

Abstract
Panton-Valentine leukocidin (PVL) is a Staphylococcus aureus (S. aureus) toxin that binds to and kills human neutrophils, resulting in the formation of neutrophil extracellular traps (NETs). Some individuals colonized with PVL-positive S. aureus (PVL-SA) suffer from recurring infections whereas others are asymptomatically colonized. We found that neutrophils from affected patients express higher levels of CD45, one of the PVL receptors, and are more susceptible to killing at a low concentration of recombinant PVL than control neutrophils. We verified that PVL induces the formation of NETs and provide genetic and pharmacological evidence that PVL-induced NET formation is independent of NADPH-oxidase and reactive oxygen species (ROS) production. Through NET proteome analysis we identified that the protein content of PVL-induced NETs is different from NETs induced by mitogen or the microbial toxin nigericin. The abundance of the proteins cathelicidin (CAMP), elastase (NE), and proteinase 3 (PRTN3) was lower on PVL-induced NETs, which were inefficient in killing S. aureus. Neutrophils from patients that suffer from recurring PVL-positive infections may be more sensitive to PVL-induced NET formation, which may impair their ability to combat the infection.
Introduction
S. aureus is a widespread Gram-positive pathogen that causes infections in skin and soft tissue (SSTI), osteoarticular, airway, and the blood stream in humans and animals. Nasal colonization with S. aureus is found in approximately 50% of humans, with strong age variation, and may be asymptomatic provided that skin and mucous membrane barriers are intact [1,2]. Pathogenicity of S. aureus strains depends on a variety of virulence factors, among those, pore-forming leukocidins such as PVL that target neutrophils (also called polymorphonuclear leukocytes, PMN), and macrophages to evade phagocytosis and intracellular killing (reviewed in [3]). Over the last two decades an increasing number of outbreaks of SSTI in close communities [4–12] and in other health care settings [13–24] were caused by S. aureus strains that produce PVL (PVL-SA).
Although recurrent deep skin abscesses and furunculosis are the most frequent manifestations of PVL-SA infections, other pathologies are also linked to these strains. These include (a) worldwide reports of severe invasive infections - often complicated by thrombotic events - in previously healthy young individuals [25–27], (b) breast abscesses in lactating women [28] and transmission to offspring that can cause life-threatening infections [29], and (c) necrotizing pneumonia [30], a severe manifestation, often fatal within days of hospital admission [31,32]. In some cases, pneumonia is preceded by viral airway infections, like influenza, parainfluenza [31,32] and SARS-CoV2 [33].
During outbreaks, identical PVL-SA strains have been obtained from individuals with asymptomatic nasal colonization as well as from patients with severe SSTI or invasive infections [11], suggesting that host factors may explain susceptibility to, and severity of, PVL-SA infections. Although studies in African Pygmies suggest that genetic variations in C5ARI may be associated with PVL-SA colonization [34], other factors that allow PVL-SA colonization or infections are not known.
The binding specificity of PVL defines both its host- and cell tropism. PVL is a two-component toxin consisting of LukF-PV and LukS-PV, which bind to the human panleukocyte receptor CD45 [35], and the human complement 5a receptors CD88/C5aR and C5L2, respectively [36]. As a result, human phagocytes are the major target of PVL. Upon binding, the subcomponents hetero-oligomerize into an octameric membrane spanning pore [37,38], which drives cell death. Neutrophils express higher levels of both C5a receptors than monocytes [36] and are more sensitive to PVL mediated killing than both monocytes and macrophages [39], suggesting they are the major target of PVL. Importantly, neutrophils form an important cellular host defense against S. aureus [40] and patients with impaired production of reactive oxygen species (ROS) in phagocytes are particularly sensitive to S. aureus infections (see review [41]). Neutrophils clear invading S. aureus through phagocytosis and by undergoing NETosis, a cell death process which results in the formation of neutrophil extracellular traps (NETs). NETs consist of an externalized web of chromatin decorated with antimicrobial peptides and proteases and are toxic to bacteria [42].
Depending on the stimulus, NET formation may require ROS formation [43]. A common mediator of ROS-dependent NET formation is NADPH-oxidase, which produces superoxide (O2−) that spontaneously dismutates to hydrogen peroxide (H2O2). Myeloperoxidase (MPO) converts hydrogen peroxide into highly reactive hypochlorous acid (HOCl). ROS production allows for the release of neutrophil proteases from granules, protease-mediated chromatin decondensation and the final lysis of the plasma membrane [44–46]. Two S. aureus toxins that kill neutrophils and result in NET formation are γ-hemolysin AB and PVL [47–49]. Interestingly, S. aureus also produces nucleases to escape from NETs [50]. Whether NET formation is a beneficial or detrimental neutrophil response, both for the host as well as for S. aureus, remains unclear and may be context dependent [48].
In this study, we show that neutrophils from patients with a history of recurrent PVL-SA infections express higher levels of the PVL receptor CD45 than healthy controls and make more NETs in response to a low concentration of PVL. Furthermore, PVL-induced NET formation is NADPH-oxidase independent. Consistently, neutrophils isolated from chronic granulomatous disease (CGD) patients, which have a mutation in the gene encoding one subunit of the NADPH oxidase complex, make NETs in response to this toxin [51]. PVL-induced NETs showed quantitative proteomic differences to NETs produced after PMA or nigericin stimulation. PVL NETs contained a lower abundance of multiple antimicrobial proteins and did not kill S. aureus. Our results suggest that PVL-receptor expression may mediate susceptibility to symptomatic S. aureus infection and that NET-formation induced by this toxin serves as an offensive strategy to preemptively neutralize the neutrophil.
