

Abstract
Hereditary hemorrhagic Telangiectasia (HHT) also known as Rendu-Osler-Weber syndrome is a rare genetic disease relying on mutations affecting components of TGF-β/BMP signaling pathway in endothelial cells. This disorder is characterized by vascular dysplastic lesions or arterio-venous malformations prone to rupture and ensuing hemorrhages are thought to be responsible for iron deficiency anemia. Along with Activin receptor-like kinase ALK1, Endoglin (CD105) is involved in the vast majority of reported HHT cases. Unlike Type-1 TGF-β Receptor ALK1, Endoglin is not endowed with Ser/Thr kinase activity but serves as an ancillary receptor that modulates TGF-β receptor complex signaling. In this report, we characterized zebrafish endoglin locus and demonstrated that it produces two phylogenetically conserved protein isoforms using a distinctive alternative splicing mechanism. This preliminary work served as a base to the establishment of a Crispr/Cas9 engineered Endoglin mutant line from which functional analysis revealed that Endoglin deficiency results in massive fish death during the course from juvenile stage to adulthood. Endoglin deficient fish develop a cardiomegaly resulting in heart failure and hypochromic anemia which both stem from chronic hypoxia. A combination of histological analysis and confocal imaging evidenced structural alterations of the developing gill and its underlying vascular architecture that tally with respiratory distress and hypoxemia/hypoxia occurrence. Finally, phenylhydrazine treatment demonstrated that lowering hematocrit/blood viscosity constraint strongly alleviates heart failure and enhances survival of Endoglin deficient fish. Altogether our data support the notion that zebrafish Endoglin is crucial for gill proper development and respiratory function and that further studies using zebrafish in general and this endoglin mutant in particular will provide crucial hints of molecular and cellular events altered in HHT for the development of new therapeutic strategies.
Introduction
Endoglin (CD105) belongs to the Bone Morphogenetic Protein and Transforming Growth Factor-beta BMP/TGF-β receptor superfamily and binds TGF-β1, TGF-β3, BMP9 and BMP10 1,2. Like betaglycan (TGBR3), this single-pass transmembrane protein works as an ancillary receptor by modulating the signaling activity of the heterotetrameric receptor complex constituted of type I and type II serine-threonine kinase receptors which phosphorylate R-Smads which eventually associates with Co-Smad (Smad4) to form a complex that regulates the transcription of target genes via Smad Binding Elements (SBE). Endoglin is mainly expressed in endothelial cells (EC) and Endoglin deficient mice die around E10.5 from cardiovascular defects associated with improper vascular smooth cell coverage3. In addition to endothelial cell expression, in mammals, Endoglin is also expressed in neural crest cells and derivatives such as vascular smooth muscle cells responsible for blood vessel stabilization and vascular tone control and in Hematopoietic Stem cells (HSCs) as they emerge from intraaortic clusters to later being restricted to HSCs with long term repopulating capacities (LT-HSCs)4–6.
In humans, inactivating mutations in endoglin (ENG) gene are responsible for type 1 Hereditary Hemorrhagic Telangiectasia (HHT) while HHT2 is due to mutations affecting ACVRL1 (ALK1) a type I TGF-β receptor specifically expressed in endothelial cells. Together, HHT1 and HHT2 account for approximately 85% of diagnosed HHT cases7. Endoglin and Alk1 independent HHT cases rely on mutations affecting protein function of BMP9, a high affinity ligand for ALK1, microprocessor RNAse III Drosha and additional loci yet to be characterized while mutations in SMAD4 are causative of Juvenile Polyposis-HHT 8–11.
Also known as Rendu-Osler-Weber syndrome, HHT is a rare inherited autosomal dominant genetic disorder characterized by a wide array of symptoms among which mucocutaneous telangiectasias and recurrent epistaxis are the foremost diagnosis cues. These angiodysplastic lesions or arteriovenous malformations (AVMs) also often affect organs such as the gastrointestinal tract, the lungs, the liver and the brain. They consist of dilated veins prone to rupture under mechanical strains. These AVMs resulting from the loss of the arteriolar-capillary plexus connect venous vessels directly to arteries exposing the former to arterial-type blood flow mechanics12.
Although much concern arise from brain, liver or lung AVMs which can have life-threatening outcomes, massive blood loss from hemorrhages affecting the gastrointestinal tract or from recurrent epistaxis episodes are also important issues as it results in iron deficiency anemia that requires attentive medical management by way of regular iron replenishing cures and/or blood transfusion13.
Congestive heart failure constitutes another critical clinical manifestation of HHT although considered fairly rare. Cases seem to be mostly associated with liver AVMs and chronic anemia of iron deficiency type14–17
Albeit different in terms of incidence in specific organs such as brain or liver which is much higher in HHT1, all HHT forms rely on mutations affecting genes working on a common TGF-β/BMP pathway and recent findings regarding Drosha indicate that non-classical TGF-β/BMP pathways are also involved18. This suggests that while this pathway is indisputably at the core of the pathology, tissue specific cues or vascular bed specificities might provide a terrain that will somehow trigger or favored the onset of the pathology. In humans, HHT relies on hemizygous mutations and accordingly, mice carrying a single knockout Endoglin allele do spontaneously develop HHT-like phenotypes with, however, varying penetrance depending on genetic background19. This collectively refers to as the second hit concept by which additional alterations would be required for the development of the pathology. Consistent with this concept, angiogenic/inflammatory environments have been found to be potent drivers of AVM formation in both heterozygous knockout Endoglin and ALK1 mouse models20.
Several recent studies using BMP9/10 blocking antibodies, inducible tissue specific Endoglin, ALK1 and Smad4 knockout mouse models as well as Endoglin genome editing in zebrafish have provided important insights into the cellular and molecular mechanisms governing AVM formation which results from inappropriate signaling responses to blood flow including EC polarization, venous/arterial identity maintenance, proliferation and pericyte recruitment21–25. In keeping with this, in HUVECs, laminar shear stress triggers Endoglin association with ALK1 and potentiates ALK1 signaling pathway in response to endogenous serum BMP9/10 concentration21.
In this work, we examined the consequences of Endoglin loss in postembryonic stage zebrafish. Using a CRISPR/Cas9 engineered endoglin mutant line, we demonstrated that homozygous mutants massively die from congestive cardiomyopathy from about 1 month old accompanied with iron deficiency anemia (IDA). We found that this pathological condition sets just before 15dpf when hypoxia and cardiac stress markers start increasing in mutants and coincide with heart chamber enlargement which involves cardiomyocyte proliferation. Data from histology and imaging of blood vessel organization during gill development strongly suggest that hypoxia stems from improper gill functioning due to Endoglin direct activity in this organ. Controlling hematocrit/blood viscosity triggered by chronic hypoxia markedly lessens hypoxic response, cardiac stress to result in markedly enhanced survival of Endoglin deficient fish. Thus, by reproducing important features of HHT, our zebrafish model of Endoglin deficiency will provide the ground for future detailed molecular analysis that will be important for both the identification of HHT altered signaling pathways and the development of new treatments.
Results
Zebrafish endoglin locus, transcripts and expression characterization
In an effort to develop tools that would be useful to address Endoglin expression and function in zebrafish, we first aimed at the characterization of endoglin locus and associated transcripts in this organism. We conducted 5’ and 3’ RACE experiments and obtained complete and trustful sequences unavailable at the time this work was initiated. This allowed to reconstruct exon-intron organization of zebrafish endoglin gene, essential for gross mapping of transcriptional regulatory regions. Although such a structure has recently been published25 it slightly differs from ours (Figure 1a). We identified 3 additional exons. Two non-coding exon, one very short located the utmost 5’ positioning Endoglin Transcriptional Start Site (TSS) and one 3’ containing most of Endoglin 3’UTR. The third identified exon (exon 13 in our nomenclature) corresponds to an alternate exon which introduces an early termination codon resulting in an Endoglin protein isoform with a very short cytosolic domain (Figure 1b and Figure 1c). Noteworthy, a similar isoform is also expressed in mammals26. By contrast to Long isoforms, alignments revealed little or no residue conservation in the cytosolic tail of Short isoforms (Figure 1d). RT-PCR analysis demonstrated that both variants are expressed in embryonic and adult tissues, endoglin messenger coding for the short isoform being markedly less abundant (Figure 1b). Similar analysis performed on developmentally staged embryos indicated that with exception of a marginal maternal contribution, the zygotic expression of endoglin coincides with early somitogenesis and gradually expands in later stages with a somewhat stable Long isoform/Short isoform ratio (Figure 1e). Whole-mount in situ hybridization using an endoglin antisense riboprobe detected endoglin expression in developing blood vessels with the highest endoglin expression mostly associated with veins (Figure 1f). In line with early findings in mammals, our data also confirmed the results from a recent study in zebrafish and suggested of a conserved Endoglin function throughout the whole vertebrate phylum25.

