P and BC in modeled structures based on force-field energies relative to the far greater solvation contribution claimed under our binding free energy paradigm.

(A) Stereo view of the free pore volume between the surface of manually docked astemizole (green surface) and the pore surface (gray surface) viewed from the antechamber entrance looking along the pore axis toward the selectivity filter located deep within the cavity. This volume serves as the putative desolvation path between the leading edge of the associating blocker and antechamber. (B) Same as A, except as a cutaway viewed perpendicular to the pore axis, with the entrance at the top and selectivity filter at the bottom of the figure. The putative desolvation path is annotated by the yellow arrow pointing from the leading edge of astemizole to the antechamber. The blocker association direction is annotated by the green arrow.
Instead, we propose that the buried blocker binding geometries observed in the hERG and Nav1.5 cryo-EM structures are relevant to equilibrium radioligand binding (RLB) assays and docked models (the latter of which are biased toward maximal interatomic contacts and minimum force-field energies), but not non-equilibrium conditions in vivo or in patch clamp assays preformed at the native gating frequency. We showed previously that %hERG inhibition is poorly correlated among 7,231 compounds tested in RLB and QPatch assays, consistent with a variable, condition-dependent blocker binding paradigm (the observed/modeled fully buried versus partially buried states assumed in our model). Furthermore, the need for specifically positioned polar blocker groups capable of replacing the H-bonds of H-bond enriched protein solvation is obviated by the lack of HOVs within the pore (corresponding to “gatekeeper” solvation). As such, multiple binding geometries of a given blocker are conceivable under both equilibrium and non-equilibrium conditions. Our results are consistent with the high tolerance of hERG for diverse blocker chemotypes conforming to a general Y-shaped motif or subset thereof. In the absence of H-bond enriched pore solvation, blocker association free energy costs are relegated to the BP desolvation cost, which are non-attenuated in the absence of H-bond replacements within the pore. No specific conformational or pharmacophore requirement exists for blocker binding. As such, hERG blockade represents a form of non-specific binding that depends principally on:
Steric shape/size pore complementarity (including the energetic preference for complementary blocker and pore conformations).
Total blocker desolvation cost (including the availability of a desolvation path between the pore and bulk solvent).
Solubility (the gain in free energy between the solvated and unsolvated forms, which is typically enhanced by incorporation of one or more basic groups in BP).
Permeability (blocker desolvation and resolvation costs vis-à-vis membrane desolvation cost).
General hERG safety criteria suggested by our findings
The holistic optimization of primary target activity, permeability, solubility, and mitigation of hERG and other off-target activities, depends on the correct understanding of blocker structure-kinetic and structure-free energy relationships leading to the arrhythmic tipping point of hERG occupancy under native cellular conditions. Numerous studies aimed at predicting binding geometries and interactions between hERG and diverse blocker chemotypes via ligand- and structure-based methods have been attempted over the last few decades by ourselves and other workers [8, 30]. Mitigation nevertheless remains a largely trial-and-error proposition due to heavy reliance on:
Equilibrium data that does not apply to binding sites undergoing high frequency buildup and decay cycles.
Status quo data-driven free energy models that are based on interatomic contacts (van der Waals, electrostatic, H-bonds, π- π, π-cation, hydrophobic, hydrophilic, etc.) rather than solvation fields (the putative horse’s mouth of SAR).
The arrhythmic tipping point resides at a dynamic hERG occupancy of ∼50% [5, 6] in simulations that we performed previously using a modified version of the O’Hara-Rudy model of the undiseased ventricular action potential [31, 32]. Cellular arrhythmia in the form of atypical depolarizations can occur stochastically during transient or sustained occupancy incursions at or above this level. Occupancy amplitude increases in both cases as blocker exposure approaches the intracellular free Cmax, and decays with clearance-driven cytoplasmic efflux. Measured IC50 weighted toward k-b < the channel deactivation rate and kb < the channel activation rate may result in overestimated potency of both trappable and non-trappable blockers. Arrhythmic occupancy by trappable blockers depends on blocker free Cmax relative to hERG IC50 (accumulating to the 50% level at exposures ≈ the true blocker IC50 [5, 6]), where the rate of fractional occupancy buildup depends on the slower of the antechamber and pore association steps. Trappable blocker dissociation is “paused” during the closed channel state, which exists during the approximately last two-thirds of the cardiac cycle (noting that decay of the bound state prior to channel deactivation results when k-b exceeds the channel closing rate). Arrhythmic occupancy by non-trappable blockers, which builds and decays within each channel gating cycle, is governed by the following contributions:
Free intracellular blocker exposure > hERG IC50 (concentration-driven) and/or kb approaching the rate of channel activation (kb-driven), where kb depends largely on the cost of expelling H-bond enriched blocker solvation in the absence of polar pore replacements. Free intracellular exposure, in turn, depends on:
Free plasma exposure, which depends on solubility, absorption, clearance, distribution, and dynamic plasma protein binding (PPB).