Materials and Methods
Patient and control characteristics
Patient demographic and clinical data are summarized in Supplementary Table 1. Controls were matched for gender and age. All study participants provided written informed consent and were free of infections at the time of blood withdrawal. The study was approved by the local Ethics committee (EA2/003/19). Healthy control samples were collected according to the approval and guidelines of the local ethics committee (EA1/0104/06).
Neutrophil isolation, experimental conditions, and inhibitors
Human neutrophils were isolated by layering whole blood over Histopaque-1119 (Sigma) followed by a discontinuous Percoll gradient (Amersham Biosciences) as previously described [45]. Alternatively, they were also isolated using the direct human neutrophil isolation kit (EasySep, StemCell Technologies) following manufacturer’s instructions. All experiments were performed in Seahorse XF RPMI medium (SF-RPMI, Agilent) supplemented with 2 mM glutamine, 10 mM HEPES, 1 mM glucose and 0.1% human serum albumin (HSA) at pH 7.4 except where mentioned. The stimuli used to induce NET formation were phorbol 12-myristate 13-acetate (PMA, Sigma), PVL (equal amounts of S. aureus recombinant LukS and LukF (Bioservices), and nigericin (InvivoGen). We used the following inhibitors: Gö6983 (PKCi, Tocris) BAPTA-AM (Thermo Fisher Scientific), pyrocatechol (Sigma), 4-Aminobenzoic acid hydrazide (ABAH, Cayman chemical), neutrophil elastase inhibitor (NEi, MedChemExpress), Diphenyleneiodonium chloride (DPI, Calbiochem), AEBSF (Sigma), allopurinol (Sigma), Apamin (Sigma), 2,4-Dinitrophenol (DNP, Sigma) and 2-[[4-(trifluoromathoxy) phenyl]hydrazinylidene] propanedinitrile (FCCP, Abcam).
Neutrophil markers
1×106/ml neutrophils were fixed for 15 min using 4% PFA and washed to PBS supplemented with 0.1% HSA. Cells were incubated with anti-CD63-PE, anti-CD66b-APC (Miltenyi Biotec), 5 μg/ml anti-CD45-AlexaFluor 647 (Santa Cruz Biotechnology), 10 μg/ml CD88 (S5/1)-FITC (Santa Cruz Biotechnology) and 10 μg/ml anti-human C5L2-APC (BioLegend) antibodies for 30 min in the dark, washed thereafter to PBS supplemented with 0.1% HSA and measured on a CytoFLEX (BeckMan Coulter) or MACSQuant (Miltenyi Biotec).
ROS measurement
Purified neutrophils were seeded at 105 cells per well in a 96-well plate and incubated for 30 min with the indicated inhibitors, followed by incubation for 10 min with 50 μM luminol and 1.2 units/ml horseradish peroxidase at 37°C prior to stimulation with indicated stimuli. Luminescence was measured over time in a VICTORX luminometer (Perkin Elmer) [43].
Neutrophil lytic cell death assay
105 neutrophils were seeded in a 96-well plate and incubated with the appropriate inhibitors for 30 min, followed by incubation with 50 nM cell-impermeable DNA dye SYTOX Green (ThermoFisher Scientific) for 5 min at 37°C, prior to stimulation with indicated stimuli. Fluorescence was recorded once per hour for 4h using a Fluoroskan Ascent (ThermoFisher Scientific).
NET staining
105 neutrophils were seeded on glass coverslips in a 24-well plate and incubated with or without appropriate inhibitors followed by stimulation with indicated stimuli for 4 h at 37°C. Cells were fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature. Cells were washed with PBS, permeabilized with 0.5% Triton X-100 for 1 min and incubated in blocking buffer (3% normal goat serum, 3% cold water fish gelatin, 1% bovine serum albumin and 0.05% Tween-20 in PBS) for 30 min. Neutrophils were then stained using antibodies detecting elastase (Merck Millipore, 481001) and a subnucleosomal complex of Histone 2A, histone 2B and chromatin (Losman et al., 1992; in-house generated). The secondary antibodies used were goat anti-mouse Alexa Fluor 488 (Invitrogen, A11029) and goat anti-rabbit Alexa Fluor 647 (Invitrogen, A21245) followed by staining with DAPI (0.1 μg/ml, Invitrogen). Finally, the samples were mounted using antifade mountant (ProLong Diamond Antifade mountant, ThermoFisher Scientific). Images were acquired using a Leica TCS SP8 confocal microscope.
For live NET imaging, cells were resuspended in Agilent XF RPMI medium (Agilent) at pH 7.4, supplemented with 0.1% Human Serum Albumin, 10 mM Glucose (Sigma Aldrich), 2 mM L-Glutamine (Gibco), 20 mM HEPES (Gibco) 500 nM SYTOX Green (Thermo Fischer) and 2.5 μM DRAQ5 (Biostatus) and seeded at a density of 5×105 cells per well into μ-slide 8 well ibiTreat dishes (ibidi). Cells were stimulated with final concentrations of 100 nM PMA and 10 nM PVL. Images were collected at 2 min intervals on a Leica TCS SP8 confocal microscope equipped with a climate chamber at 37°C and with a Leica HC PL APO 20x/0.75 IMM CORR CS2 objective using glycerol immersion [43].
Phagocytosis assay
1×106/ml neutrophils were incubated with 10×106/ml AlexaFluor 488-labeled Escherichia coli (E. coli) particles (ThermoFisher Scientific) in RPMI supplemented with 5% human pooled serum (Sigma) for 30 min on a rotator. Cells were fixed for 15 min using 4% PFA and separated from non-phagocytosed particles by centrifugation at 200x g for 10 min. Cells were washed with PBS supplemented with 0.1% HSA and phagocytosis was quantified by flow cytometry on a Cytoflex (Beckman Coulter) or MACSQuant (Miltenyi Biotec).