Characterization of zebrafish Endoglin chromosomic organization, transcripts, expression pattern. (a) Zebrafish endoglin gene exon-intron structure on chromosome 5. Coding Exons are in black, non-coding exons are in grey. (b) RT-PCR analysis of endoglin splice variants. Endoglin Long (arrowhead) and short isoform (asterisk) coding mRNAs are detected in both whole embryo and adult tissue such as brain. (c) Protein alignment of zebrafish Endoglin isoforms. Alternate amino acids of Endoglin short isoform are depicted in bold. Transmembrane domain amino acids are in red. Endoglin transmembrane domain is highlighted in red. (d) Alignment of Human, mouse and zebrafish Endoglin short isoform reveals little or no evolutionary amino acid conservation of zebrafish isoform with respect to mammalian counterparts. Identical and similar residues are highlighted by black and grey shades respectively. (e) RT-PCR analysis of endoglin splice variants during zebrafish early development. Asterisk indicates endoglin variant producing Endoglin short isoform. Arrowhead points to endoglin variant producing Endoglin Long isoform. Note that Endoglin is faintly detected as early as one-cell stage indicative of a marginal maternal contribution whereas endoglin zygotic expression onset coincides with early somitogenesis. (f) Whole-mount in situ hybridization using dig-labelled endoglin antisense RNA probe on 24 hpf wild-type zebrafish embryo detects specific endoglin expression in developing blood vessels. Inset, close-up of trunk region showing endoglin differential expression between dorsal aorta (DA) and posterior cardinal vein (PCV). Arrowheads point to sprouting intersegmental vessels. Right panel, Close-up of head region showing mostly venous-associated endoglin expression, ACeV, Anterior cerebral vein; MCeV, Middle cerebral vein; PHBC, Primordial hindbrain channel; PMBC, Primordial midbrain channel; OV, optic vein,
Crispr/Cas9 generation of endoglin knockout zebrafish
In order to address Endoglin function in zebrafish, we used a Crispr/Cas9 approach based on a guide RNA overlapping with endoglin ATG located on exon 2 injected along with a Cas9 recombinant protein into wild-type embryos. To avoid potential deleterious effects of Endoglin inactivation such as observed in mouse, fish from 2 different injection setups (1-2 cell-stage (early) versus 4-16 cell-stage (late)), intended that late-stage injection would result in higher mosaicism and higher survival, were raised to adulthood. Analysis of germline presence of indels in sperm samples or in clutches resulting from mating with wild type fish detected indels in 18% (2/11) and 94% (32/34) of fish resulting respectively from early and late injections indicating that late-stage injection indeed circumvent lethality. Interestingly, numerous fish did not recover from anesthesia/sperm sampling procedure and massive bleeding from the gills was systematically observed in these fish. Retrospective analysis revealed that, by contrast with fish recovering normally from this rapid non-invasive sampling procedure, bleeders were actually indel carriers. We, however, managed to obtain a single clutch from one male which died on re-sampling confirmation procedure from which we derived the line analyzed in this study. This founder (F0) transmitted to 26.4% of the progeny (27/102) a unique indel consisting in a 2bp deletion and a base change of either C>A or T>A thus destroying endoglin translation initiation ATG codon and nucleotide −1 and −2 of the Kozak sequence (Figure 2a). Among F1, some fish later found to carry a mutated allele died at a juvenile stage (3-4 weeks) and some other showed overt redness later on but the vast majority developed normally and were indiscernible from wild-type siblings. Taken together these data suggested that endoglin mutation would be correlated with late development and/or survival issues in zebrafish.

Endoglin deficiency results in congestive heart failure in zebrafish. (a) Sanger sequencing chromatograms of gRNA targeted region on endoglin exon 2 in wild-type (eng+/+), heterozygous (eng+/-) and homozygous (eng-/-) endoglin mutants. Complementary gRNA sequence is indicated below chromatograms. (b) Imaging of 72hpf eng-/- and siblings reveals profound modification of blood flow pattern in eng-/-. Analysis was performed in Tg(kdrl:GFP), Tg(gata1:mRFP) transgenic background to highlight blood vessels and erythrocytes respectively. Pictures shown are representative of data obtained from siblings (n=10) and eng-/- (n=8). Bar, 500µm. (c) Kaplan-Meyer representation of the survival of eng-/-, eng+/-and eng+/+ siblings. +/+ vs -/- and +/- vs -/- P<0.0001, +/+ vs +/- ns P=0.7576 Log-rank (Mantel-Cox) Test. +/+ n=45, +/-=104, -/-=56. Red arrows point to individuals assigned to a subgroup of 8 sibling fish with overall redness and showing Surface Respiratory Behaviour and later identified to carry a single mutant allele. (d) Analysis of genotype influence over sex ratio in 3 months plus individuals. eng-/- population exhibits a stark bias towards male gender. Graph combines data from 2 complete independent experiments and two additional ones only focused on eng-/- fish. Total number of individuals analyzed per genotype is indicated above bars. (e) Representative morphology of 30-day-old eng-/- and siblings. Note enlarged cardiac area and overall paleness in eng-/- fish. Bar, 1 mm. (f) Hematoxyllin/Eosin staining of histological section of heart area reveals dramatic enlargement and structural alteration of the ventricule, hypochromic red blood cells and swollen surrounding tissue. Bar, 100 µm. (Abbreviations: a, atrium; ba, bulbus arteriosus; v ventricule).
Endoglin deficient zebrafish massively die from congestive heart failure
These observations along with Endoglin established role in HHT and midgestation lethality in homozygous knockout mice prompted us to analyze the effect of endoglin mutation in incrosses of heterozygotes fish. By 3dpf, we observed a strong modification of blood flow pattern in a dilated DA/CA–CV/PCV loop as well as a poor perfusion of ISVs (Figure 2b). Quantitative RT-PCR demonstrated that endoglin messenger abundance was reduced by 36.6% and 26.2% in eng-/- compared to wild-type and siblings, respectively, which could reflect nonsense mRNA decay induced by deleterious mutation. Altogether our results confirmed earlier report by Sugden et al. (2017) published during the course of this analysis and strongly suggested that the mutation we created also corresponds to a null allele. Therefore, homozygous mutants (eng-/-) were then systematically identified and separated from siblings at 3dpf. Siblings and eng-/- were raised along to adulthood with daily monitoring and dead fish were collected for detailed genotyping. Data processed in Kaplan-Meier survival plots showed a high mortality rate of eng-/- fish (Figure 2c). Indeed, while 63.5 % of eng+/+ and 59.6 % of eng+/- survived up to 5 months only 16.8 % of eng-/- managed to reach this age. Data indicated that eng-/- started to die massively by the age of 1 month-old and displayed a median survival of 44 days. Although a non-negligible fraction of eng-/- fish survived the 5 months considered period, survival figures underestimated the severity of the phenotype as over half (7/13) of eng-/- surviving fish never reached adulthood and stalled in a 11-17mm length range that precludes macroscopic gender determination. This is in stark contrast with siblings which completed growth and exhibited proper gender-associated traits. Surprisingly, eng-/- individuals reaching adulthood were found to be almost exclusively phenotypic males (Figure 2d). Because zebrafish sex is not assigned chromosomally, whether this reflects a genuine bias towards males or a higher sensitivity of females to Endoglin deficiency should be considered (see below). Macroscopic examination of eng-/- fish at 30dpf i.e. the very onset of lethality revealed that the vast majority of fish exhibited a crimson red enlarged cardiac area whereas the rest of the body appeared markedly paler than siblings (Figure 2e). We also noticed that, by contrast with siblings, eng-/- were hyperventilating and displayed a marked surface respiratory behavior (SRB). To get a better insight into the cardiac issue, we analyzed histological sections of the cardiac region stained with hematoxylin-eosin (Figure 2f). From these, the ventricule of eng-/- appeared dramatically oversized. By contrast with sibling whose red blood cells exhibited a characteristic deep orange color resulting from eosin reaction with hemoglobin, those in eng-/- did not present such a staining revealing a decrease in their hemoglobin content. Surrounding connective tissue also appeared loose indicating that edema, an accompanying symptom of heart dysfunction, was also taking place. Altogether this data demonstrated that Endoglin deficient fish develop congestive heart failure as well as anemia with lethal outcome.
A single endoglin mutant allele is sufficient to induce pathology
Based on macroscopic analysis, 3 month old or older eng-/- surviving fish fell into 3 classes of phenotype severity: 1) fish with arrested growth, hyperventilating and with recurrent enlarged cardiac area, 2) adult size fish hyperventilating with enlarged cardiac area to various degrees, 3) fish macroscopically asymptomatic (Supplementary figure 2). Interestingly, a fraction of heterozygotes was also found to be symptomatic i.e. hyperventilating with occasionally enlarged cardiac area (Supplementary figure 1). Symptomatic siblings later assigned as heterozygous by genotyping were observed around one month old and raised apart to monitor the evolution of phenotypes and life expectancy. Collective data from two independent experiments indicated that the frequency of symptomatic eng+/- averaged 14% ± 3% of total eng+/-(16/114). In addition to hyperventilation and SRB, their most striking feature was their marked ruddy complexion which persisted as they completed growth to adulthood as well as the occasional presence of dilated surface vessel reminiscent of telangiectasias (Supplementary figure 3). Although the number of such animals was too low to significantly affect heterozygotes overall survival rate, we did observe a higher frequency of death events in this group (Figure 2c, arrowheads) resulting in an estimated 5-month survival of 37.5% (6/16). Thus, similar to HHT1 mouse model, a single mutated endoglin allele results in pathological conditions with limited penetrance and are in line with the “second hit” concept, in zebrafish as well.
