

Abstract
Peripheral neuropathy is a frequent complication of type 2 diabetes mellitus (T2DM), of which the pathogenesis is not fully understood. We investigated whether human islet amyloid polypeptide (hIAPP), which forms pathogenic aggregates that damage islet β-cells in T2DM, is involved in T2DM-associated peripheral neuropathy. In vitro, hIAPP incubation with sensory neurons reduced neurite outgrowth. Transgenic hIAPP Ob/Ob mice, an established animal model for T2DM, as well as hIAPP mice, which have elevated plasma hIAPP levels but no hyperglycaemia. Both transgenic mice developed peripheral neuropathy as evidenced by pain-associated behavior and reduced intra-epidermal nerve fibers (IENF), suggesting hIAPP is a mediator of diabetic neuropathy. Intraplantar and intravenous hIAPP injection in WT mice induced long-lasting mechanical hypersensitivity and reduced IENF, whereas non-aggregating murine IAPP or mutated hIAPP (Pramlintide) did not have these effects, and were not toxic for cultured sensory neurons. In T2DM patients, significantly more hIAPP oligomers were found in the skin compared to non-T2DM controls. Thus, we provide evidence that hIAPP is toxic to sensory neurons, and mediates peripheral neuropathy in mice. The presence of hIAPP aggregates in skin of humans with T2DM supports the notion that human IAPP is a potential driver of T2DM neuropathy in man.
Introduction
Type 2 diabetes mellitus (T2DM) is the most common type of DM, and is characterised by insulin resistance and β-cell dysfunction (1). Diabetic peripheral neuropathy (DPN) is a frequent, debilitating complication of DM, affecting ∼50% of T2DM patients (2). The pathogenesis of this complication is not fully understood as control of hyperglycaemia is not sufficient to attenuate DPN. Moreover, neuropathy can be present in the pre-diabetic state, i.e. when hyperglycaemia has not yet developed. Thus, apparently hyperglycaemia is not the only cause of DPN (3).
Pancreatic islet amyloid is a characteristic histopathological feature of T2DM, found in 90% of T2DM patients. Human islet amyloid polypeptide (hIAPP) is the main component of these amyloid deposits. IAPP, or amylin, is a 37 amino acids polypeptide hormone that belongs to the calcitonin gene-related peptide (CGRP) family. It is co-secreted with insulin by the pancreatic islets β-cells. As a monomer, hIAPP is a soluble protein that controls blood glucose levels by enhancing satiety, and reducing gastric emptying and glucagon release (4). In T2DM, hIAPP is produced at large quantities. At high concentrations hIAPP forms toxic aggregates (oligomers and amyloid fibrils), leading to β-cell death and possibly damage in other tissues (5). In contrast, mouse IAPP (mIAPP) has a different amino acid sequence that prevents β-sheet formation and formation of toxic aggregates (6). hIAPP amyloid deposits are not exclusively found in the pancreatic islets of T2DM patients, but can also occur in other organs/tissues such as brain, heart and kidney (7).
Considering that peripheral neuropathy is a common complication of amyloid diseases such as familial amyloid polyneuropathy (FAP) (7), that T2DM is an amyloid disease (4, 7), and that hIAPP aggregation is not restricted to the pancreas, we investigated whether hIAPP is involved in the development of DPN.
Results and Discussion
To investigate whether hIAPP has direct neurotoxic effects on primary sensory neurons in vitro, we treated cultured mouse sensory neurons with various concentrations of hIAPP (0.1-1000 nM) for 24h. At concentrations of 100 and 1000 nM hIAPP, neurite outgrowth was reduced by 12% compared to vehicle (Fig. 1A). These results are in agreement with other amyloid neuropathy diseases such as FAP, where mutant amyloidogenic transthyretin (TTR) amyloid inhibits the neurite outgrowth of sensory neurons (8).

(A) Sensory neurons were cultured and treated for 24h with different concentrations of hIAPP. The average neurite length/neuron was assessed and expressed as the percentage of the average neurite length/neuron of vehicle-treated neurons (n=8; n represents a DRG culture of one mouse). (B) Non-fasting blood glucose levels in wild type (WT; n=8) and hIAPP Ob/Ob (n=5-8) mice. (C) Mechanical threshold of the plantar surface of WT (n=8) and hIAPP Ob/Ob (n=7) mice measured with the Von Frey test. (D) Number of nerve fibers crossing from dermis to epidermis in WT and hIAPP Ob/Ob mice at an age of 15 weeks. (E) Representative images of paw skin of these mice stained for the pan neuronal marker PGP 9.5 and collagen IV (Scale bar: 20 μm). (A) One-way ANOVA with Sidak’s test, **p < 0.01. (B,C) 2-way ANOVA with Sidak’s test,***p < 0.001. (D) unpaired t test, *p < 0.05. (B-D) experiments were performed in male mice.