Mass spectrometry (MS)
Human neutrophils from three different healthy donors were seeded in 6-well tissue culture plate to a density of 3×105 cells per well (SF-RPMI without HSA) and then stimulated with 50 nM PMA, 10 nM PVL, or 15 μM nigericin for 4 h to induce NETs. As a control, neutrophils were incubated in medium for 4 h. Media was carefully removed followed by a wash with fresh media to remove unbound proteins. NETs were collected by treatment with lysis buffer (1% SDS, 50 mM HEPES pH 8, 10 mM tris-(2-carboxyethyl)phosphine (TCEP), 40 mM Chloroacetamide and protease-inhibitor cocktail) and subsequent scraping.
All samples were subjected to SP3 sample preparation [52]. Briefly, proteins were denatured, reduced and alkylated, and subsequently digested with Trypsin and Lys-C proteases. TMT 11plex (Pierce) labelling was used for peptide multiplexing and quantification. Samples were mixed, desalted using solid phase extraction (Seppak 1cc/50 mg, Waters), and fractionated using basic reversed phase fractionation on a quaternary Agilent 1290 Infinity II UPLC system equipped with a Kinetex Evo-C18 column (150 × 2.1 mm, 2.6μm, 100 Å, Phenomenex). Fractions were concatenated into 8 final samples, dried down and resuspended in 2% acetonitrile, 0.1% trifluoroacetic acid (TFA) prior MS analysis. All samples were analyzed on an Orbitrap Q Exactive HF (Thermo Scientific) that was coupled to a 3000 RSLC nano UPLC (Thermo Scientific). Samples were loaded on a pepmap trap cartridge (300 μm i.d. x 5 mm, C18, Thermo) with 2% acetonitrile, 0.1% TFA at a flow rate of 20 μL/min. Peptides were separated over a 50 cm analytical column (Picofrit, 360 μm O.D., 75 μm I.D., 10 μm tip opening, non-coated, New Objective) that was packed in-house with Poroshell 120 EC-C18, 2.7 μm (Agilent). Solvent A consists of 0.1% formic acid in water. Elution was carried out at a constant flow rate of 250 nL/min using a 180-minute method: 8-33% solvent B (0.1% formic acid in 80% acetonitrile) within 120 minutes, 33-48% solvent B within 25 minutes, 48-98% buffer B within 1 minute, followed by column washing and equilibration. The mass spectrometer was operated in data-dependent acquisition mode. The MS1 survey scan was acquired from 375-1500 m/z at a resolution of 120,000. The top 10 most abundant peptides were isolated within a 0.7 Da window and subjected to HCD fragmentation at a normalized collision energy of 32%. The AGC target was set to 2e5 charges, allowing a maximum injection time of 78 ms. Product ions were detected in the Orbitrap at a resolution of 45,000. Precursors were dynamically excluded for 45 s. Raw files were processed with Proteome Discoverer 2.3 (Thermo Scientific) using SEQUEST HT for peptide identification. Peptide-spectrum-matches (PSMs) were filtered to a 1% false discovery rate (FDR) level using Percolator employing a target/decoy approach. The protein FDR was set to 1%. Further data processing was carried out in R and Perseus (v. 1.6.2.3). Only proteins identified with at least two peptides were included in the analysis. All contaminant proteins were filtered out. A three-step normalization procedure was applied. First, the total intensity of each TMT channel was normalized to correct for mixing errors. Next, the common channel in both TMT sets was used for internal reference scaling [53] in order to correct for batch effects. Afterwards the data was normalized applying trimmed mean of M values (TMM) using the edgeR package in R [54]. Proteins were filtered on those detected in at least two of three replicate experiments. Remaining undetected (NA) values were replaced with the sample-wise minimum abundance as an estimation of the limit of detection. Differential protein abundances were calculated using the limma package in R [55]. Principle component analysis and calculation of Euclidean distance between proteome samples were performed using scaled log2(abundance) values.
NET killing assay
Isolated neutrophils were resuspended in RPMI supplemented with 10% human serum (Sigma). 106 neutrophils were seeded in a flat-bottom 96-well plate, and NETosis was induced for 3 h at 37 °C, 5% CO2 with 100 nM PVL or 200 nM PMA. S. aureus (USA300) in mid-logarithmic phase was resuspended in RPMI with 10% human serum and added to each well at a multiplicity of infection of 0.1. Bacteria were spun down for 5 min at 1500 x g and incubated for 1 h at 37 °C after which the NETs were treated with 2 U Micrococcal nuclease from S. aureus (Sigma) for 10 min at RT. Subsequently, the samples were serially diluted in DPBS and plated on trypticase soy agar plates. The plates were incubated overnight at 37 °C and colony-forming units were counted.
For DNA quantification, 106 neutrophils were seeded in plates and NETosis was induced as described above. NETs were digested with 2 U micrococcal nuclease for 10 min at RT, and subsequently with 0.5 mg/ml of proteinase K (Invitrogen) for 1 h at 50°C. The samples were vortexed and the DNA concentration was determined by measuring absorbance at 260 nm on a Nanodrop 2000 spectrophotometer (ThermoFisher Scientific).
Statistical analysis
Analysis of differential protein abundance was performed in limma (Linear Models for Microarray data) in R, which has been shown to outperform t-tests in detecting significant changes in protein abundance [56]. Protein-wise linear models were fit and batch-corrected using the formula (0 + condition + batch) and significant changes in abundance were tested using an empirical Bayes moderated t-statistic with Benjamini-Hochberg correction. Changes in abundance were considered significant at an absolute log2 fold change > 1, and an adjusted p value < 0.01.