Early detection of concomitant hypoxic and cardiac stress responses in endoglin deficient zebrafish
Hypoxia being a potent driver of cardiac remodeling involving cardiomyocyte hypertrophy in mammals and proliferation as well as hypertrophy in zebrafish27–29, we reasoned that it might trigger heart failure in our model. Supporting this hypothesis, hyperventilation and surface respiratory behavior appeared as characteristic manifestations in eng-/-. Moreover, gender bias towards males in eng-/- was also interpreted as a sign of hypoxia which was found to impair sex differentiation through an imbalance of testosterone/estradiol ratio under the control of cytochrome P450 aromatase (CYP19) via a mechanism that likely involves HIF-dependent transcriptional repression of ERRγ30–32. First, we monitored the expression of cardiac stress markers, i.e. atrial-and brain-type natriuretic peptides nppa and nppb respectively, in order to define when the heart of eng-/- start being at stake. Using RT-qPCR, we found that by 15dpf, when compared to siblings, eng-/- exhibit a 1.8 and a 3.3 fold induction of nppa and nppb respectively. These differences in expression between eng-/- and siblings then gradually expand to reach up to 49 and 223 fold change of nppa and nppb transcript abundance respectively, consistent with the gradual enlargement of eng-/- heart region over time (Figure 3a). We, thus, analyzed the expression of well accepted hypoxia-responsive genes egln3 (prolyl hydroxylase 3) and epoa (erythropoietin) in the very same samples. Again, using RT-qPCR, we found a 2.1 fold increased expression for egln3 and 1.5 for epoa by 15dpf in eng-/- fish when compared to siblings. Hypoxia responsive gene expression steadily rises to 6.8 and 14.9 fold increase of egln3 and epoa respectively in eng-/- in regard to siblings by 30dpf (Figure 3b). Since functional Hypoxia Response Elements (HREs) have been identified in the promoters of nppa and nppb genes whose expression can be induced by hypoxia33, we verified that early difference in nppa and nppb would also reflect heart response to increase workload. We thus measured and compared the ventricule volume of siblings and eng-/- fish at 10, 12 and 15dpf. We found that while this volume is similar between eng-/- and siblings at both 10 and 12dpf, it was significantly increased in mutants by 15dpf (Figure 3c). Although similar to stress markers in its trend, hypoxia response appeared far more limited in its extent thus strongly suggesting that hypoxia indeed triggers cardiomegaly in our model. Also supporting this hypothesis is the fact that in symptomatic heterozygotes cardiac region enlargement is macroscopically less frequent or severe. Finally, to define how the heart would specifically respond to hypoxic cues in Endoglin deficient fish, we performed FACS analysis to monitor relative changes in cardiomyocyte abundance. Analysis of cmlc2:GFPpos cardiomyocytes in cell suspension from whole fish revealed a recurrent increase of GFPpos/total cell ratio in 15dpf and older eng-/- indicating enhanced cardiomyocyte proliferation (Figure 3d) consistent with direct proliferative effect of hypoxia on cardiomyocytes29.

Cardiac stress, ventricule enlargement and cardiomyocyte proliferation correlate with hypoxia in Endoglin deficient fish (a) Kinetic RT-qPCR analysis of nppa and nppb (cardiac stress responsive gene), (b) egln3, epoa (hypoxia responsive gene), in 5, 10, 15, 20, 25 and 30 day-old sibling and eng-/- fish (genotype analysis using allele specific RT-PCR is presented underneath graphs). Hypoxia and cardiac stress are concomitantly detected in 15-day-old mutants. Gene of interest expression values were normalized to RPL13a. 5dpf=15 fish, 10dpf=15 fish, 15dpf=8 fish, 20dpf=5fish, 25dpf=4 fish, 30dpf=3 fish. Statistical analysis performed using ANOVA (nppa 15dpf P=0,00102395, 20dpf P=3,3852E-05, 25dpf P=1,2295E-05, 30dpf P=9,7454E-08), (nppb 15dpf P=6,2759E-06, 20dpf P=1,441E-06, 25dpf P=2,9037E-06, 30dpf P=5,8624E-06), (egln3 15dpf P=0,00141317, 20dpf P=0,00011036, 25dpf P=0,00017342,30dpf P=9,4665E-07), (epoa 15dpf P=0,02376684, 20dpf P=0,00113505, 25dpf P=0,00225646, 30dpf P=2,1172E-06), [Symbol meaning, ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001]. (c) Analysis of ventricule volume in 10, 12 and 15-day-old siblings and eng-/- fish in Tg(cmlc2:GFP) background. Volume is estimated by the simplified ellipsoid volume calculation formula V=0.523(width in mm)2(length in mm)81. Statistical analysis one-tailed unpaired t test. * P=0.0183. (d) Representative Flow cytometry analysis of cardiomyocytes (cmlc2:GFPpos) representation in whole organism cell suspension from 5, 10, 15, 20 ad 25dpf fish. Cell suspensions at 5, 10, 15, 20 and 25dpf were prepared from pool of 26, 18, 9, 7 and 7 sibling or eng-/- fish, respectively. Bottom graph represents fold change in CMLC2:GFPpos cells in eng-/- relative to siblings at the same stage.
Hypochromic Anemia is not the primary trigger of heart failure in Endoglin deficient fish
As anemic mutant fish such as riesling34, merlot/chablis35 or retsina36 develop cardiomegaly as the result of cardiac compensation for oxygen transport, we sought to define whether hypochromic anemia observed in eng-/- fish could account for hypoxia detected as early as 15pdf in these fish. We thus performed a series of o-dianisidine staining in a time course that spaned over the onset of hypoxia and cardiac stress i.e 3, 5, 10 and 15dpf, to evaluate hemoglobin content. As shown in Figure 4, no dramatic drop in staining intensity could be observed between eng-/- and siblings groups at any given time point with, inversely, a trend to enhanced stainings in eng-/- group at 15dpf which would likely reflect increased epoa levels at this time point. This results indicated that anemia is not the primary cause of hypoxia but a late secondary acquired feature of Endoglin deficiency that will undoubtedly enhance hypoxia.

Anemia is not responsible for hypoxia in Endoglin deficient larvae. Hemoglobin assessment in eng-/- and siblings does not reveal early anemia. Representative Whole-mount o-dianisidine staining of 3, 5, 10 and 15dpf eng-/- and siblings. Numbers in upper right part of pictures indicate the number of fish with similar pattern out of total number analyzed. Bar, 500µm.
Endoglin deficient fish exhibits defects in gill blood vessel development
Since in eng-/- fish increased epoa levels and cardiac compensation fails to correct hypoxia, we hypothesized that chronic hypoxic condition might reflect respiratory system issues in eng-/- fish. This hypothesis was strongly supported by systematic bleedings from the gills observed in F0 indel carriers during sperm sampling. We thus compared gill architecture between sibling and eng-/-. Analysis of histological sections from 30dpf fish revealed that eng-/- gills were structurally abnormal. Lamellae were unusually short and crooked and supplying blood vessels were markedly enlarged in particular in the fourth branchial arch (AA6) (Figure 5a). We thus compared sibling and eng-/- gill vascular architecture at 10, 12 and 15dpf i.e. before and at the early onset of hypoxia in Tg(kdrl:GFP), Tg(flt-1:Tomato) transgenic background (Figure 5b). Although gill vasculature is an arterio-arterial system37, we found that Flt-1 arterial marker was mainly if not exclusively associated with afferent vascular branches, whereas kdrl promoter appeared evenly active in both afferent and efferent parts of the gill vascular system. As early as 10dpf, differences could already be noticed: vascular loops (afferent/efferent) are shorter in mutants (arrowheads) and the efferent artery of the fourth branchial arch is substantially dilated (asterisk). This differences persisted and exacerbated later on and by 15dpf while pan-vascular endothelium kdrl reporter expression remained essentially unchanged in its extent, we observed a dramatic loss of Flt1 reporter activity in the afferent arterial gill vascular system suggesting that arterial identity might be affected. Finally, to ascertain Endoglin direct role in gill formation, we analyzed Endoglin expression in this tissue by whole-mount in situ hybridization on wild-type fish. Endoglin messenger was indeed detected in developing gills of 10dpf larvae although its expression appeared rather faint, it followed branchial archs pattern and was also detected in the paired anterior dorsal aortas. Expression strengthened and spread out by 12 and 15dpf indicating that Endoglin expression was not restricted to major arteries supplying and collecting blood but also in filamental arteries and lamellae (supplementary figure 4a and 4b). Notably, Endoglin expression appeared absent in heart at these stages indicating that heart failure in eng-/- fish arise from extrinsic issues. These data showed that, in eng-/- fish, important alterations of gill vasculature worsening with time take place ahead of hypoxia onset thus strongly suggesting that defective gill function results in hypoxemia leading to tissue hypoxia.
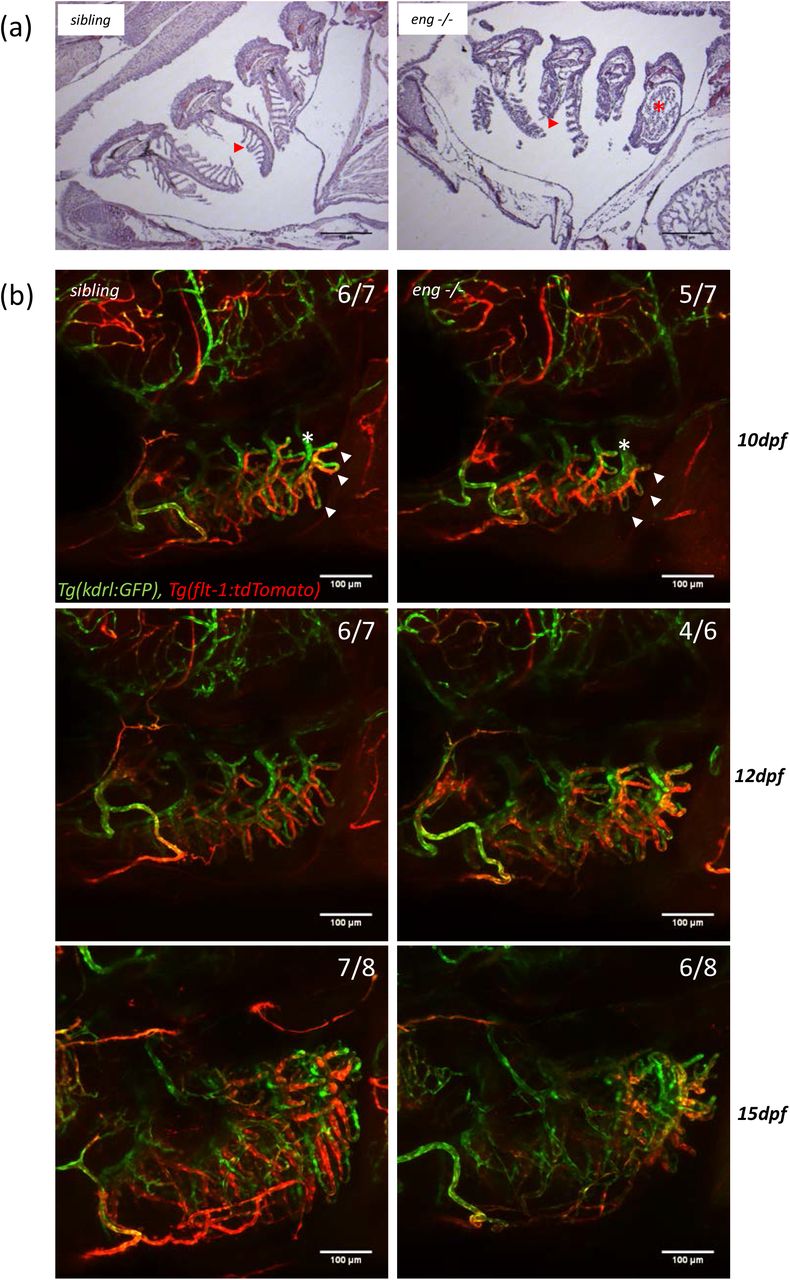
Abnormal branchial vascular development in Endoglin deficient fish underlies structural impairment of gills. (a) Representative histological sections of 30-day-old sibling and eng-/- fish gills stained with hematoxylin/eosin. Note poorly developed and organized lamellae (arrowhead) and dramatically enlarged artery (asterisk) on branchial arch 4 (AA6) in eng-/- fish gills compared to siblings. Bar, 100 µm. (b) Kinetic analysis of gill vascular development between 10, 12 and 15-day-old sibling and eng-/- zebrafish in Tg(kdrl:GFP, flt1:tdtomato) background. Note reduced length of AFA (Afferent Filamental Artery) (GFPpos, dtTomatoHigh) and EFA (Efferent Filamental Artery) (GFPpos, dtTomatolow/neg) (arrowheads) and enlarged EBA (Efferent Branchial artery) (asterisk) as early as 10dpf. Note overall loss of Flt-1 reporter signal in 15dpf eng-/- fish. Bar, 100 µm.