To investigate whether hIAPP drives T2DM neuropathy in vivo, we used a hIAPP transgenic mouse model of T2DM (hIAPP Ob/Ob). These obese mice become insulin resistant and hyperglycaemic, and develop pathogenic amyloid deposits in pancreatic islets due to overproduction of hIAPP by the β-cells (4, 9). hIAPP Ob/Ob mice have significantly elevated blood glucose levels from 7 weeks until at least 14 weeks of age (Fig. 1B). To test if hIAPP Ob/Ob mice develop pain-associated behavior such as allodynia (mechanical hypersensitivity), we used the Von Frey test, which measures the withdrawal response to light mechanical stimulation. hIAPP Ob/Ob mice had allodynia from 7 weeks of age until at least 14 weeks, as compared to WT mice (Fig. 1C). Moreover, hIAPP Ob/Ob mice of 15 weeks of age also had reduced intra-epidermal nerve fibers (IENF) in the plantar skin compared to WT mice of the same age (Fig. 1D/E). Thus, hIAPP Ob/Ob mice not only have the metabolic characteristics of T2DM, but also suffer from DPN.
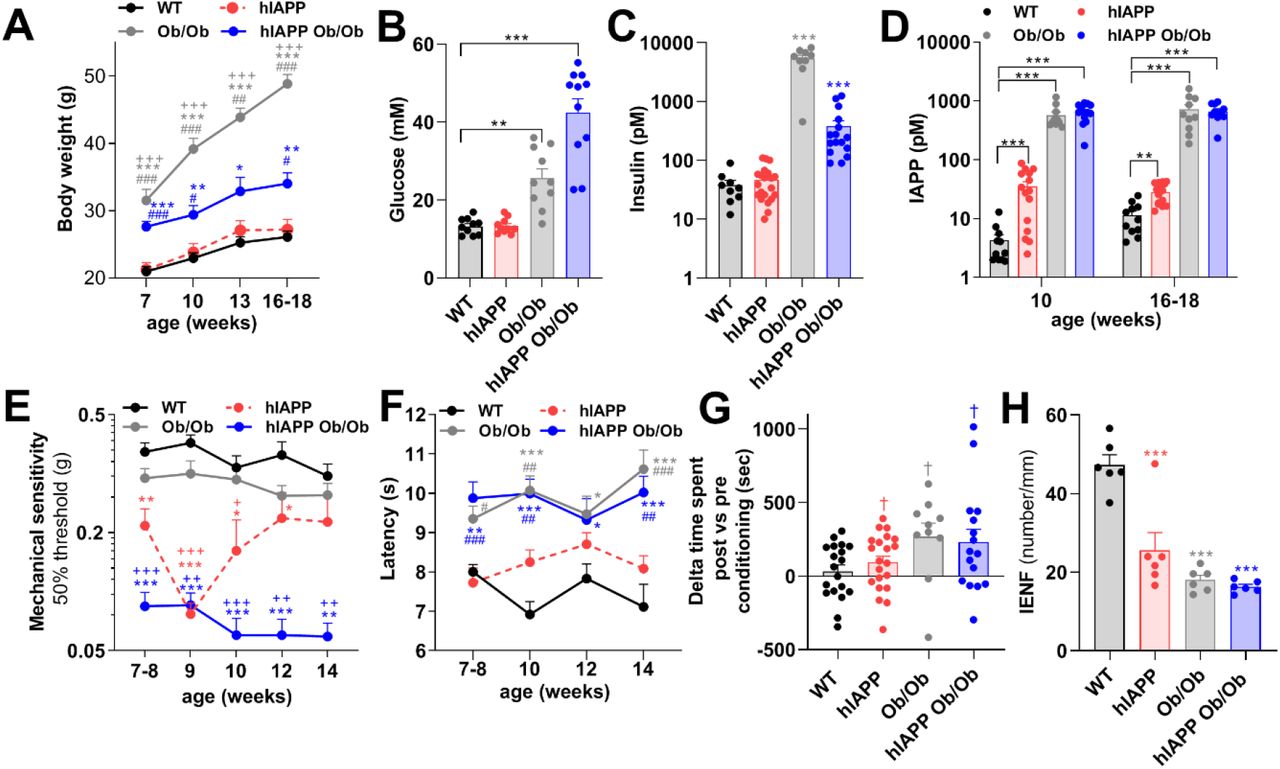
hIAPP Ob/Ob mice have three characteristics that may contribute to the development of DPN, i.e. hyperglycaemia, obesity and production of amyloidogenic IAPP (3, 7). To assess whether hIAPP independently of hyperglycaemia or obesity induces neuropathy, we also measured pain-associated behaviors in non-obese hIAPP mice and in non-transgenic obese mice (Ob/Ob). Notably, Ob/Ob and hIAPP Ob/Ob mice have elevated body weight and increased plasma glucose levels, whereas these parameters are normal in hIAPP mice and comparable to those of WT mice (Fig. 2A/B). Furthermore, plasma insulin levels are elevated in hIAPP Ob/Ob and Ob/Ob mice, but normal in hIAPP mice (Fig. 2C). Thus, hIAPP mice, in contrast to Ob/Ob and hIAPP Ob/Ob mice, do not have diabetes mellitus. However, hIAPP mice, just like hIAPP Ob/Ob mice, have increased plasma levels of IAPP (mIAPP plus hIAPP). Ob/Ob mice also have increased plasma levels of IAPP (but only mIAPP), compared to WT mice at the age of 10 weeks and at ∼4 months of age (Fig. 2D). Male and female mice did