Euler diagrams of significant changes across samples were visualized with the eulerr package [57]. Significant proteins were clustered by pattern across conditions using k-means clustering with indicated number of clusters. Heatmaps were produced using pheatmap package [58].
R scripts for proteome analysis and visualization are available upon request and will be made publicly available upon publication.
Data are represented as mean±SD unless otherwise stated.
MS data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [59] partner repository with the dataset identifier PXD025702.
Reviewer account details:
Username: reviewer_pxd025702{at}ebi.ac.uk
Password: Yr3cVMPZ
Results
PVL induces NETs efficiently in neutrophils from patients with recurrent PVL-SA infections
To determine why some individuals show increased susceptibility to recurrent PVL-SA infections, we assessed neutrophil responses in these patients. To verify that PVL induced cell death results in NETs [48,49], we stimulated control and patient neutrophils with 1 nM PVL for 4 h and stained them with the DNA dye DAPI and immuno-labeled with anti-NE and anti-chromatin antibodies (Figure 1A). As a control we used PMA, a well characterized mitogen that induces NADPH-oxidase dependent NETosis [60]. As shown by confocal microscopy, PVL and PMA induced NET formation in both control and patient neutrophils. Interestingly, 0.1 nM PVL killed neutrophils isolated from patients more efficiently than PMNs isolated from healthy controls (Figure 1B) as measured with a cell impermeable DNA dye. Patient and control neutrophils were equally susceptible to make NETs in response to PMA.

(A) Representative confocal images of control and patient neutrophils, either naïve or stimulated with 50 nM PMA or 1 nM PVL, stained for DNA (blue), neutrophil elastase (NE, red) and chromatin (green). Scale bar represents 10 μm. (B) Purified primary neutrophils from patients suffering from recurrent PVL-SA infections (n=19) and healthy controls (n=19) were either left untreated or stimulated with PMA (50 nM) or PVL (0.1 nM, 1 nM or 10 nM) for 4 h and cell death was quantified by adding the cell-impermeable DNA dye SYTOX Green and measuring fluorescence. Boxplot indicates SYTOX fluorescence relative to naïve neutrophils at 4 h. (C) CD45, C5L2, CD88, CD63 and CD66b were immunolabelled on neutrophils from patients and healthy controls. The fluorescence was measured and indicated as log-transformed MFI. (D) CD45 or (E) C5L2 fluorescence of patient neutrophils relative to control was plotted against SYTOX fluorescence of patient neutrophils relative to control after incubation with 0.1 nM PVL for 4 h. (B) Unpaired Wilcoxon signed rank test with Bonferroni post-hoc test, * P<0.05. (C) Paired t-test, *** P<0.001. (D-E) Spearman’s correlation test where the coefficient of correlation (ρ) and probability (p) are indicated. A best-fit line indicates the trend.
Given the binding specificity of PVL we checked the expression of CD45, CD88, and C5L2, as well as the neutrophil activation markers CD63 and CD66b on control and patient neutrophils by flow cytometry. Patient neutrophils expressed higher levels of CD45 (adjusted p value = 0.000473), and there was no difference in CD88. Furthermore, a subset of patients expressed increased C5L2 levels compared to control neutrophils (Supplementary Figure S1A), but this trend was not consistent for all patients (Figure 1C). Interestingly, the difference in expression of C5L2 and possibly CD45 in patients compared to controls correlated with the difference in NET formation in response to 0.1 nM PVL (ρ = 0.4491 for CD45, ρ = 0.7105 for C5L2) (Figure 1D and 1E). Differences in expression levels of CD88, CD63 or CD66b did not correlate with differences in NET formation (Supplementary Figure S1B-D).
Neutrophils isolated from control and patient phagocytosed opsonized Escherichia coli particles with equal efficiency; the proportion of neutrophils phagocytosing and the average quantity of phagocytosed bacteria per cell were similar in both neutrophil populations, suggesting normal phagocytic capacity of patient neutrophils (Supplementary Figure S1E).
Taken together, our data show that neutrophils from patients with recurrent PVL-SA infections express higher levels of the PVL receptor CD45 and make more NETs in response to 0.1 nM PVL when compared to control neutrophils.
PVL induces NETs independent from ROS
NADPH-oxidase dependent superoxide formation is essential in NETs induced by some stimuli [43], prompting us to investigate the NETosis pathway initiated by PVL. We first tested superoxide generation in healthy primary neutrophils stimulated with different concentrations of PVL or PMA. PVL stimulation resulted in weak superoxide production at all concentrations (Figure 2A). In contrast, PMA induced a significant superoxide burst.