Phenylhydrazine induced anemia in wild-type fish fails to mimic Endoglin deficiency related heart failure
To define how hypoxia specifically contribute to heart failure that gradually sets in eng-/- fish, we induced anemia in wild-type fish through the use of phenylhydrazine, a versatile model of hypoxia, that should allow us to trigger and maintain hypoxic conditions over time through repeated treatment. To closely match the onset of hypoxia observed in eng-/- fish, treatment was applied every other day from 14dpf with 3 different phenylhydrazine (phz) concentrations. This regimen was provided up to 75dpf then fish were allowed to recover. First, we assessed hypoxic and cardiac stress response in one month-old fish. Surprisingly, while hypoxia markers indeed gradually increased with phz concentration and seem to plateau at 5µg/ml phz concentration (Figure 6a), only phz harshest conditions led to a notable increase in cardiac stress markers expression (Figure 6b), suggesting that nppa and nppb would be at best weak hypoxia responsive genes in zebrafish. In one month-old wild-type fish treated with 5µg/ml phz, cardiac stress extent reached a 2.33 ±0.16 and 6.51 ±0.84 fold activation for nppa and nppb respectively while hypoxic response was induced by 3.14 ±0.1 and 2.82 ±0.2 folds for egln3 and epoa respectively. Thus, while hypoxic response in 1 month-old phz treated and eng-/- seemed roughly in the same range of magnitude, cardiac stress in phz treated fish appeared overtly marginal (see Figure 3a, 3b and 7a for comparison). Macroscopically, we observed a strict correlation between the extent of heart region enlargement and cardiac stress markers levels but both appeared rather limited compared to eng-/- at the same age (Figure 6a). Consistent with these mild features, phz treatment had no significant effect on survival at this point. Prolonged treatment eventually affected survival in a dose-dependent manner but still stayed marginal (Figure 6c). Decline in survival set in by about 2 month of age that is ∼45 days after the start of phz treatment which happened to be significantly later than in eng-/-. Furthermore, analysis of sex-ratio at 3 month, after one month phz free for recovery, did not reveal male gender bias one would have indeed expected from a hypoxia-related effect (Figure 6d). Collectively, these results show that although chronic hypoxic conditions can be achieved efficiently through the use of phz, chronic hypoxia per se does not result in cardiac issue reminiscent of that observed Endoglin deficient fish.

Phenylhydrazine-induced hypoxia fails to mimic Endoglin deficiency heart failure condition. (a) Representative pictures of one-month-old wild-type fish non-treated (ctrl) or treated with 5µg/ml phenylhydrazine (phz). Note overt paleness of gills area indicative of anemia (asterisk) and slightly dilated heart region indicative of limited cardiomegaly (arrowhead). Bar, 1mm. (b) RT-qPCR analysis of egln3, epoa (hypoxia responsive gene) and nppa and nppb (cardiac stress responsive gene) expression in one-month-old wild-type fish non-treated (ctrl) or treated with 1.25, 2.5 and 5µg/ml phenylhydrazine. Relative quantities of target genes are expressed as ΔΔCT using rpl13a as reference. Samples are pools of 5 fish. One-tailed unpaired t test was used for statistical analysis: egln3 ctrl vs phz1.25 P= 0.0002, ctrl vs phz2.5 or phz5 P< 0.0001, phz1.25 vs phz2.5 P= 0.0027, phz1.25 vs ph5 P< 0.0001 and phz2.5 vs phz5 P= 0.0814; epoa ctrl vs phz1.25 P= 0.0171, ctrl vs phz2.5 P= 0.0003, ctrl vs phz5 P< 0.0001, phz1.25 vs phz2.5 P= 0.0023, phz1.25 vs ph5 P= 0.0002 and phz2.5 vs phz5 P= 0.1629; nppa ctrl vs phz1.25 P= 0.1151, ctrl vs phz2.5 P= 0.0147, ctrl vs phz5 P< 0.0001, phz1.25 vs phz2.5 P= 0.0506, phz1.25 vs ph5 P= 0.0007 and phz2.5 vs phz5 P= 0.2041; nppb ctrl vs phz1.25 P= 0.2358, ctrl vs phz2.5 P= 0.0080, ctrl vs phz5 P< 0.0001, phz1.25 vs phz2.5 P= 0.0389, phz1.25 vs ph5 P< 0.0001 and phz2.5 vs phz5 P= 0.0013 (c) Analysis of phenylhydrazine treatment effect over fish survival. Kaplan-Meyer representation of the survival of non-treated (NT) and 1.25, 2.5 and 5µg/ml phz treated wild-type fish. Note the dose dependent effect of phz concentration over survival but absence of deleterious effect at early juvenile stage. NT vs phz1.25 P=0.0114, NT vs phz2.5 P=0.0051, NT vs phz5 P<0.0001, phz1.25 vs phz5 P=0.0113, phz2.5 vs phz5 P=0.0310 and phz1.25 vs phz2.5 P=0.71219 (ns) Log-rank (Mantel-Cox) Test. (d) Analysis of phz treatment influence over sex ratio in 3-month-old individuals. Note absence of gender bias at any phenylhydrazine concentration.