not differ in these parameters (Fig. S1 A-C). Thus, hIAPP mice provide a model to study the effects of hIAPP independently of hyperglycaemia. Intriguingly, hIAPP mice, similar to hIAPP Ob/Ob mice, have lower mechanical thresholds in the Von Frey test as compared to WT mice from age 7 weeks onwards (Fig. 2E). Ob/Ob mice also showed mechanical hypersensitivity with this test compared to WT mice, but this allodynia was less severe than the results observed in hIAPP mice at 9/10 weeks of age, and hIAPP Ob/Ob mice from 7-14 weeks. Female hIAPP mice had slightly stronger allodynia than males (Fig. S1D). To further explore sensory deficits we also assessed sensitivity of the mice to heat stimuli, by measuring the latency time of paw withdrawal upon heat stimulation. Ob/Ob and hIAPP Ob/Ob mice had an increased latency in this test as compared to WT mice (Fig. 2F), indicating thermal hyposensitivity. In contrast, hIAPP mice had a normal response to heat stimulation, comparable to that of WT mice (Fig. 2F).
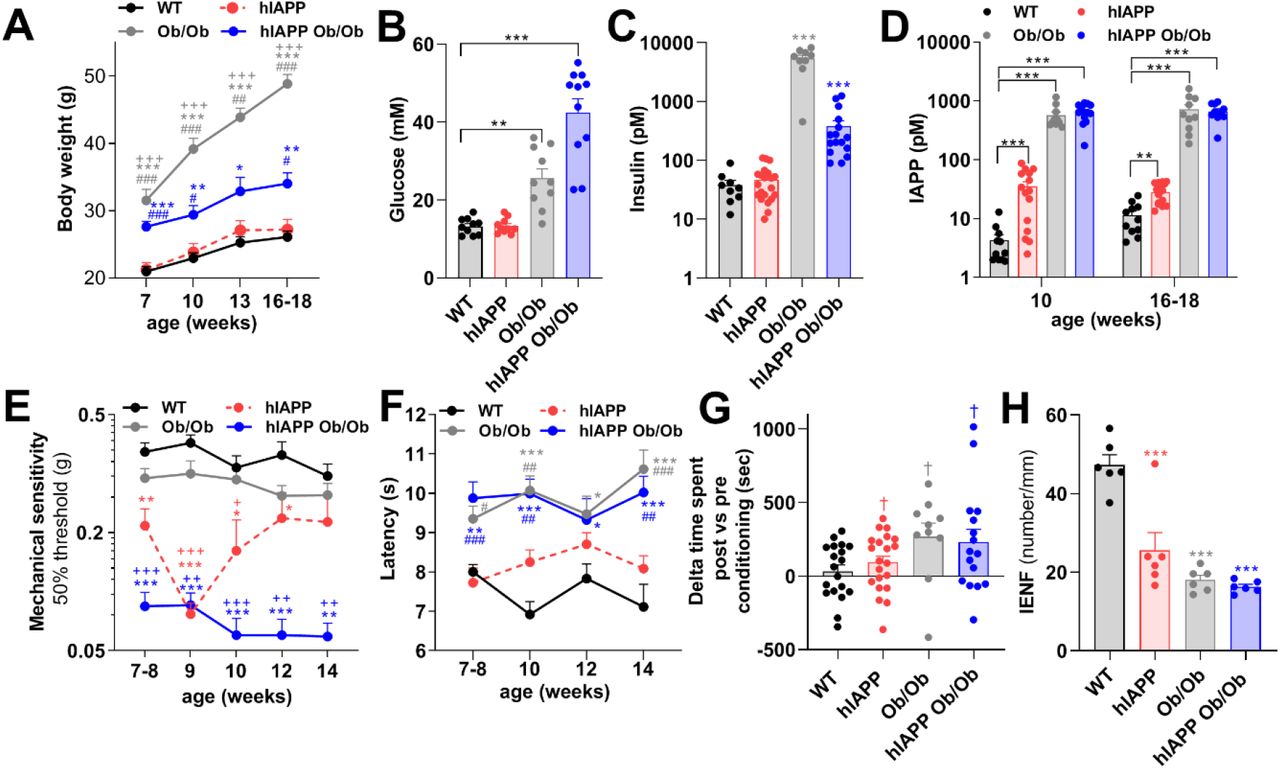
(A) Body weight of WT (n=12), hIAPP (n=15), Ob/Ob (n=10), and hIAPP Ob/Ob (n=15) mice. (B) Non-fasting plasma glucose level, (C) Non-fasting plasma insulin level, (B/C; mice age: 16-18 weeks), and (D) Non-fasting plasma IAPP levels in WT, hIAPP, Ob/Ob and hIAPP Ob/Ob mice. (E) Mechanical threshold of WT (n=11), hIAPP (n=23), Ob/Ob (n=21), hIAPP Ob/Ob (n=14) mice. (F) Thermal sensitivity of WT (n=11), hIAPP (n=15), Ob/Ob (n= 12) and hIAPP Ob/Ob (n=10) mice. (G) Conditioned place preference to reveal the presence of pain. Mice were conditioned with gabapentin for 3 consecutive days and time spent in the conditioning compartment between the post and pre-conditioning phases was determined in WT, hIAPP, Ob/Ob and hIAPP Ob/Ob mice at 15-17 weeks of age. (H) Quantification of IENF of the hind paw of mice at 16-18 weeks of age. All experiments were performed with male and female mice. (A, D-F, H) Two-way ANOVA with Tukey’s test; *p < 0.05,**p < 0.01, ***p < 0.001 vs WT; +p < 0.05, + +p < 0.01, +++p < 0.001 vs Ob/Ob mice,, #p < 0.05, ##p < 0.01, ###p < 0.001 vs hIAPP. (B-C) One-way ANOVA with Tukey’s test; **p < 0.01,***p < 0.001. (G) one sample t test, †p<0.05.