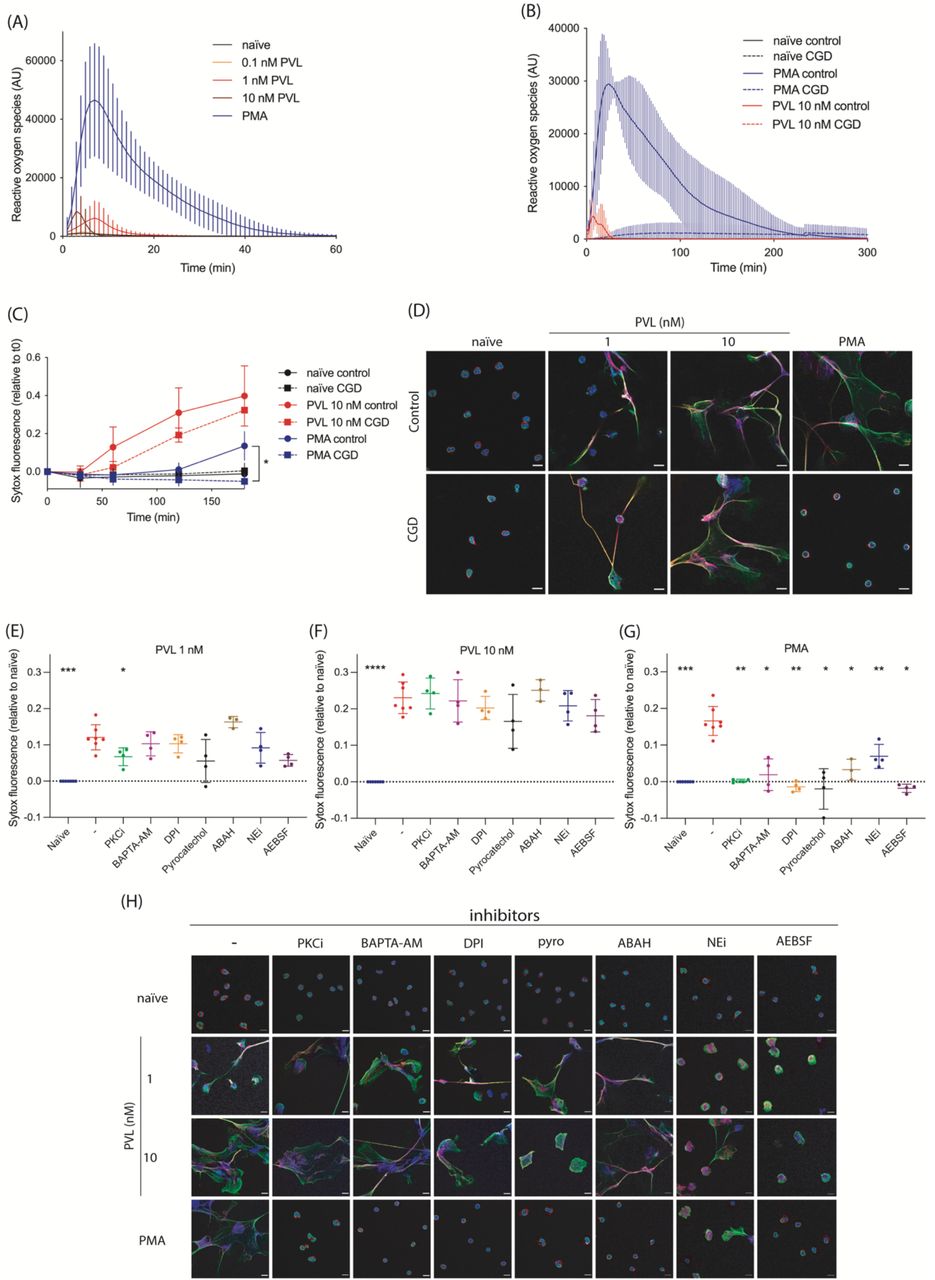
(A-B) Reactive oxygen species (ROS) formation was quantified in control (A n=14, B n=3) or CGD human primary neutrophils (n=3) stimulated with PMA (50 nM) or PVL (0.1 nM, 1 nM or 10 nM). (C) Control or CGD neutrophils (n=3) were stimulated with PMA (50 nM) or PVL (10 nM) for 3h and cell death was quantified using the cell impermeable DNA dye SYTOX Green. Indicated is SYTOX Green fluorescence relative to t = 0 min. (D) Representative confocal microscopy images of control and CGD patient neutrophils either naïve or stimulated with PMA (50 nM) or PVL (1 nM or 10 nM) and stained for DNA (blue), NE (red) and chromatin (green). Scale bar represents 10 μm. (E-G) Healthy human neutrophils were treated with PKC inhibitor Gö6983 (PKCi, 1 μM), BAPTA-AM (10 μM), DPI (1 μM), pyrocatechol (30 μM), ABAH (500 μM), NEi (20 μM) and AEBSF (100 μM) for 30 min, before stimulating with (E) PVL 1 nM, (F) PVL 10 nM or (G) PMA 50 nM for 4h. We quantified cell death with SYTOX Green and the fluorescence signal relative to naïve is indicated. (H) Representative confocal microscopy of naïve neutrophils or after stimulation with PMA or PVL (1 nM or 10 nM) in the presence or absence of indicated inhibitors, and stained for DNA (blue), NE (red) and chromatin (green). Scale bar represents 10 μm. (C) Two-way ANOVA with Tukey’s post-hoc test. *P<0.05. (E-G) Mean ±SD of four independent experiments. *P<0.05, ** P<0.01, *** P<0.001, one-way ANOVA with Dunnett’s multiple comparison test.
We quantified ROS production as well as NET formation in neutrophils from CGD patients. As expected, CGD neutrophils failed to generate superoxide in response to PVL or PMA (Figure 2B). However, unlike PMA, PVL induced extracellular DNA release (Figure 2C) and NET formation (Figure 2D and supplementary videos S1-S8) in CGD neutrophils. These data show that while PVL stimulation weakly activates NADPH-oxidase, NET formation is independent from the produced ROS.