Hypoxia-induced erythropoiesis is detrimental to endoglin deficient fish
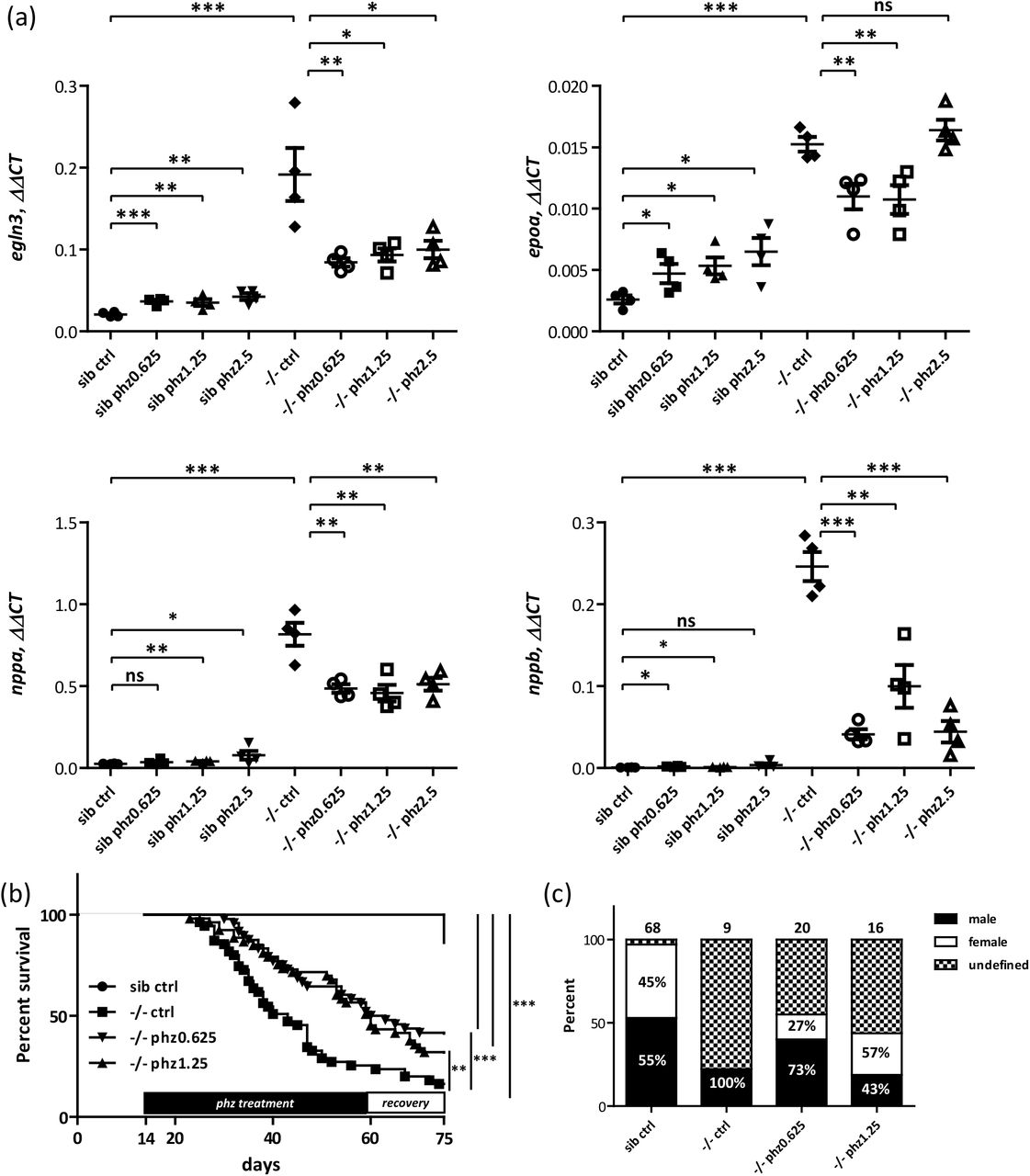
Since phz induced chronic hypoxia failed to mimic Endoglin related heart failure, we wondered whether, hematocrit/blood viscosity that should be increased in endoglin mutant as the result of both enhanced erythropoieisis induced by high epoa levels and natriuresis (hemoconcentration), could contribute to these discrepancies. Supporting this hypothesis, 1) histological sections of 30dpf eng-/- kidney, the definitive hematopoïesis organ in fish, revealed dramatically increased cellularity when compared to siblings, indicative of reactive erythropoiesis, 2) blood smears from 25 and 30dpf eng-/- reveal high content in erythrocytes, most with immature shape and abnormal staining when compared to siblings and 3) direct measurement in adult eng-/-surviving fish revealed increased hematocrit when compared to wild-type adult fish (Supplementary figure 5a, 5b and 5c respectively). To modulate hematocrit/blood viscosity parameter, we treated eng-/- and siblings with phenylhydrazine at concentrations that showed only minimal effect on hypoxia and cardiac stress in wild-type fish following the same timetable as for wild-types (Figure 7a). Consistent with results on wild-type fish, these low phz concentrations showed no overt macrocopical effect on siblings compared to their untreated counterparts. One-month-old Phz treated eng-/-, by constrast, appeared overall healthier with less prominent cardiomegaly (not shown). We used, again, qPCR to measure both hypoxia and cardiac stress in these different settings. Similar to results presented in Figure 3a and 3b, eng-/- group exhibited a mean 9.3 fold egln3, 5.9 fold epoa, 31.4 fold nppa and 344 fold nppb, increase over siblings group. On siblings, phz, regardless its concentration, induces modest increases in egln3 (1.7, 1.7 and 2.0 fold increase over non-treated siblings for fish treated with phz at 0.625, 1.25 and 2.5µg respectively), epoa (1.8, 2.0 and 2.5 fold increase over non-reated siblings for fish treated with phz at 0.625, 1.25 and 2.5µg respectively), nppa (1.4, 1.5 and 3.0 fold increase over non-treated siblings for fish treated with phz at 0.625, 1.25 and 2.5µg respectively) and nppb ((2.5, 1.7 and 5.1 fold increase over non-treated siblings for fish treated with phz at 0.625, 1.25 and 2.5µg respectively) expression levels. In stark contrast, on mutants, phz reduces nppa (mean 18.7, 17.6 and 19.7 fold increase over non-treated siblings group for mutant fish treated with phz at 0.625, 1.25 and 2.5µg/ml respectively) and nppb (mean 57.8, 139.6 and 61.9 fold increase over non-treated siblings group for mutant fish treated with phz at 0.625, 1.25 and 2.5µg/ml respectively) induction levels. These data strongly supported our working hypothesis that hematocrit/ blood viscosity would be an important determinant of cardiac stress response. Surprisingly, though, we observed an accompanying reduction of hypoxic response markers egln3 (mean 4.1, 4.5 and 4.8 fold increase over non-treated siblings group for mutant fish treated with phz at 0.625, 1.25 and 2.5µg/ml respectively) epoa (mean 4.2, 4.1 and 6.3 fold increase over non-treated siblings group for mutant fish treated with phz at 0.625, 1.25 and 2.5µg/ml respectively) expression indicating that heart failure would worsen oxygenation. To define whether phz treatment would provide long-term benefit on eng-/- fish health, we evaluated its effect in survival experiments. eng-/- fish were treated with either 0.625µg/ml or 1.25µg/ml phenylhydrazine every other day from 14dpf onward then allowed to recover from 60dpf to 75dpf. As in Figure 2c, survival of eng-/- is severely impaired compared to siblings with a 16.4% survival at 75dpf (Median survival=43 days), while phz treated eng-/- groups exhibited markedly enhanced survivals with 41.7% for phz0.625 group and 32.1% for phz1.25 group (Median survival of phz0.625 and phz1.25 group equal 61.5 and 60 days, respectively) (Figure 7b). Further highlighting phz relieving effect over Endoglin deficiency associated symptoms, phz treatments allowed higher fractions to reach adulthood and corrected gender bias toward male (Figure 7c). These results show that, in eng-/- fish, hypoxia response mechanism fails to work as compensation mechanism but brings the pathology to a higher degree.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phenylhydrazine treatment efficiently alleviates pathological conditions induced by Endoglin deficiency in zebrafish. (a) RT-qPCR analysis of egln3, epoa (hypoxia responsive gene) and nppa and nppb (cardiac stress responsive gene) expression in one-month-old siblings (sib) versus eng-/ fish untreated (ctrl) or treated with 0.625, 1.25 and 2.5µg/ml phenylhydrazine (phz). Relative quantities of target genes are expressed as ΔΔCT using rpl13a as reference. Samples are pools of 4 to 5 fish. The correct genotype of samples was verified beforehand by RT–PCR (not shown). One-tailed unpaired t test was used for statistical analysis: egln3: sib ctrl vs -/- ctrl P= 0.0009, -/- ctrl vs -/- phz0.625 P= 0.0084, -/- ctrl vs -/- phz1.25 P= 0.0127, -/- ctrl vs -/- phz2.5 P= 0.0179; epoa: sib ctrl vs -/- ctrl P< 0.0001, -/- ctrl vs -/- phz0.625 P= 0.0060, -/- ctrl vs -/- phz1.25 P= 0.0068, -/- ctrl vs -/- phz2.5 P= 0.1530; nppa: sib ctrl vs -/- ctrl P< 0.0001, -/- ctrl vs -/- phz0.625 P= 0.0022, -/- ctrl vs -/- phz1.25 P= 0.0030, -/- ctrl vs -/- phz2.5 P= 0.0045; nppb: sib ctrl vs -/- ctrl P< 0.0001, -/- ctrl vs -/- phz0.625 P< 0.0001, -/- ctrl vs -/- phz1.25 P= 0.0018, -/- ctrl vs -/- phz2.5 P< 0.0001. (b) Phenylhydrazine treatment enhances Endoglin deficient fish survival. Kaplan-Meyer representation of the survival of siblings and eng-/- non-treated (ctrl) or treated with phenylhydrazine at 0.625 or 1.25 µg/ml. siblings vs -/- ctrl or -/-phz1.25 or -/- phz0.625 P<0.0001, -/- vs -/- phz1.25 P=0.0039, and -/- vs -/-phz0.625 P=0.0009 Log-rank (Mantel-Cox) Test. siblings n=66, -/- n=55, -/-phz1.25 n=53 and -/-phz0.625 n=48. (c) Analysis of phenylhydrazine treatment influence over sex ratio in 2.5 months plus individuals. Phenylhydrazine treatment increase the fraction of fish reaching adulthood and correct gender bias towards male associated with Endoglin deficiency in fish. Numbers above bars represent the numbers of surviving fish analyzed. Percent of males and females are indicated inside bars, note that fish of undefined gender have been excluded from calculation.
Altogether our results demonstrated that Endoglin deficiency in zebrafish induces heart failure through both direct and indirect effects of chronic hypoxia which stems from gill dysfunction. Hypoxia is certainly reinforced by hypochromic anemia and massive hemorrhages from the intestinal tract (Supplementary figure 6).
Discussion
In the present work, using a CRISPR/Cas9 approach, we engineered an endoglin mutant zebrafish line and analyzed the functional consequences of Endoglin deficiency in post-embryonic stage zebrafish. We found that mutant fish massively die from congestive heart failure induced by chronic hypoxia/hypoxemia resulting from gill dysfunction. We also found evidence of anemia and observed intestinal hemorrhages that surely further worsen the cardiac pathology. Finally, we found that health and survival could be strongly enhanced by regular treatment with hemolysis inducing agent, phenylhydrazine demonstrating that increased hematocrit/ blood viscosity induced by hypoxia is the main driver of heart failure in Endoglin deficient fish.