Cell permeability, which depends on plasma concentration and mutual membrane and blocker desolvation costs.
The fraction of blocker bound to membranes, lysosomes, and intracellular non-hERG off-targets.
k-b approaching the rate of channel deactivation, which depends largely on the rate of channel closing or blocker/pore resolvation costs that are qualitatively proportional to the number of ULOVs surrounding P and BP (whichever is faster).
Knowledge of blocker trappability is therefore essential for assessing concentration-occupancy relationships based on IC50 measurements. Furthermore, data models unwittingly generated using mixtures of trappable and non-trappable blockers may not be meaningful (noting that trappable and non-trappable analogs may even occur within the same chemical series, as evidenced by the propafenone analogs reported by Windisch et al. [20]).
Optimal hERG occupancy is achieved within a Goldilocks zone of solubility, permeability, and binding for both trappable and non-trappable blockers, as follows:
Solubility is proportional to the H-bond free energy of H-bond enriched solvation, as reflected in HOV number and size (noting that all HOVs fall deep within the tail of the Gaussian distribution of water counts tabulated by WATMD). Poor solubility reduces the free blocker concentration, whereas high solubility equates to high desolvation cost. H-bond enriched solvation is further enhanced by basic groups, which additionally speed kb (as a function of increasing pKa) due to electrostatic attraction with the negative field within P. Since blocker potency is typically enhanced by basic groups, it follows that the electrostatic kb-speeding contribution of such groups typically outweighs the kb-slowing desolvation contribution.
Permeability is proportional to the desolvation cost of H-bond enriched blocker solvation, which additionally depends on polar groups for replacing the H-bond enriched solvation of membrane phospholipid head groups (the rationale underlying the Pfizer Rule of 5). The on-rate to the antechamber is proportional to intracellular blocker concentration. High desolvation costs corresponding to HOVs located anywhere on blocker surfaces slow permeation, as does the lack of polar groups that facilitate membrane desolvation (again, which is qualitatively consistent with the Rule of 5).
The overall asymmetric distribution of HOVs among BP and BC blocker moieties is consistent with our proposed binding mode, in which BP (exhibiting the lower desolvation cost/lower association free energy barrier) projects into the pore, while BC (exhibiting the higher desolvation cost/higher association free energy barrier) remains within the solvated antechamber. HOVs on the BP moiety of most blockers (except fexofenadine) are positioned around the basic group (when present), which is typically incorporated for solubility enhancement purposes, and in some cases, therapeutic target binding. The Redfern Classes are correlated qualitatively with the basic group and HOV position/desolvation cost along the longitudinal BP axis, as follows:
A lower desolvation cost is incurred when positioned near the proximal end of BP in blocker Classes 1-3 (corresponding to the pore entrance region).
A higher desolvation cost is incurred when positioned near the middle or distal end of BP in blocker Classes 4-5 (projecting deeper within the non-polar pore environment).
The Goldilocks zone of pro-arrhythmic hERG occupancy thus depends on optimal solubility + permeability + free intracellular exposure + fast antechamber kon + fast pore kb, as reflected in HOV sizes and distributions among the BP and BC regions. Our WATMD results are qualitatively consistent with the aforementioned dependencies. The Class 1-2 blockers in our dataset (almokalant, ibutilide, terfenadine) (Figures 3A-C) lack large HOVs on the BP moiety but contain significant numbers of ULOVs needed to slow k-b (in addition to ULOVs distributed around the surface of P). Conversely, HOVs are predicted at the BP positions of Class 4 blockers (fexofenadine, propafenone, and desipramine). The t-butyl acid group located on the distal end of BP explains the weak hERG activity of fexofenadine, whereas the weaker potencies of propafenone and desipramine can be explained by the more distal position of the basic group on BP relative to that of Class 1-2 blockers. The HOV positions in all of these cases reside deeper along the pore axis, consistent with higher desolvation costs at these positions.