Given the known involvement of various neutrophil proteins in NET formation, we asked if PVL-induced NET formation could be blocked pharmacologically by the PKC inhibitor Gö6983 (PKCi), the calcium chelator BAPTA-AM, the NADPH-oxidase inhibitor DPI, the ROS scavenger pyrocatechol, the myeloperoxidase inhibitor ABAH, the neutrophil elastase inhibitor NEi, or the pan serine protease inhibitor AEBSF (Figure 2E-F and 2H). We observed that PKCi weakly inhibited NET formation induced by 1 nM PVL (Figure 2E), whilst none of the other inhibitors, including inhibitors of ROS-producing enzymes (DPI and ABAH) or ROS scavengers (pyrocatechol), blocked NETosis induced by 1 or 10 nM PVL (Figure 2E-F and 2H). Notably, neutrophils pre-treated with NEi or AEBSF and stimulated with 1nM PVL formed smaller NETs (Figure 2H). These data suggest that PVL-mediated NET formation is ROS- and protease-independent. As a control, we confirmed [43] that PMA induced NET formation was blocked by all inhibitors tested (Figure 2G-H).
Mazzoleni et al. [49] suggested that small conductance calcium-activated potassium (SK) channels and alternative ROS sources, such as mitochondria or xanthine oxidase, mediate PVL-induced NETosis. To verify this, we pre-treated neutrophils with the xanthine oxidase inhibitor allopurinol, the SK channel inhibitor apamin, and the mitochondrial uncouplers DNP and FCCP, before stimulating with either PMA or PVL. The SK channel inhibitor NS8593 was toxic to neutrophils in our hands and was excluded from our experiments. At 1 nM, but not at 10 nM PVL stimulation, allopurinol inhibited NET formation, whilst mitochondrial uncoupling with FCCP partially inhibited NET formation. (Supplementary Figure S2A-B). These findings were supported by confocal imaging (Supplementary Figure S2D). None of the inhibitors affected PMA-induced NET formation (Supplementary Figure S2C). Given that PVL induces NET formation in CGD patients despite a complete lack of ROS (Figure 2B), and the inability of the ROS scavenger pyrocatechol to inhibit PVL induced NETosis, the inhibition of NETs by allopurinol treatment or mitochondrial uncoupling appears to be independent of ROS production. This is further supported by the observation that DPI, which not only inhibits NADPH-oxidase but all flavin-containing proteins including xanthine oxidase [61] and mitochondrial complex I [62], did not block PVL induced NETosis (Figure 2E-F). Finally, the inability of ROS-targeting inhibitors to block NETosis induced by 10 nM PVL indicates that PVL-induced NETosis is independent from ROS production.
PVL-induced NETs lack enrichment of key antimicrobial proteins
Since PVL probably induces NETs through a non-canonical pathway we analyzed the proteomes of NETs induced by PVL, PMA, and nigericin, an NADPH-oxidase independent NET inducer [43]. We analyzed NETs from three independent donors by quantitative mass spectrometry and compared them to the proteome of unstimulated neutrophils. Principal component analysis revealed that PVL-, PMA- and nigericin-induced NETs each have distinct proteomes (Figure 3A). Interestingly, PVL NETs appear to cluster between naïve neutrophils and NETs induced by PMA and nigericin, suggesting their proteome composition is intermediate between naïve cells and classical NETs. Measurement of the Euclidean distance between samples confirmed that PVL NETs are less distinct from naïve neutrophils than are PMA- or nigericin-stimulated NETs (Supplementary Figure S3A).

(A) Principal component analysis of the proteomes of naïve neutrophils, and PVL-, PMA-, and nigericin-induced NETs. Analysis was performed on scaled log2-abundance of all proteins detected in at least 2/3 replicates. (B) Euler diagram showing the number and distribution of significant Differentially Abundant Peptides (logFC > 1, adjusted p < 0.01) on NETs compared to naïve neutrophils. Areas are proportional to DAP set size. (C) Clustered heatmap showing fold change (log2) values on each NET sample compared to naïve neutrophils of significant DAPs from (B). Clustering was performed by k-means algorithm with k=4 clusters. (D-E) Top 25 most-abundant DAPs that are significantly differentially abundant on PVL-induced NETs compared with PMA-(D) and nigericin-induced NETs (E). Point size and fill color represent average abundance across samples and adjusted p value, respectively. Proteomes were made from n=3 samples per condition from independent donors. DAP significance for each comparison was determined by a threshold of |[log2 fold change]| > 1 and adjusted p value < 0.01.
We detected 2458 distinct proteins in the combined NET samples (present in at least two of three replicate experiments). To examine which proteins are driving the differences between the NET proteomes, we performed a differential enrichment analysis using a 2-fold change in protein abundance with an adjusted p value below 0.01 as a cut-off. We found significant differences in relative protein abundance between all three NET stimuli when compared with naïve neutrophils (Figure 3B and Supplementary Figure S3A). Fewer differentially abundant proteins (DAPs) were detected between PVL-induced NETs and naïve PMNs (194 significant) compared to PMA-(449) and nigericin-induced NETs (670). These data suggest that the specific enrichment or depletion of proteins during NETosis is lower or incomplete in PVL-treated neutrophils.
Examination of the fold changes of NET-specific DAPs revealed that most DAPs have the same pattern of enrichment or depletion across all types of NETs compared to naïve neutrophils (459 with negative logFC, 294 positive logFC, Figure 3C). Regression of global DAP fold changes (Supplementary Figure S3B) revealed that though most proteins have the same enrichment pattern across NETs, the fold-changes are reduced in PVL-compared to PMA- and nigericin-induced NETs (slopes = 0.64, and 0.49 respectively), while PMA and nigericin NETs have equivalent DAP fold-changes (slope = 1).