From our molecular work on zebrafish endoglin locus characterization, we demonstrated that two distinct endoglin transcripts are expressed via the use of an alternate exon which introduces an early stop of traduction leading to two Endoglin proteins with unique cytosolic sequences. Of much interest, similar isoforms are expressed in mammals26. However, in the latter, Endoglin short isoform is produced by an intron retention mechanism involving Splicing Factor 2 (ASF/SF2)38. Zebrafish endoglin overall similarity with mammal orthologs is fairly low but the cytosolic region of the long isoform is remarkably homologous to that of mammals which interacts with proteins such as GIPC, β-arrestin2 and NOS339–41. Such homology, by contrast, is absent in the short isoform and suggests that Endoglin short isoform cytosolic domain has no protein binding function. This assertion is further supported by the wide diversity of sequence of short Endoglin cytosolic domain which fluctuates in mammals according to the position or the absence of translation stop in retained intron38. Conservation despite evolutionary distance underscores that both Endoglin isoforms should be essential although Endoglin short isoform seems absent in reptiles and birds. Still, very little is known about the role of Endoglin short isoform. In mammals, it was shown to be induced in senescent endothelial cells and EC-specific Endoglin short isoform overexpressing mice were found hypertensive and insensitive to NO synthesis inhibitor L-NAME42. Endoglin also exists as a soluble form (S-Endoglin) resulting from the cleavage of ectodomain by MMPs such that it can exert paracrine or remote activity using for instance exosomes as vehicle43. High S-Endoglin levels correlates with preeclampsia and induce hypertension by inhibiting TGF-β induction of NOS dependent vasodilatation44. This suggests that Endoglin lacking cytosolic domain might prevent normal NOS3 activation in response to TGF-β. Surprisingly, a recent report tends to exclude S-endoglin working as a ligand-trap towards BMP945. Further work will be necessary to establish whether S-Endoglin is actually produced in zebrafish but sequence analysis indicates that the ectodomain Glu-Leu residues, located in the close vicinity of the transmembrane domain and essential to MMP14 cleavage in mammals, are conserved46.
In zebrafish, by contrast with mammals, Endoglin participation in ALK1 signaling pathway seems highly questionable. Besides a marginal overlap between expression patterns during development, striking differences in lethality timing between ALK1 and Endoglin mutants (7-10 vs 30dpf, respectively) rather argue against a function on a shared pathway47. More importantly, ALK1 mutants exhibit enlarged cranial blood vessels due to impaired endothelial cell ability to migrate against blood flow47–49. These defects are phenocopied by embryos injected with morpholinos targeting either ALK1 or both bmp10 and bmp10-like demonstrating that BMP10s signal through ALK1 to control cranial blood vessel caliber50. This phenotype is, by contrast, overtly absent in Endoglin mutants and morphants (not shown) indicating that Endoglin is dispensable for BMP10-ALK1 signaling in this respect. Conversely, our Endoglin mutant exhibits the very same phenotype as one reported earlier from an independently produced mutant line (i.e. accelerated blood flow in a simple DA/CV vascular loop) which is not mentioned for ALK1 deficient zebrafish embryos indicating that Endoglin association with ALK1 pathway is not restricted to a specific vascular bed either. ALK5 seems neither an alternate candidate receptor as both alk5a and alk5b are overtly absent in axial blood vessels by the time Endoglin deficiency induces changes in flow pattern51. Besides, in mammals, although ALK5 is co-expressed with ALK1 in endothelial cells in vitro, evidences are accumulating that this would not be true in vivo52. Recent data, in mammals, suggest that Endoglin could act outside of TGFβ/BMP signaling pathways. Endoglin was indeed found to interact with VEGF-R2 and promoted its signaling in response to VEGF53.
Further investigation will definitely be required to define whether, in zebrafish, phenotypes induced by Endoglin deficiency could be related to this specific signaling. This, however, does not rule out that ALK1-Endoglin interaction might be required at later stages. Unfortunately, early lethality of zebrafish ALK1 mutants precludes analysis of its function in gill vasculature proper development and maintenance that will be necessary to definitely reach a conclusion on this matter. It is nonetheless interesting to note that ALK1 conditional knockout in adult mice results in high output heart failure and anemia54. In line with these results, EC-specific endoglin knock-out in adult mice, also results in high-output heart failure induced by AVM formation in the pubic symphysis. The high VEGF levels expressed in this fibrocartilaginous structure stimulate EC proliferation through an exacerbated VEGF-R2 receptor signaling in Endoglin deficient condition55. Thus, provided technical issues regarding both conditional and tissue-specific expression could be overcome, it will be very interesting to assess whether constitutively active ALK1 or VEGF-R1 (Flt-1) would rescue heart issue in Endoglin deficient zebrafish.
Our results demonstrate that in zebrafish endoglin deficiency elicits a chronic hypoxic response. In normoxia, HIFs (HIF-1a/HIF-2a) are hydroxylated on specific proline located in their Oxygen-dependent Degradation Domain (ODD) by prolyl hydroxylases (PHDs) enzymes which use O2, 2-oxoglutarate, ascorbate and ferrous iron ions (Fe2+) as cofactors. Hydroxylated HIFs are recognized by the Von Hippel Lindau protein (pVHL) component of E3 ubiqutin ligase complex leading to HIFs polyubiquitylation and ultimately HIFs degradation by the proteasome. Conversely, hypoxia hampers PHD activity leading to HIFs stabilization, nuclear translocation and transcription factor complex formation with HIF-1β/ARNT and regulation of specific sets of genes through the binding of specific DNA elements (HRE)56. In addition to hypoxia, this pathway is activated by physio-pathological conditions such anemia and iron deficiency which are all intimately connected57.
Iron deficiency leads eventually to anemia due to the essential role of iron in the formation of haem complex, an obligatory cofactor of hemoglobin in the transport of oxygen. Iron depleted diet was found to repress iron reabsorption inhibitor Hepcidin hepatic expression by a direct HIF-dependent transcriptional repression mechanism in response to HIF stabilization under such a regimen58. In keeping with this, long used hypoxia mimetics desferrioxamine and cobalt or nickel chloride all inhibit Prolyl hydroxylase (PHD) by interfering with ferrous iron59.
Endoglin deficient fish exhibit features highly reminiscent of vhl zebrafish mutants which models Cuvash syndrome60. These mutations abolish vhl ability to associate with HIF factors and lead to the systemic constitutive activation of HIF-dependent pathways in normoxic conditions. Vhl mutant fish exhibit exacerbated erythropoiesis in response to Epo overexpression which results in a form of polycythemia with immature and hypochromic characteristics. Mutant fish also exhibits a hyperventilation phenotype which stems from the direct action of Epo on respiratory neurons and develop congestive heart failure, edema and eventually die by 11dpf60,61.
In mouse, liver-specific deletion of VHL results in HIF-mediated Hepcidin repression and Epo induction leading to excessive (polycythemia) microcytic and hypochromic erythropoiesis. Thus, despite converging mechanisms set to mobilize iron and red blood cell production, conditions of chronic HIF stabilization seem to end up invariably in an imbalance between Epo and iron levels58.
Chronic hypoxic response induces cardiomegaly in both fish and mammals although the cellular mechanisms, that leads to increased cardiac muscle mass as an adaptive strategy to cope with increased blood demand, appears highly divergent. Unlike mammals where cardiomyocytes undergo cell size increment (hypertrophy), fish cardiomyocytes are able to re-enter cell cycle after dedifferentiation62. Consistent with this, our data, although quite indirectly, show that cardiomyocyte proliferation is enhanced in Endoglin deficient fish in a timetable that matches hypoxia. The links between hypoxia and cardiomyocyte proliferation are still poorly understood but recent reports have shed light as to how HIF activation induces cardiomegaly through cardiomyocyte hypertrophy and cardiac remodeling. Hence, Hypoxia Inducible Mitogen Factor (HIMF), a non-canonical ligand of Calcium Sensing Receptor (CaSR) was found to control in vitro and in vivo cardiomyocyte hypertrophy by activating HIF, CaN-NFAT and MAPKs pathways63,64. Interestingly, cardiac stress hypertrophic markers, β-MyHC and atrial natriuretic peptide nppa were found regulated by HIF-1α under HIMF control. HIF activity would also preserve cardiac function by limiting cardiac remodeling resulting from iron deficiency induced anemia through a non-hematopoietic Epo-EpoR signaling axis65. Published data suggest that in zebrafish both hematopoietic and non-hematopoietic Epo-EpoR signaling are remarkably conserved66. By contrast, HIMF potential orthologs while present in the genome of some fish i.e. Erpetoichthys calabaricus, are, to date, absent in zebrafish databanks but this will definitely require specific experimental work before this factor could be removed from the equation.
The differences in the onset of hypoxic response between vhl and endoglin mutants, suggest that Endoglin does not directly tamper with the molecular mechanisms that control HIF activation. Moreover, Endoglin deficient fish anemia is not causative of hypoxia but most likely the consequence of chronic HIF activation and the result of uncontrolled Epo-induced erythropoiesis and our data strongly argue in favor of a faulty respiratory system. Fish extract water dissolved oxygen through their gills, a highly complex respiratory organ composed of multiple functional units called lamellae. Bridging afferent and efferent circulation, lamellae are thin flat vascular sinusoids of endothelial and non– endothelial pillar cells ensheathed by a simple layer of pavement epithelial cells37. In zebrafish, gills lamellae were found to form around 12-14dpf to reach their definitive adult morphology by about 4 weeks67. They develop on a scaffold of vascular loops composed of afferent and efferent parts linking ventral aorta to lateral and dorsal aorta through branchial arches. Since structural and molecular alterations of the vascular network of the gill are observed in Endoglin deficient zebrafish before or at the very onset of lamellae formation, it is reasonable to conceive that these defects would directly hamper lamellae proper development and function of this water-blood interface. Our data hence indicate that hypoxia stems from gill dysfunction that will result in hypoxemia leading to poor tissue oxygenation triggering hypoxic response. It is, thus, not surprising that mutant fish will start to decline by 30dpf which corresponds to the time when gill structure reaches its fully operative shape in normal zebrafish and that variability in gill dysfunction severity allows fish to survive longer periods, not excluding, though, that blood vessel defects affecting other organs resulting in inappropriate perfusion would reinforce hypoxia.