The data reported by Redfern et al. (Table 1) suggests that arrhythmic occupancy levels are achieved at the maximum end of the reported free Cmax range, rather than the minimum end of the reported IC50 range for most of the Class 1-2 drugs in our study (with the exception of cisapride and terodiline) (Table 3 and Figure 9). Cisapride is a known non-trappable Class 2 blocker [19], the high pro-arrhythmicity of which is necessarily due to fast kb and/or exposure escalation far above the therapeutic level. Since this drug does not achieve the putative ∼50% arrhythmic hERG occupancy level at the maximum reported IC50 and Cmax (Tables 1 and 3), the former explanation is more likely (noting that fast kb is consistent with the 2 nM lowest reported IC50). The Class 2 pro-arrhythmicity of terodiline (predicted to be trappable based on our overlay model [4]) likewise achieves the putative arrhythmic hERG occupancy level at the maximum reported Cmax and minimum IC50 (Tables 1 and 3). Interestingly, verapamil exhibits sub-arrhythmic occupancy for all IC50-Cmax combinations in Table 3, suggesting that the lack of pro-arrhythmicity of this drug is only partially attributable to concurrent Cav1.2 blockade.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
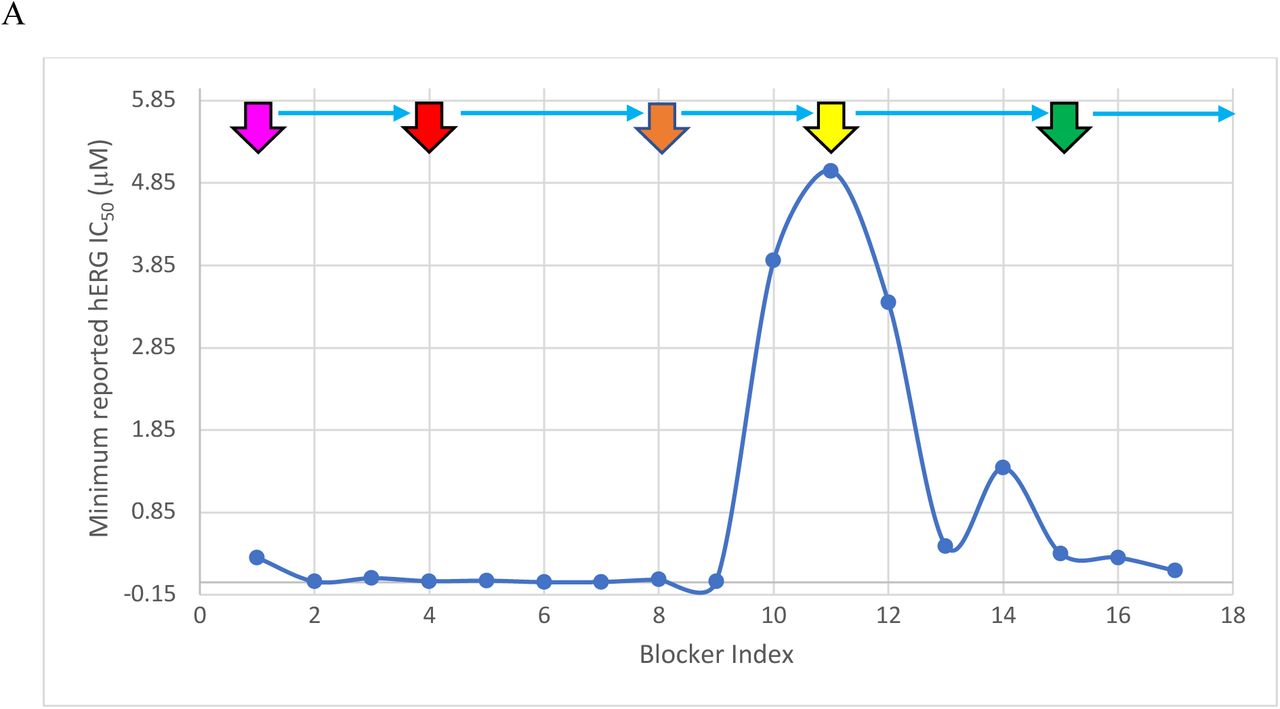
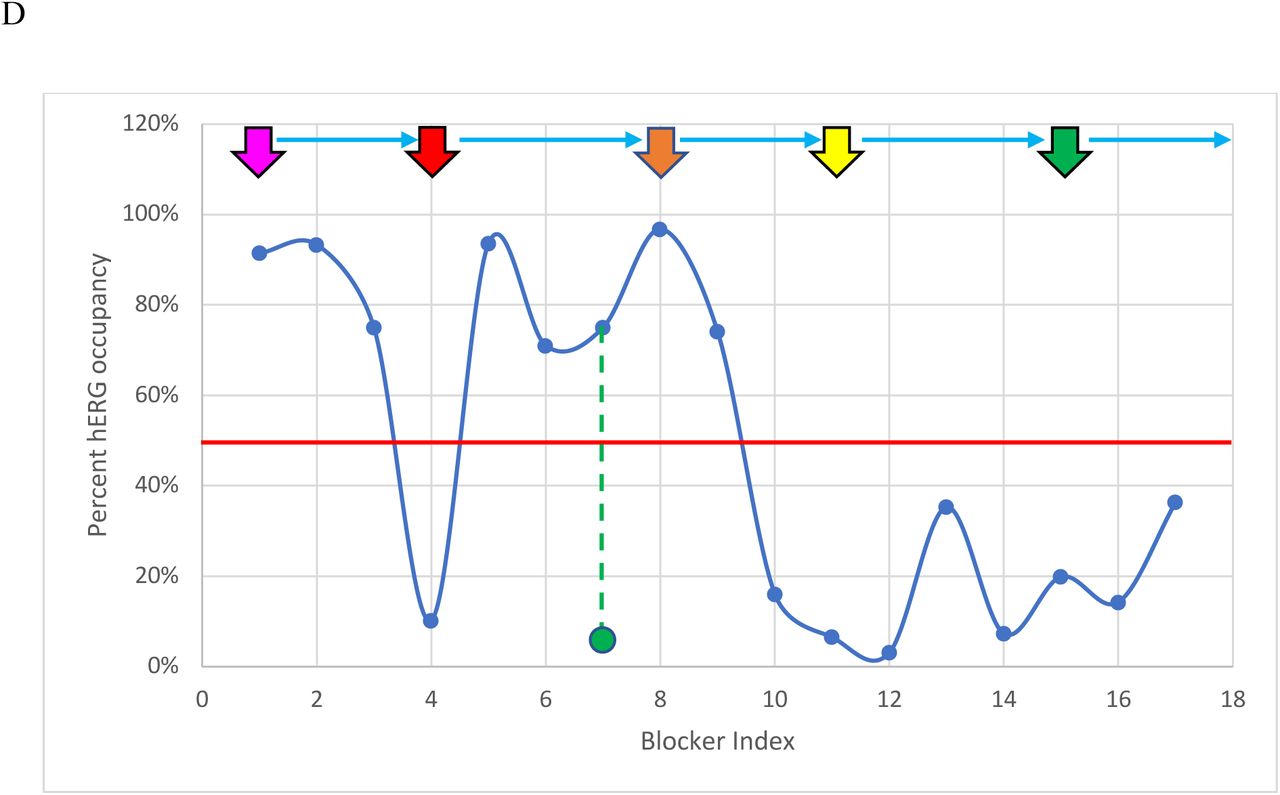