K-means clustering of DAP fold change patterns supported this trend, and further identified a cluster of proteins specifically enriched on nigericin induced NETs (Figure 3C cluster two, orange, and Supplementary Table 2). To look more closely at the differences between PVL-induced NETs and PMA- or nigericin-induced NETs, we performed pair-wise comparisons between PVL and PMA or PVL and nigericin (Supplementary Figure S3C and S3D) and ranked the 25 most abundant DAPs (Figure 3D and 3E, respectively). Among these, key neutrophil proteins NE (ELANE), PRTN3, and CAMP, were all less abundant on PVL-induced NETs compared with PMA- or nigericin-induced NETs. In contrast, we found cytoskeleton proteins such as actin (ACTB), myosin (MYH9), and tubulin (TUBA1B) to be more enriched on PVL NETs. Our data suggest that PVL induces NETs with a different NET proteome assembly when compared with PMA or nigericin, resulting in a lack of enrichment of key antimicrobial proteins on PVL NETs.
PVL-induced NETs are less bactericidal
Given the differential enrichment of antimicrobial proteins in PMA-compared to PVL induced NETs, we hypothesized that their bactericidal activity is different. We incubated methicillin-resistant S. aureus (MRSA) with either PMA- or PVL induced NETs for 1 h, harvested the bacteria, and quantified the surviving colony forming units. PVL-induced NETs did not kill S. aureus, whilst PMA-induced NETs did (Figure 4A). We verified that both stimuli at these concentrations released similar amounts of NETs by nanodrop DNA quantification (Figure 4B). These results show that PVL NETs have a lower antimicrobial potential against MRSA than PMA NETs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Primary human neutrophils were stimulated with PMA (200 nM) or PVL (100 nM) for 3 h to induce NETs. MRSA was incubated with NETs for 1 h and CFU was quantified. Bacterial viability is expressed as a percentage of CFUs normalized to MRSA incubated in the absence of NETs. (B) The DNA content of PMA and PVL induced NETs at 3 h was quantified by spectrophotometry. (A) mean ± SD of five or four independent experiments. **p<0.01, **** p<0.0001, two-tailed paired t test.
Discussion
The reporting of PVL-SA infections has increased in the past few decades. Interestingly, human host factors that mediate susceptibility or severity of PVL-SA infections are not known. To identify these factors, we determined functional responses of neutrophils, a major cellular target of PVL, in response to PVL in patients that suffer from recurrent PVL-SA infections. We identified increased NET formation in patient neutrophils upon stimulation with a low PVL concentration. (Figure 1B), and a higher expression of CD45 and potentially C5L2, two of the toxin’s receptors (Figure 1C), in patient neutrophils compared to healthy controls. Furthermore, the increase in CD45 and C5L2 expression correlated with more NET formation induced by PVL (Figure 1D-E).
LukS-PV and LukF-PV have different binding affinities to their respective receptors (C5L2 and CD88 for LukS-PV, CD45 for LukF-PV), and the low binding affinity of LukF-PV to CD45 suggests that varying expression levels of CD45 are unlikely to modulate pore formation [35]. In contrast, LukS-PV binding affinity to C5aR or C5L2 is much higher and therefore their expression may modulate sensitivity to PVL toxin. Given that neutrophils express C5L2 less abundantly than C5aR, it was surprising to find that differential expression of C5L2 correlates with an increased sensitivity of patient neutrophils to PVL (Figure 1E) [36]. However, the binding affinities of LukS-PV to C5aR and C5L2 and the relative contribution of C5L2 to PVL mediated cell death is not known. Whether there is a genetic predisposition in patients with recurrent PVL-SA remains to be determined and we are currently investigating avenues to study this.
C5aR localizes to the plasma membrane, rendering it accessible for extracellular PVL toxin. However, there are conflicting reports on the localization of C5L2 since it has been detected both intracellularly and on the plasma membrane [63,64]. We detected C5L2 on the plasma membrane through flow cytometry (Figure 1C), suggesting that the expression is not exclusively intracellular. However, much is unknown concerning the regulation of C5L2 and C5aR expression and receptor shuttling and it is unclear how this regulation might affect targeting by PVL [63,64].
Pore formation by PVL is described to exert various cellular effects, intracellular calcium flux, ATP-release into the extracellular environment, induction of apoptosis and cellular lysis [65]. These various phenotypes likely depend on the toxin concentration a cell encounters. We therefore characterized NETosis at different PVL concentrations.
NET formation induced by PMA or Candida albicans is ROS-dependent and is blocked by compounds that scavenge ROS or inhibit ROS [43]. Other stimuli, like nigericin and calcium ionophores induce NETs through a ROS independent mechanism [43]. PVL induces the formation of NETs independently of NADPH-oxidase and MPO activity, two enzymes that produce ROS. This is evidenced by two observations (1) CGD neutrophils do not produce ROS upon stimulation with PVL (Figure 2B), but make NETs (Figure 2C-D) and (2) DPI, an inhibitor of flavin-containing proteins such as NADPH-oxidase, xanthine oxidase [61], and cytochrome C [62,66], the ROS scavenger pyrocatechol, and the MPO inhibitor ABAH all fail to inhibit PVL-induced NET formation (Figure 2E-H).