The specific loss of zebrafish arterial marker Flt-1, here found specifically associated with gill afferent blood vessels, is difficult to explain given how little knowledge regarding transcriptional regulation of this promoter is available in zebrafish, especially for stages past embryonic development for which Tg(−0.8flt1:tdTomato) artery-restricted activity appears as organ-dependent68. Other markers will definitely be required to define whether Flt-1 reporter loss reflects a genuine change in endothelial cell identity. However, since Endoglin has been shown to be required for a proper response to haemodynamic cues, we could legitimately hypothesize that this might reflects high blood flow strains associated with the afferent circulation21,25.
Treating Endoglin deficient fish with hemolysis inducer phenylhydrazine according to a timetable that matches hypoxia and cardiac stress has a strong impact on fish health. Enhanced survival clearly demonstrates that it protects from heart failure. This beneficial effect of phenylhydrazine, a drug historically used to cure polycythemia vera69, suggests that a form a polycythemia is taking place in Endoglin deficient fish that puts the heart at stake. Interestingly, in HHT, polycythemia is a complication of pulmonary AVM which, by mixing arterial and venous blood (right-to-left shunting), results in hypoxemia eventually triggering erythropoiesis (for review70,71). Thus, despite structural difference between fish and human cardiorespiratory systems, Endoglin deficiency appears to results in similar pathological situations.
Based on zootechnical considerations, zebrafish has become a choice organism to perform screens. Hence, by reproducing important features of HHT, both heterozygous and homozygous Endoglin mutant fish will give us the opportunity to address the 2nd hit issue by screening for drugs able to induce symptoms at a higher frequency, while on the other hand homozygotes will serve to identify molecules able to rescue both embryonic and post-embryonic phenotypes.
Experimental procedures
Zebrafish lines and maintenance
Wild type AB zebrafish (Danio rerio), Tg(myl7:EGFP) here referred to as Tg(cmlc2:EGFP), Tg(−0.8flt1:RFP) here referred to as Tg(flt1:Tomato), Tg(kdrl:GFP), Tg(−8.1gata1a:mRFP) here referred to as gata1:RFP were raised and maintained in housing systems (Aquatic Habitat and Zebtec Techniplast). Embryos were raised in housing systems from 3dpf onward. Embryos were fed several times a day from 5dpf with dry food (Gemma Micro, Skretting) supplemented with brine shrimp (Artemia salina) nauplii by 9dpf. Regimen usually allow fish to reach adulthood by 8-10 weeks. Embryos were staged according to kimmel et al72 and blood vessels nomenclature refers to Isogai et al73. All experiments were performed in accordance with the 2010/63/EU Directive and the ARRIVE guidelines74. Procedures used in this study have been evaluated and approved by Ethical Committee (#036) as part of an authorized project registered by the French Ministère de l’Enseignement Supérieur de la Recherche et de l’Innovation under APAFIS number #23822-2020052717421507 v3 to be conducted under E. Lelièvre responsibility. Animals were housed in a zebrafish facility registered under Agreement number #A3417237.
Crispr/Cas9 generation of endoglin mutant
Guide RNA was designed using TEFOR’s CRISPR/Cas9 Assistant CRISPOR using zebrafish endoglin Exon 2 as template. Retained gRNA was selected among candidates for its position relative to Eng first codon and absence of predicted off-targets nearby a PAM sequence. See Figure 2a for gRNA sequence. Briefly, 1 nl of a mixture composed of endoglin targeting gRNA S. pyCas9-3NLS recombinant protein in 20mM HEPES ph7.5, KCl 150mM was injected into 4-to 16-cell-stage wild type AB embryos. Cas9 recombinant protein and gRNAs were purchased from TACGENE. Fish were raised to adulthood and indels carriers were screened by T7 Endonuclease I (T7EI) assay following PCR amplification from crude genomic DNA. T7EI positive samples were sequenced and analyzed using TIDE software (https://tide.nki.nl/). Selected founder F0 candidates were crossed with wild type AB and progeny F1 raised to adulthood. Indels carriers were identified using T7EI assay and direct sequencing of PCR product. A single endoglin mutant line was maintained for further analysis. F1, F2 and F3 mutation carriers were then crossed with transgenic reporter lines maintained in AB background. Throughout this study Endoglin mutation was maintained in a heterozygous status to prevent the potential selection of individuals able to cope with Endoglin deficiency.
Genotyping
Routine genotyping was performed on crude genomic DNA prepared from caudal fin clips or in the case of survival experiments pieces from dead larvae or juvenile using an allele specific PCR strategy using the following primers EngWTfwd 5’-ACAGACGAATCTACAGCCGACAT-3’, EngMutfwd 5’-AGAACAGACGAATCTACAGCCGAA-3’ and Engrev 5’-AGCATGTTTTAACAAGACGGCAG-3’ and HotGoldStar PCR mix (Eurogentec).
Survival
Siblings and mutants embryos were screened based on modified blood flow pattern in DA - PCV and poor ISV perfusion phenotypes previously described25 and introduced along in housing systems at 3dpf. Larvae were fed several times a day with dry food from 5dpf onward supplemented artemia with live preys by 9dpf. Dead fish were systematically collected and store at −20°C awaiting genotyping to confirm sibling vs eng-/- status and discriminate wild-type form heterozygous. Kaplan-Meier survival plots and Log-Rank (Mantel-Cox) statistical analysis were generated using Prism GraphPad software. For survival studies of fish under phz treatment, survival was evaluated from 14dpf at the start of phz treatment. Dead fish were collected daily and genotype of both dead and surviving fish was assessed by PCR according to the procedure described above at the end of experiment.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed essentially as previously described75. Briefly, wild type AB fish embryos or larvae were fixed overnight at 4°C in 4% PFA, pH 9.5. Digoxygenin-labeled Endoglin antisense riboprobe was synthesized from Endoglin partial cDNA obtained from 5’ RACE (nucleotide 1 to 1454) cloned into pBSSK(-) vector. Samples were Pre-hybridized overnight at 65°C and hybridization was performed overnight at 65°C in presence of 0.4 ng/nl endoglin antisense probe. Probe was detected using alkaline phosphatase conjugated anti-digoxygenine Fab fragments (Roche) and NBT/BCIP mix reagent mix (Roche).
5’ and 3’ RACE
For endoglin 5’ RACE we adopted a template-switch strategy based on a previously described procedure76. Briefly, 36hpf total RNA (1 µg) were retrotranscribed from either 5’-CATGCTGCTGGTGGGTGTGCT-3’ or 5’-CCATAAAGCACCGGTGAGCAGAA-3’ endoglin specific reverse oligonucleotides (500 nM) using RevertAid H minus reverse Transcriptase (Thermo Scientific) 10 U/µl in 1X RevertAid H minus Buffer supplemented with 2U/µl RNAse OUT (Thermo Scientific), 1mM dNTPs (ref), 2 mM MgCl2 at 50°C for 1 hour. The template-switch reaction was then conducted in presence of Template Switch oligonucleotide (1 µM) and 3mM MnCl2 and 4U/µl RevertAid H minus reverse transcriptase for 90 minutes at 42°C. Reverse transcriptase was finally inactivated by a 10 minutes at 70°C step. Then using Template Switch RTs (50 ng total RNA equivalent) as template together with shortened U_sense oligonucleotide 5’-GTCGCACGGTCCATCGCAG-3’ and endoglin-specific oligonucleotides 5’-CCATAAAGCACCGGTGAGCAGAA-3’ and 5’-GTTTATCCTTTTGACGCCGCAGAG-3’ as primers, PCR reactions were performed using Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific) following manufacturer’s instructions. PCR products were then cloned into pBSSK and sequenced on both strands using T3 and T7 primers.
Endoglin 3’ RACE was performed using GeneRacer kit (Thermo Scientific) following manufacturer’s recommendations. Briefly, 36hpf total RNA (1 µg) were used in RT reactions containing 2.5 µM of GeneRacer Oligo dT primer and 10 U/µl of SuperScript III RT. PCR reactions were then conducted as described above using endoglin-specific forward primer 5’-CCTGGGACCTCGAATGTGCTGTAA-3’ and GeneRacer 3’ primer. One percent of whole PCR was then used as template in nested PCR reactions using endoglin-specific forward primer 5’-CTGGGCATAGCGTTCGGAGGATT-3’ and GeneRacer 3’ Nested primer. PCR products were then cloned blunt into pBSSK vector and sequenced on both strands using T3 and T7 primers.
Total RNA extraction, semi-quantitative and qRT-PCR analysis
Total RNA was extracted from staged embryos, sibling and eng-/- fish at specific ages as pool using Nucleospin RNA kit (Macherey-Nagel) or NucleoZOL (Macherey-Nagel) following manufacturer’s instructions. Equal amounts (500 ng or 1 µg) of total RNA were retrotranscribed using High Capacity cDNA Reverse Transcription kit (Applied Biosystems) following manufacturer’s instruction. Endoglin variants were amplified by semi-quantitative PCR using Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific) or HotGoldStar PCR mix (Eurogentec) using 50ng total RNA equivalent RT reaction and Endoglin EngExon12fwd 5’-CTGGGCATAGCGTTCGGAGGATT-3’, EngExon10fwd 5’-CCTGGGACCTCGAATGTGCTGTAA-3’ EngExon15rev 5’-CATGCTGCTGGTGGGTGTGCT-3’ specific primers and Housekeeping gene beta-actin bactinfwd 5’-CCTGGAGAAGAGCTATGAGCTG-3’, bactinrev 5’-ATGGGCCAGACTCATCGTACTC-3’ primers as control. When necessary, the genotype of samples was verified by RT-PCR using EngWTfwd, EngMutfwd (see sequence above) along with dEngExon5rev 5’-CGTTGGTGACGGATGTGACT-3’ according to the semi-quantitative PCR procedure described above. qPCR reactions were carried out using 1.25ng total RNA equivalent RT reaction in sensiFAST SYBR No-Rox mix (Bioline) in presence of 600 nM of each primer. Reactions were assembled in triplicates in 384- well plates using Labcyte Echo 525 Liquid Handler and PCR reaction was performed using Roche LightCycler 480 available at MGX - High throughput qPCR facility. Target expression values were normalized to RPL13a and Relative expression was determined using ΔΔCT method. Error bars are +/- SEM. In kinetic analysis, results were presented as fold change over siblings group for each time point. dreegln3fwd 5’-TGGGAAAAAGCATTCGTGCG-3’, dreegln3rev 5’-CGGCCATCAGCATTAGGGTT-3’, dreepoafwd 5’-CCATTACGCCCCATCTGTGA-3’, dreepoarev 5’-GTGACGTTCGTTGCAATGCT-3’, drenppafwd 5’-GACACAGCTCTGACAGCAACA-3’, drenpparev 5’-TCTACGGCTCTCTCTGATGCC-3’, drenppbfwd 5’-TGTTTCGGGAGCAAACTGGA-3’, drenppbrev 5’-GTTCTTCTTGGGACCTGAGC-3’, drerpl13afwd 5’-CGCTATTGTGGCCAAGCAAG-3’, drerpl13arev 5’-TCTTGCGGAGGAAAGCCAAA-3’. dreengfwd 5’-AGACGGAGAACGGGACAGAA-3’, dreengrev 5’-TCACCACAGACTTGTTCGCC-3’. One tailed unpaired t test was used for statistical analysis.