Parameter distributions for the blockers listed in Table 1 annotated by Redfern Class (magenta = Class 1, red = Class 2, orange = Class 3, yellow = Class 4, and green = Class 5). Mizolastine (omitted from our WATMD dataset) was added to increase the representation of Class 5 blockers. (A) Distribution of the minimum reported hERG IC50. (B) Distribution of the maximum reported Cmax. (C) Distribution of the ratios of the minimum reported hERG IC50/maximum reported Cmax. (D) Equilibrium hERG occupancy calculated using (max Cmax)/(max Cmax + min IC50), with the 50% arrhythmic level predicted in our previous work [6] denoted by the horizontal red line. The pro-arrhythmic effects of verapamil (blocker 17) are widely believed to be attenuated by co-blockade of Cav1.2 channels. It is apparent from Table 3 and Figure 3D that verapamil approaches but does not exceed the putative arrhythmic 50% hERG occupancy level at the minimum reported IC50 and maximum reported Cmax, suggesting that co-blockade of the outward hERG and inward Cav1.2 currents is only partially responsible for the safety of this drug. Sertindole is an outlier in the Redfern dataset, having reported incidences of arrhythmia, despite falling well below 50% hERG occupancy at the maximum reported Cmax and minimum reported hERG IC50. The calculated equilibrium hERG occupancy of cisapride (blocker 6) is likely exaggerated due to the known non-trappability of this compound. The calculated solvation field of terodiline is more similar to the Class 4 blockers desipramine and imipramine than Class 2 blockers, consistent with calculated occupancy = 1.7% at the highest reported IC50 = 0.7 μM (green circle).