A recent study by Mazzoleni et al. suggested that ROS derived from mitochondria or xanthine oxidase are involved in PVL-induced NETosis [49]. We observed that mitochondrial uncoupling by DNP or FCCP as well as xanthine oxidase inhibition by allopurinol partly inhibited PVL-induced NET formation (Supplementary Figure 2A-C). Xanthine oxidoreductase consists of two isoforms, xanthine oxidase and xanthine dehydrogenase. Allopurinol inhibits both isoforms, which affects purine metabolism. Moreover, allopurinol itself generates superoxide radicals upon inhibition of xanthine oxidase [67]. Therefore its inhibitory effect on PVL-induced NET formation may be independent from xanthine oxidase inhibition or ROS formation. How allopurinol exerts its inhibitory effects on PVL-induced NET formation is still unclear. Furthermore, given that both DPI and the ROS scavenger pyrocatechol were unable to inhibit PVL-induced NET formation (Figure 2E-H), we hypothesize that the inhibitory effect of allopurinol and FCCP is independent from their effects on ROS production. Notably, inhibition of NET formation by these inhibitors was seen at 1 nM PVL, but was lost when stimulating with 10 nM PVL. It may be that PVL at high concentrations is able to induce NET formation without the involvement of neutrophil components interrogated by the inhibitors used in our study, whilst at low PVL concentrations some components may contribute to NET formation.
Neutrophil proteases were not essential for PVL-induced NET formation, although we did observe that formed NETs appeared smaller upon protease inhibition (Figure 2H). Neutrophil elastase is known to contribute to chromatin decondensation during NADPH-oxidase dependent NETosis [44,68] and this decondensation may occur during PVL-mediated NET formation as well (Supplementary Videos S3-4).
Interestingly, different NETosis mechanisms lead to NETs with specific compositions. We find differences in protein abundances of NETs induced by PVL, PMA, or nigericin, and that specifically PVL NETs appear to be less distinct from unstimulated neutrophils (Figure 3A). Several antimicrobial proteins are less abundant in PVL-induced NETs, when compared to PMA- or nigericin-induced NETs (Figure 3D-E). In contrast, cytoskeletal proteins are more abundant on PVL NETs. A previous report also identified a similar enrichment of cytoskeletal proteins in spontaneously lysed neutrophils [69]. Given PMA or nigericin-induced NET formation involves an intracellular release of granular components [68], this ‘mixing-of-the-bag’ may allow for efficient tethering of granular components to the chromatin backbone and degradation of the cytoskeleton. Cell lysis occurs 2-3 h after stimulation with PMA (Figure 2C and Supplementary Videos S1-S8), while PVL-induced NET formation occurs swiftly, with cell lysis occurring within the first 2 h after stimulation. We speculate that granular components may be lost to the extracellular environment during this rapid lysis, resulting in a less bactericidal NET (Figure 4A). These data suggest that NETs induced by different stimuli may display different functional characteristics. Leukocidins such as PVL may have evolved to elicit a harmless form of NET formation to promote survival of the invading bacteria.
NETs have at least three described functions, they are antimicrobial [42], procoagulant [70], and activate the immune system [71,72]. PVL-SA infections may be associated with a higher risk for thrombotic events. In addition to being less antimicrobial, it would be interesting to investigate if PVL-induced NETs also differ in their procoagulant or immune activation functions.
In conclusion, our observation of (a) differentially expressed PVL receptors in individuals with recurrent or severe PVL-SA infections and (b) functionally different NET formation after neutrophil exposure to PVL in contrast to other stimuli may explain specific clinical features and interindividual differences in PVL-SA infections. We propose that overexpression of PVL receptors makes a PVL-SA infected patient more prone to this disarmed NET formation, preventing efficient clearance of the invading bacteria. In turn, this could provide a competitive advantage to PVL-SA, facilitating colonization and recurrent infections. Further investigations are necessary to verify this finding and find potential treatment or prevention strategies for these infections.
Author Contributions
HJ, AZ, RK, and GM designed the study
HJ, DC, and GM performed experiments and analyzed the data
HVB and RK cared for the patients
AL, LGH, RL, JTS, MSS, MS, and HVB have interpreted the data and revised the manuscript for important intellectual content
CJH analyzed the mass spectrometry data
HJ, AZ, RK, and GM wrote the manuscript
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
We thank Christian Frese from the Proteomics Research Platform at the Max Planck Unit for the Science of Pathogens for proteomic analysis.
We thank Alf Herzig for visualizing PVL-induced NET formation in healthy and CGD neutrophils in real time by confocal microscopy.
List of abbreviations
- CFU
- Colony forming unit
- CGD
- Chronic granulomatous disease
- DAP
- Differentially abundant proteins
- DAPI
- 4’,6-diamidino-2-phenylindole
- DHR
- Dihydrorhodamine
- DNP
- Dinitrophenol
- DPBS
- Dulbeccos’s phosphate-buffered saline
- DPI
- Diphenyleneiodonium chloride
- FCCP
- Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- FDR
- False discovery rate
- HCD
- Higher-energy collisional dissociation
- HEPES
- Hydroxyethyl-Piperazine-Ethane Sulfonic Acid
- Hlg
- γ-Hemolysin
- HAS
- Human serum albumin
- MFI
- Mean fluorescent intensities
- MPO
- Myeloperoxidase
- MS
- Mass spectrometry
- NE
- Neutrophil elastase
- NEi
- Neutrophil elastase inhibitor
- NETs
- Neutrophil extracellular traps
- PBS
- Phosphate-buffered saline
- PFA
- Paraformaldehyde
- PKC
- Protein Kinase C
- PMA
- Phorbol 12-myristate 13-acetate
- PMN
- Polymorphonuclear leukocytes
- PSMs
- Peptide-spectrum-matches
- PVL
- Panton-Valentine leukocidin
- PVL-SA
- PVL-positive S. aureus
- ROS
- Reactive oxygen species
- RT
- Room temperature
- S. aureus
- Staphylococcus aureus
- SDS
- Sodium Dodecyl Sulfate
- SSTI
- Skin and soft tissue infections
- TCEP
- Tris-(2-carboxyethyl)phosphine
- TFA
- Trifluoroacetic acid
- TMM
- Trimmed mean of M values
- TMT
- Tandem Mass Tag
References