To assess whether any of the mice had spontaneous pain, mice of ∼4 months were conditioned with the neuropathic painkiller gabapentin according to a conditioned place preference paradigm (10). Male and female Ob/Ob, hIAPP Ob/Ob and hIAPP mice, but not WT mice, had a place preference after conditioning with gabapentin compared to preconditioning (Fig. 2G), indicating spontaneous pain.
Peripheral neuropathy may lead to loss of IENF. To evaluate whether nerve fibers in the plantar skin of the hind paws were affected in mice of the different genotypes, skin samples were stained for the PGP9.5 marker. Interestingly, the number of IENF was reduced in male and female hIAPP mice, hIAPP Ob/Ob, and Ob/Ob mice compared to WT mice (Fig. 2H). Overall, these data indicate that human IAPP, in the absence of hyperglycaemia and obesity, is sufficient to induce signs of peripheral neuropathy, though some signs of neuropathy are more pronounced in hIAPP mice when they are hyperglycaemic (hIAPP Ob/Ob).
Peripheral and central administration of IAPP affects the sensory nervous system in rodents, and causes endothelial dysfunction, vessel wall disruption, and neurological deficits (11). To test whether hIAPP administration to WT mice is sufficient to induce pain-associated behaviors, hIAPP (40 μg/kg and 4 μg/kg) was injected intravenously in WT mice. A single intravenous hIAPP injection at 40 μg/kg, but not at 4 μg/kg, reduced mechanical thresholds for 4 days (Fig. 3A). Intravenous hIAPP injection did not affect thermal sensitivity (Fig. S2A). The hIAPP dose used results in maximal plasma levels of hIAPP that are approximately 1000 times higher than IAPP plasma concentrations in hIAPP mice (up to 100 pM) and/or humans with T2DM (up to 42 pM) (12). Since hIAPP has a very short half-life (few minutes) (13), these data indicate that a short but strong rise in systemic hIAPP concentration is sufficient to cause long-lasting changes in peripheral sensory neurons.