Equilibrium hERG occupancy (predicted from the Hill equation defined above) as a function of maximum and minimum reported Cmax and hERG IC50. The Redfern Classes correlate poorly with reported max IC50 at min Cmax, and best with reported min IC50 at max Cmax. However, max IC50 and max Cmax are also well-correlated with Redfern Class (bolded values), with the exception of cisapride and terodiline (italicized values), suggesting that arrhythmic occupancy is driven more by Cmax escalation above the therapeutic level than by potency (noting that sertindole is an across the board outlier, which is also shown in italics).
A general hERG mitigation strategy suggested by our findings
Potent hERG blockers reside within a Goldilocks zone of H-bond depleted and enriched solvation corresponding to optimal solubility, permeability, and hERG binding. Successful hERG mitigation depends on the extent to which drug-hERG and drug-target Goldilocks zones are separable. Blocker conformational preferences and possible rearrangements of the blocker-bound pore, which could contribute significantly to potency, solubility, and permeability, were neglected in the present work due to the inadequacy of current force-field-based conformational searching methods that overweight and underweight the interatomic contact and solvation free energy contributions, respectively (to be addressed in future work).
Our proposed canonical “BP-in/BC-out” blocker binding model, together with WATMD, can be used to predict the pore-binding moiety of a given blocker, and qualitatively guide mitigation to hERG occupancies << 50% at the highest plausible Cmax by:
Achieving the highest therapeutic target occupancy at the lowest possible exposure (the biggest bang for the buck) via “kinetically tuned” drug-target binding [33]. Low efficacious/therapeutic exposure affords the greatest possible safety margin for hERG and all off-target liabilities, many of which may track with target activity.
Disruption of blocker trappability via incorporation of a bulky group within the putative constriction zone in the closed channel state (as we reported previously [4]).
Disruption of P-BP shape compatibility (e.g., putatively exemplified by ebastine versus terfenadine).
Introducing strategically positioned polar groups on BP aimed at increasing the desolvation cost of this substituent and slowing kb without slowing the primary target kon (noting that increased polarity at other blocker positions, as reflected in decreased logP) is likely insufficient for hERG mitigation.
Exploring structure-solubility and permeability relationships, which are likewise governed by solvation free energy and membrane desolvation/resolvation costs.
Optimal positioning and pKa of a basic group on BP used for solubility (noting that increased solubility as a f(pKa) results in higher desolvation cost, but also speeds kb electrostatically).
Conclusion
This work is a follow-on to our previous work in which we postulated that hERG blockers are first captured by the intracellular antechamber contained within the CNBH and C-linker domains of the channel, followed by the projection of a single R-group (BP) into the open pore (P) [4]. As described in our previous work [4] and by others [19, 20], trappable blockers remain bound within closed channels, whereas non-trappable blockers are expelled during channel closing. Blocker occupancy is powered primarily by desolvation of H-bond depleted solvation of P, BP, the pore-facing surface of BC, and the peri-pore region of the antechamber (which slows k-b). The key determinants of hERG blockade consist of:
Steric size/shape complementarity between BP and P, where BP is typically a non-polar/weakly polar rod-shaped moiety of almost any chemical composition. The antechamber volume is sufficient for capturing large chemotypes that need not fully fit within P.
Second order buildup of the antechamber-resident blocker population (governed by on-rate = kc · [free intracellular blocker] · [free antechamber]), followed by first order buildup of the P-BP population (governed by kb · [antechamber-bound blocker]).
A Goldilocks zone of BP solvation free energy needed to simultaneously achieve solubility and low desolvation cost of P and BP during association (which applies to both therapeutic target and all forms of off-target binding). P is predicted by both WaterMap and WATMD to contain H-bond depleted and bulk-like solvation, which incurs no desolvation cost during association and a resolvation cost during dissociation equal to the total free energy of H-bond depleted solvation expelled during association.
A basic group residing somewhere within BP that is typically used to improve solubility (traded off against a higher desolvation cost). Electrostatic attraction between P and such groups speeds kb more than the additional desolvation cost slows it.
Blocker trappability, in which arrhythmic hERG occupancy is more likely achievable by trappable blockers whose occupancy accumulates across multiple APs.
Successful hERG mitigation is necessarily achieved by slowing kb via increased polarity and desolvation cost of BP (within the Goldilocks zone of solubility, permeability, and target binding), pKa attenuation that maintains solubility (and in some cases efficacious target occupancy), avoidance of trappability, and disruption of blocker-pore shape complementarity (e.g., ebastine).
References