Tissue clearing and deep imaging of whole zebrafish
To get access to in situ dimensions of siblings and eng-/- heart ventricule at 10, 12 and 15dpf, Clutches from crosses between eng+/-,Tg(cmlc2:GFP) and eng+/- were screen at 24dpf to sort out GFPpos individuals and at 72dpf to discriminate siblings from eng-/-. Fish were raised as described earlier and collected at indicated times by excess of ethyl 3-aminobenzoate methanesulfonate (MS222) (320 μg/ml) and fixed overnight in 4% PFA. Whole zebrafish were deigmented and labeled according to the protocol described in Frétaud et al.77 using Rabbit anti-mCherry (Rockland, 600-401-P16) and Chicken anti-GFP (ThermoFisher Scientific, A10262) prior to Alexa594 goat anti-rabbit (ThermoFisher, A11012) and Alexa488 goat anti-chicken (ThermoFisher, A11039). Before imaging, larvae were cleared by incubation in RIMS78 overnight at RT. Larvae were mounted under #1 coverslips in RIMS supplemented with 0.8 % low gelling agarose. Images were acquired with a Leica SP8 confocal microscope using a HCX IRAPO L 25X/0,95NA water immersion objective (#11506340, Leica microsystems).
Confocal imaging
Clutches from crosses between eng+/-, Tg(kdrl:GFP) and eng+/-, Tg(flt1:Tomato) were screen at 24dpf to sort out double positive GFP/Tomato embryos and then at 72hpf to discriminate siblings from eng-/-. Fish were raised as described earlier and collected at indicated times by excess of MS222 (320 μg/ml) and fixed overnight in 4% PFA. Samples were rinsed in 1XPBS then store at 4°C in 1XPBS, 0.05% sodium azide until imaging. Fixed zebrafish were mounted in 0.7% low-melt agarose in Fluorodish cover-glass bottom culture dish (WPI) and imaged on a Zeiss LSM510 confocal microscope. Projections of Z-stack were performed using Fiji software.
Blood vessel perfusion imaging
Three-day-old GFPpos and dsRedpos sibling and eng-/- from crosses between eng+-/-, Tg(kdrl:GFP) and eng+-/, Tg(gata1:dsRed) fish were anesthetized mounted in 0.7% low melt agarose, MS-322 (160µg/ml) and imaged on Zeiss AXIO Zoom.V16 mounted with Zeiss AxioCam MRm with Zeiss HXP 200C illuminator set on minimal power using fixed exposure parameters (GFP 1sec and CY3 1.5sec) to obtain red blood cells traces revealing perfusion extent. When necessary, similar post-acquisition treatments were applied to images.
Flow cytometry
GFPpos siblings and eng-/- fish from crosses between eng+-/- and eng+-/-, Tg(cmlc2:EGFP) fish were sorted at 3dpf and raised along as described above. At 5, 10, 15, 20 and 25dpf fish were collected as pool of 26, 18, 9, 7 and 7 fish respectively and dissociated in 0.25% Trypsin-EDTA (Gibco, ThermoFisher Scientific) and 8mg/ml Collagenase from Clostridum histolyticum (Sigma-Aldrich) according to the procedure described by Bresciani et al79. Dissociated cells were fixed overnight in 4% PFA at 4°C, then rinsed thrice in PBS and store at 4°C in PBS, 0.05% sodium azide until analysis. 250000 cells were analyzed on a FACSCanto cytofluorimeter (Becton Dickinson) at each indicated time point and genotype to evaluate cardiomyocytes (GFPpos) representation in whole fish cell suspensions. FACs profiles were validated using cardiomyocytes enriched cell suspensions prepared from GFPpos isolated Tg(cmlc2:EGFP) hearts.
Histology
Fish were euthanized by excess MS222 (320 μg/ml), rinsed in PBS and fixed overnight in 4% PFA. Samples were decalcified using 0.35M EDTA and dehydrated gradually in methanol. Paraffin embedding was performed by RHEM (Experimental histology Network of Montpellier) Histology Facility. Seven micrometers sagittal sections were dewaxed and stained by hematoxylin/eosin. Eng WISH samples were post-fixed overnight in 4% PFA then rinsed in PBS before gradual dehydration in methanol. Paraffin embedding was performed by RHEM. Seven micrometers sagittal and transversal sections were dewaxed and counterstained with Nuclear Fast Red.
Hemoglobin staining, blood smears and hematocrit measurement
Hemoglobin content in 3, 5 10 and 15dpf siblings and eng-/- fish was assessed using o-dianisidine reagent (Alfa Aesar) according to a previously published procedure80. Briefly, embryos and larvae were stained with 0.6mg/ml o-dianisidine in 10mM sodium acetate pH4.5, 0.65% H202 and 40% (v/v) ethanol for 30 minutes at room temperature. Fish were then wash with PBS and fixed overnight in 4% PFA. The next day, fish were washed with PBS and soaked for 30 minutes in 0.8% KOH, 0.9% H202 and 0.1% Tween-20 to remove pigments. After washes with PBS, fish were scored for hemoglobin staining and pictures of representative fish were taken after mounting in 0.7% low-melt agarose.
Blood samples, obtained from cardiac puncture of MS222 anaesthetized fish were smeared on microscope slides. Wright-Giemsa staining was performed following manufacturer’s instructions (Sigma-Aldrich). For hematocrit measurement, wild-type and eng-/- adult fish were anaesthetized using MS222 (160µg/ml) and blood samples were obtained by cardiac puncture using 18 µl heparinized capillary tubes (Hirschmann). Samples were processed using Hematocrit 24 centrifuge (Hettich) and hematocrit was assessed.
Phenylhydrazine treatment
Wild-type zebrafish (about 30 fish per batch) were bathed every other day from 14dpf with 1.25, 2.5 and 5µg/ml phenylhydrazine in fish water prepared from a freshly made 5mg/ml stock solution of phenylhydrazine hydrochloride (Sigma-Aldrich). Non-treated fish served as control. Bathing volume was gradually increased to accommodate with fish growth (100 to 300ml). Fish were treated for 30 min and then allowed to recover in fresh fish water for about 1 hour and finally replaced into housing tanks and fed. Fish were subjected to this regimen up to 75dpf before treatment was definitively stopped to let fish recover and regain gender-specific hallmarks to allow for accurate sex ratio determination. To assess hypoxia and cardiac stress in fish under phz treatment, 4 batch of 5 fish of each condition were collected at 29dpf (treatment-free day) and euthanized by excess of MS222 (320 μg/ml) for total RNA extraction performed as described above. Non-treated wild-type fish were used as control. Phenylhydrazine treatment of eng-/- and siblings was performed as for wild-type (see above) but phenyhydrazine concentrations were downscaled to 0.625, 1.25 and 2.5µg/ml. Non-treated Siblings and eng-/- were used as control. At 29dpf (treatment-free day), each condition was divided into 4 samples of 4-5 fish/sample used for total RNA extraction. For phenylhydrazine effect on eng-/- survival, siblings and eng-/- fish were treated as described above using 0.625 and 1.25 mg/ml phenylhydrazine up to 2 months and were left to recover for 15 days to allow for gender determination.
Author contributions
EL. CB. YB. MF. CL. CJ Performed experiments and/or analyzed data. EL with input from CJ and KK conceived and designed the study. EL wrote the manuscript.
Data availability statement
The data underlying this article are available in the article and in its online supplementary material.
Competing interests
The authors declare no competing interest
Aknowledgments
Authors thank Mireille Rossel for invaluable technical tips and advices in the course of the work and critical reading of the manuscript, Nicolas Cubedo for sharing his expertise regarding zootechnical issues, Philippe Clair for helpful hints regarding qPCR, Stéphane Delbecq for help regarding hematocrit measurements. This work was supported by grants to Karima Kissa from Association pour la Recherche contre le Cancer (ARC), Chercheur d’Avenir - Région Languedoc-Roussillon, Fondation pour la Recherche Médicale (FRM) (FDT20150532507) and ATIP-Avenir.
Abbreviations
- ALK1 (ACVRL1)
- activin receptor-like kinase
- AVM
- arteriovenous malformation
- BMP
- bone morphogenetic protein
- EC
- endothelial cell
- HHT
- heredidary hemorrhagic telangiectasia
- phz
- phenylhydrazine
- TGF-β
- transforming growth factor β
Bibliography
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.
- 6.↵
- 7.↵
- 8.↵
- 9.
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.
- 16.
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.
- 24.
- 25.↵
- 26.↵
- 27.↵
- 28.
- 29.↵
- 30.↵
- 31.
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵