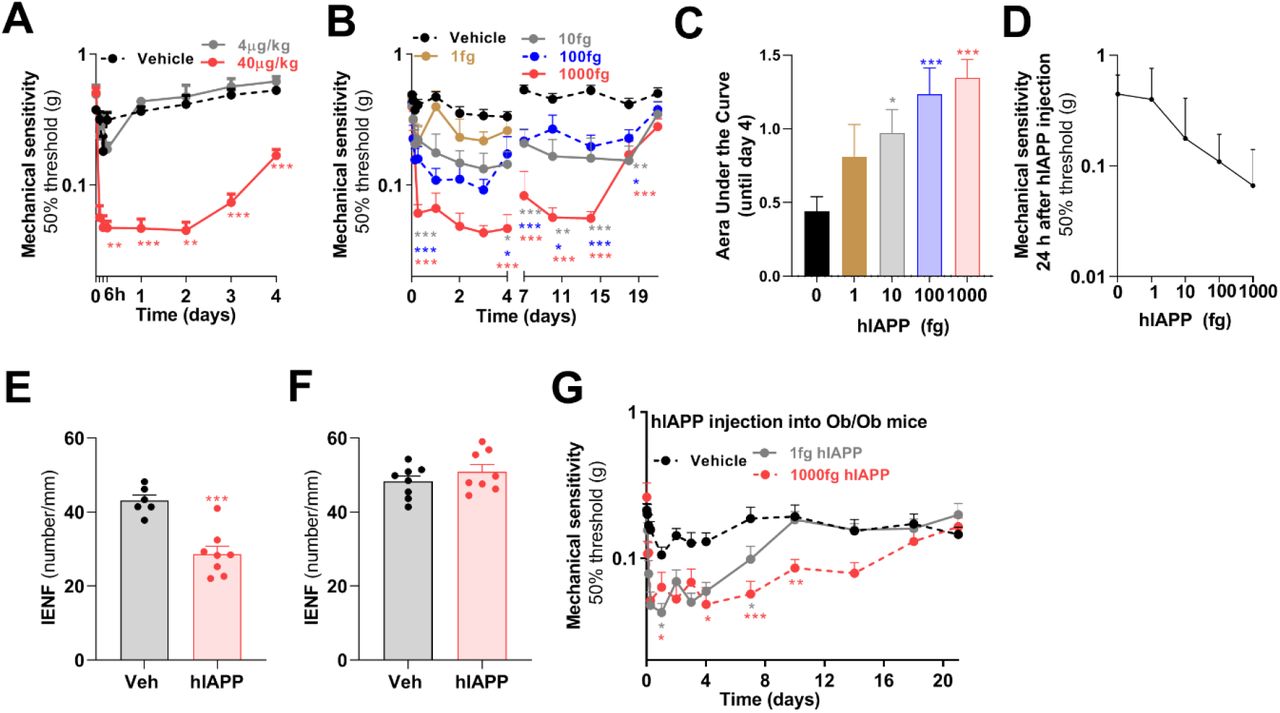
Time course of mechanical sensitivity of the hind paw after (A) intravenous injection of hIAPP (40 μg/kg, n=7 and 4 μg /kg, n=3) or saline (n=6) into WT male mice. (B) Mechanical sensitivity of the hind paw after intraplantar injection of hIAPP (1 fg, n=8; 10 fg, n=11; 100 fg, n=11; 1000 fg, n=13 or saline (n=20) into male and female WT mice. (C) Area under the curve of the reduction in mechanical threshold compare to baseline (day 0), between day 0 until day 4 after intraplantar hIAPP injection of data shown in E. (D) Dose response curve of hIAPP-induced reduction in mechanical threshold measured at 1 day after intraplantar hIAPP injection. (E) Quantification of IENF in the plantar skin of the hind paw at 6 days after intraplantar injection of 1000 fg hIAPP or saline, or (F) at 27 days after hIAPP injection (1000 fg) when hypersensitivity had resolved, n=8. (G) Mechanical sensitivity of the hind paw after intraplantar injection of hIAPP (1 fg, n=17; 1000 fg, n=12) or saline (n=17) into male and female Ob/Ob mice. (A,B,G) Two-way ANOVA with Dunnett’s test; *p < 0.05, **p < 0.01,***p < 0.001. (C) One-way ANOVA with Dunnett’s test; *p < 0.05,***p < 0.001. (E,F) Unpaired t test; ***p<0.001.
Next we tested whether local injection of hIAPP into the hind paw of WT mice could induce local signs of neuropathy. Intraplantar injection of hIAPP dose dependently reduced mechanical thresholds to Von Frey stimulation compared to vehicle injection (Fig. 3B-D). A single 5 μl injection with 1000 fg (∼50 pM), a concentration in the same range as found in blood of hIAPP mice and T2DM patients (12), reduced mechanical thresholds that persisted for at least 2 weeks (Fig. 3B), indicating sensory dysfunction. At 3 weeks after intraplantar hIAPP injection, mechanical hypersensitivity had resolved. Intraplantar injection of 1000 fg hIAPP reduced the number of IENF compared to vehicle injection at 6 days after injection (Fig. 3E), a time point were mechanical hypersensitivity was still present (Fig. S2B). One week after hIAPP-induced hypersensitivity had resolved (Fig. S2C), plantar skin IENF was indistinguishable from vehicle injected mice (Fig. 3F). Thus, the nerve fibers recover concurrent with the resolution of mechanical hypersensitivity. Because protein oligomers or aggregates may induce hypersensitivity through inducing inflammation, we evaluated expression of inflammatory cytokines in skin of the injected paws. Intraplantar injection of hIAPP did not trigger expression of interleukin 6 (IL-6), IL-1β and TNF mRNA, but increased F4/80 mRNA expression, a marker for macrophages (Fig. S2D).
Hyperglycaemia increases IAPP transcript levels and IAPP biosynthesis in pancreatic islet β-cells in human and rodents (14). Although hyperglycaemia may not be the only driver of DPN (3), it may predispose to IAPP-induced hypersensitivity because hIAPP-induced cell toxicity is exacerbated by hyperglycaemia-induced glycated insulin (15). Therefore we tested whether hyperglycaemic Ob/Ob mice are more sensitive to hIAPP-induced allodynia as compared to WT mice. Ob/Ob mice were injected intraplantar with 1 fg hIAPP, a dose that did not induce allodynia in WT mice (Fig. 3B). Intriguingly, 1 fg hIAPP increased mechanical sensitivity in Ob/Ob mice for almost 7 days (Fig. 3G). Intraplantar injection of 1000 fg hIAPP into obese mice increased mechanical sensitivity that lasted for at least 2 weeks (Fig. 3G). These data indicate that hIAPP induces allodynia in Ob/Ob mice at a dose that did not affect mechanical sensitivity in normoglycaemic WT mice, suggesting that hyperglycaemia and/or obesity aggravates hIAPP-induced allodynia. These findings possibly explain why hIAPP Ob/Ob mice have higher mechanical sensitivity than non-obese hIAPP mice.
hIAPP has 43% amino acid sequence identity with hCGRP and binds to the same IAPP/CGRP receptors (16). Therefore, hIAPP may also induce effects through direct actions on IAPP/CGRP receptors. In contrast to CGRP, hIAPP is toxic for rat insulinoma cells, hippocampal neurons and astrocytes in vitro, independent of receptors (17, 18). To evaluate whether hIAPP-induced allodynia requires IAPP/CGRP receptors, we blocked these receptors by combined intraperitoneal (i.p.) and intraplantar (i.pl.) injections of the IAPP/CGRP receptor antagonist CGRP 8-37 (ip.: 250 μg/kg; i.pl: 5 μg/5 μl) prior to intraplantar injection of hIAPP in WT mice (Fig. 4A). CGRP 8-37 completely blocked the development of CGRP-induced allodynia, in agreement with the involvement of IAPP/CGRP receptors (19). In contrast, the same dosing schedule of CGRP 8-37 did not block hIAPP-induced allodynia (Fig. 4B).
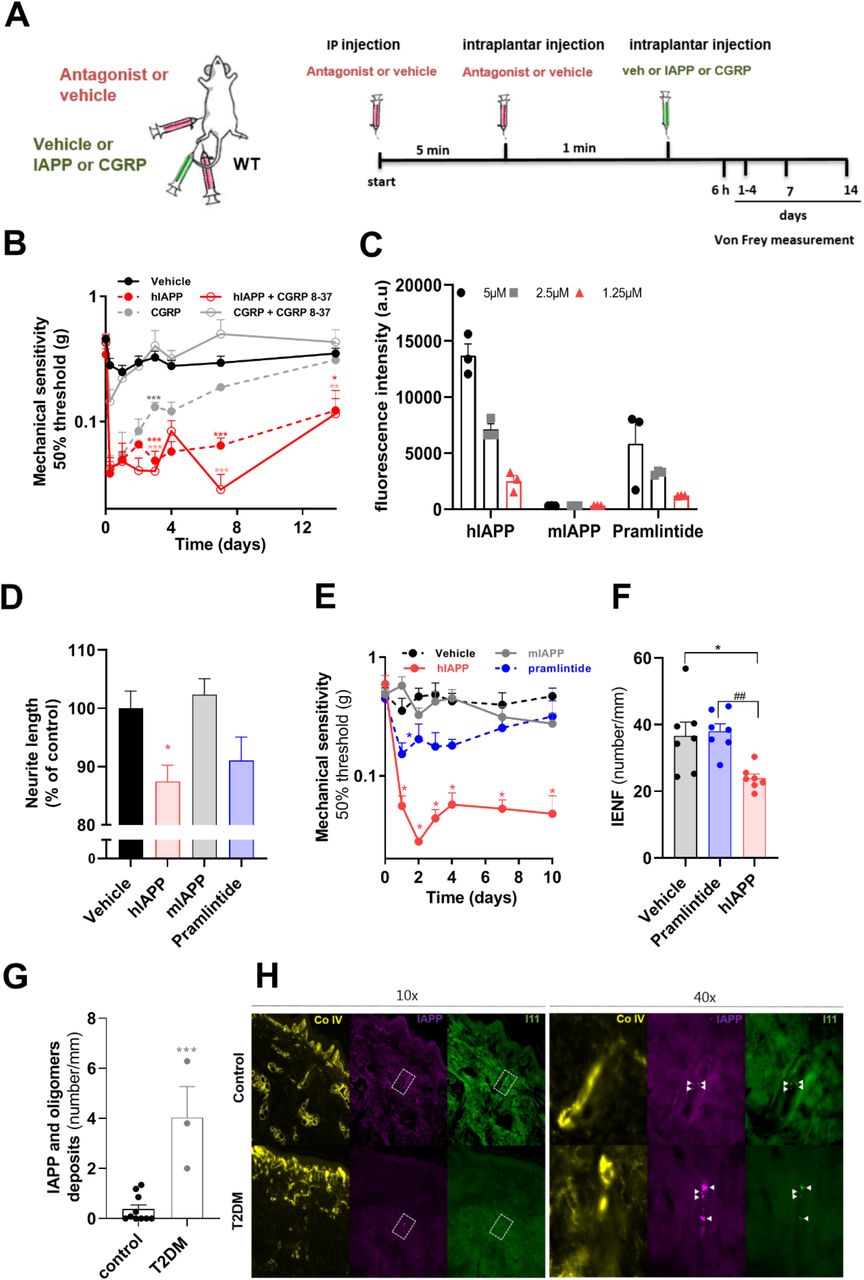
(A) Schematic diagram showing the timeline and administration routes of hIAPP, CGRP and hIAPP/CGRP antagonist. (B) Mechanical sensitivity of the hind paw of WT mice after antagonist (250μg/kg, CGRP 8-37) injection (n=13) or saline injection (n=13) prior to intraplantar injection of hIAPP (1000 fg, n=8) or CGRP injection (5 μg, n=5) in one paw and saline (n=13) in the other paw. Both male and female mice were used. Two-way ANOVA with Tukey’s test,*P< 0.05, ,**p < 0.01 ,***p < 0.001 vs vehicle. (C) Fluorescence of thioflavin T after 24h. Three concentrations (5, 2.5 and 1.25µM) of hIAPP, mIAPP and Pramlintide were tested. Fluorescence intensity indicates amount of amyloid fibrils. (D) Sensory neurons were treated with 100 nM hIAPP, mIAPP and Pramlintide for 24 hours. The average neurite length/neuron was assessed and expressed as the percentage length/neuron of vehicle-treated neurons (n=8; n represents a DRG culture of one mouse). One-way ANOVA with Dunnett’s test, *p < 0.05. (E) Mechanical sensitivity of the hind paw after intraplantar injection of 1000 fg of hIAPP, mIAPP and Pramlintide or saline in WT male mice, n=5; two-way ANOVA with Tukey’s test; *p < 0.05 vs vehicle. (F) Quantification of IENF at day 6 after intraplantar injection of 1000 fg hIAPP, Pramlintide, or saline in male and female WT mice; One-way ANOVA with Tukey’s test; *p < 0.05 vs vehicle; ##p < 0.01 vs Pramlintide). (G) Number of IAPP oligomer deposits in the dermis and sub-dermis of T2DM subjects and none-T2DM controls; Unpaired t test; ***p<0.001. (H) Representative images of the collagen IV, IAPP and oligomer staining (I11) in skin of non-T2DM (controls) and T2DM, IAPP and oligomer –positive spots are indicated by the arrowheads.
To investigate whether aggregation of IAPP is required for its neurotoxic effects we tested mouse IAPP (non-aggregating and non-amyloidogenic) and a mutant human IAPP (Pramlintide, non-amyloidogenic) in vitro. Both of these non-amyloidogenic IAPP variants activate IAPP/CGRP receptor subtypes (20). To verify their non-amyloidogenic nature, we measured amyloid fibril formation in solution using Thioflavin T fluorescence at various concentrations (1.25-5µM). These data show that mIAPP indeed does not form amyloid fibrils, whereas Pramlintide only showed minor fibril formation at the highest concentration (Fig. 4C). In contrast, hIAPP formed amyloid fibrils in solution at all concentrations tested (Fig. 4C). Treatment of cultured sensory neurons with mIAPP or Pramlintide (100 nM) did not affect neurite outgrowth in vitro, whilst amyloidogenic hIAPP reduced neurite length by 13% compared to vehicle control (Fig. 4D). In vivo, intraplantar injection of mIAPP (1000 fg) into WT mice did not affect mechanical thresholds (Fig. 4E), whilst the same dose of hIAPP reduced mechanical thresholds for at least 10 days. Intraplantar injection of Pramlintide slightly reduced mechanical thresholds, that was significantly less in magnitude and duration than that induced by hIAPP (Fig. 4E). To determine whether IAPP aggregation is required for nerve damage in vivo, we determined the plantar IENF density 6 days after injection of 1000 fg Pramlintide or hIAPP in WT mice. Injection of Pramlintide did not affect IENF density whilst the same dose of hIAPP reduced the IENF density with ∼50% (Fig. 4F). These data indicate that the ability of IAPP to form aggregates/fibrils is required for its neurotoxicity in vitro (21), and to induce allodynia and reduced IENF density in vivo. In line with these findings, Pramlintide showed some fibril formation in vitro and induced some small neuropathic-like effects in vitro and in vivo in mice. Therefore, potential neuropathic effects of Pramlintide used in clinical practice warrants further investigation.
hIAPP can accumulate in nervous tissues, e.g in hippocampal neurons of hIAPP transgenic mice and rats, and form aggregates that are associated with neurological deficits (22). hIAPP oligomers destabilize cell membranes and form pores that disappear upon formation of hIAPP mature fibrils (7, 23). In T2DM patients, small nerve fibre pathology is observed early in the development of painful DPN, sometimes even prior to presence of hyperglycaemia (24). To assess whether (more) hIAPP oligomers are present in the skin of T2DM patients with neuropathy compared to non-T2DM (Supplementary Table l), we stained skin from feet or hands for IAPP and oligomers using specific antibodies. The three T2DM patients with neuropathy had significantly more IAPP-positive oligomers in the skin compared to the 10 non-T2DM controls (Fig. 4G/H). These data suggest that potential toxic aggregates of hIAPP are present in skin of T2DM neuropathy patients.
Here we present evidence that hIAPP is a key player in the development of DPN. Diabetic and non-diabetic mice expressing hIAPP developed signs of neuropathy such as allodynia and nerve damage in the skin. Importantly, the aggregation of IAPP is required to induce these signs of neuropathy in WT mice. Thus in addition to hIAPP aggregation being associated with dysfunction and death of pancreatic β-cells in T2DM, hIAPP also damages peripheral sensory neurons, and contributes to neuropathy development. We found that hIAPP-positive oligomers are present in skin of T2DM patients, in agreement with earlier findings that IAPP oligomers are found outside of the pancreas. It remains to be determined whether IAPP aggregates in the skin and/or in other tissues potentially involved in DPN, such as axons and dorsal root ganglia, cause nerve damage and pain. How hIAPP mechanistically causes neuropathy remains unclear. However, hIAPP aggregation (with formation of oligomers and amyloid fibrils) can induce cytotoxicity though various mechanisms, including e.g. membrane disruption, impaired mitochondrial function and autophagy malfunction, that lead to cell damage in the pancreatic islets of Langerhans (23). Many of these cellular abnormalities have been implicated in the development/progression of DPN, as well as other forms of amyloid neuropathy. These pathogenic mechanisms may contribute to DPN not only by impacting neurons, but also other cell types involved in neuropathy, such as macrophages, microglia, Schwann cells and endothelial cells (7). Future studies are needed to determine which pathogenic pathways are involved in hIAPP-mediated DPN.
Until now there is no FDA approved therapy for DPN, other than the recommendation to improve glycaemic control and therapy to relieve symptoms (25). Thus, development of effective therapies for this common disease is urgently needed and blocking hIAPP (aggregation) is a novel candidate approach to treat and/or prevent DPN.
Methods
Experimental methods are detailed in the Supplementary Material.
Author Contributions
M.M.H.A., E.C.H, J.W.M.H. and N.E designed the research and performed the writing – review & editing of the paper; M.M.H.A. and S.V. performed the experiments; E.M.B and J.H.C provided human skin tissues, M.M.H.A. and S.V. analysed the data; J.W.M.H. and N.E. supervised the work.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
This work was financially supported by King Abdulaziz City for Science and Technology (KACST), Saudi Arabia. We thank Barend Elenbaas (University Utrecht, the Netherlands) for performing the thioflavin T assay and Dr. Rakez Kayed (University of Texas Medical Branch, Dept. Neurology, Galveston, USA) for providing the I11 antibody.





{kind=link}
{kind=link}
{kind=link}
{kind=link}