

Abstract
The SARS-CoV-2 B.1.1.529 variant (Omicron) contains 15 mutations on the receptor-binding domain (RBD). How Omicron would evade RBD neutralizing antibodies (NAbs) and humoral immunity requires immediate investigation. Here, we used high-throughput yeast display screening1,2 to determine the RBD escaping mutation profiles for 247 human anti-RBD NAbs identified from SARS-CoV/SARS-CoV-2 convalescents and vaccinees. Based on the results, NAbs could be unsupervised clustered into six epitope groups (A-F), which is highly concordant with knowledge-based structural classifications3–5. Strikingly, various single mutations of Omicron could impair NAbs of different epitope groups. Specifically, NAbs in Group A-D, whose epitope overlaps with ACE2-binding motif, are largely escaped by K417N, N440K, G446S, E484A, Q493K, and G496S. Group E (S309 site)6 and F (CR3022 site)7 NAbs, which often exhibit broad sarbecovirus neutralizing activity, are less affected by Omicron, but still, a subset of NAbs are escaped by G339D, S371L, and S375F. Furthermore, B.1.1.529 pseudovirus neutralization and RBD binding assay showed that single mutation tolerating NAbs could also be escaped due to multiple synergetic mutations on their epitopes. In total, over 85% of the tested NAbs are escaped by Omicron. Regarding NAb drugs, LY-CoV016/LY-CoV555 cocktail, REGN-CoV2 cocktail, AZD1061/AZD8895 cocktail, and BRII-196 were escaped by Omicron, while VIR7831 and DXP-604 still function at reduced efficacy. Together, data suggest Omicron could cause significant humoral immune evasion, while NAbs targeting the sarbecovirus conserved region remain most effective. Our results offer instructions for developing NAb drugs and vaccines against Omicron and future variants.
Main
The SARS-CoV-2 B.1.1.529 variant was first reported to the World Health Organization (WHO) on 24 November 2021. It appears to be rapidly spreading, and the WHO classified it as a variant of concern (VOC) only two days after, designating it as Omicron 8,9. An unusually large number of mutations are found in Omicron, including over 30 in the spike protein 10 (Fig. 1a). The receptor-binding domain, responsible for interacting with the ACE2 receptor, bears 15 of these mutations, including G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, and Y505H. Some of these mutations are very concerning due to their well-understood functional consequences, such as K417N and N501Y, which contribute to immune escape and higher infectivity 11–14. Many other mutations’ functional impacts remain to be investigated.
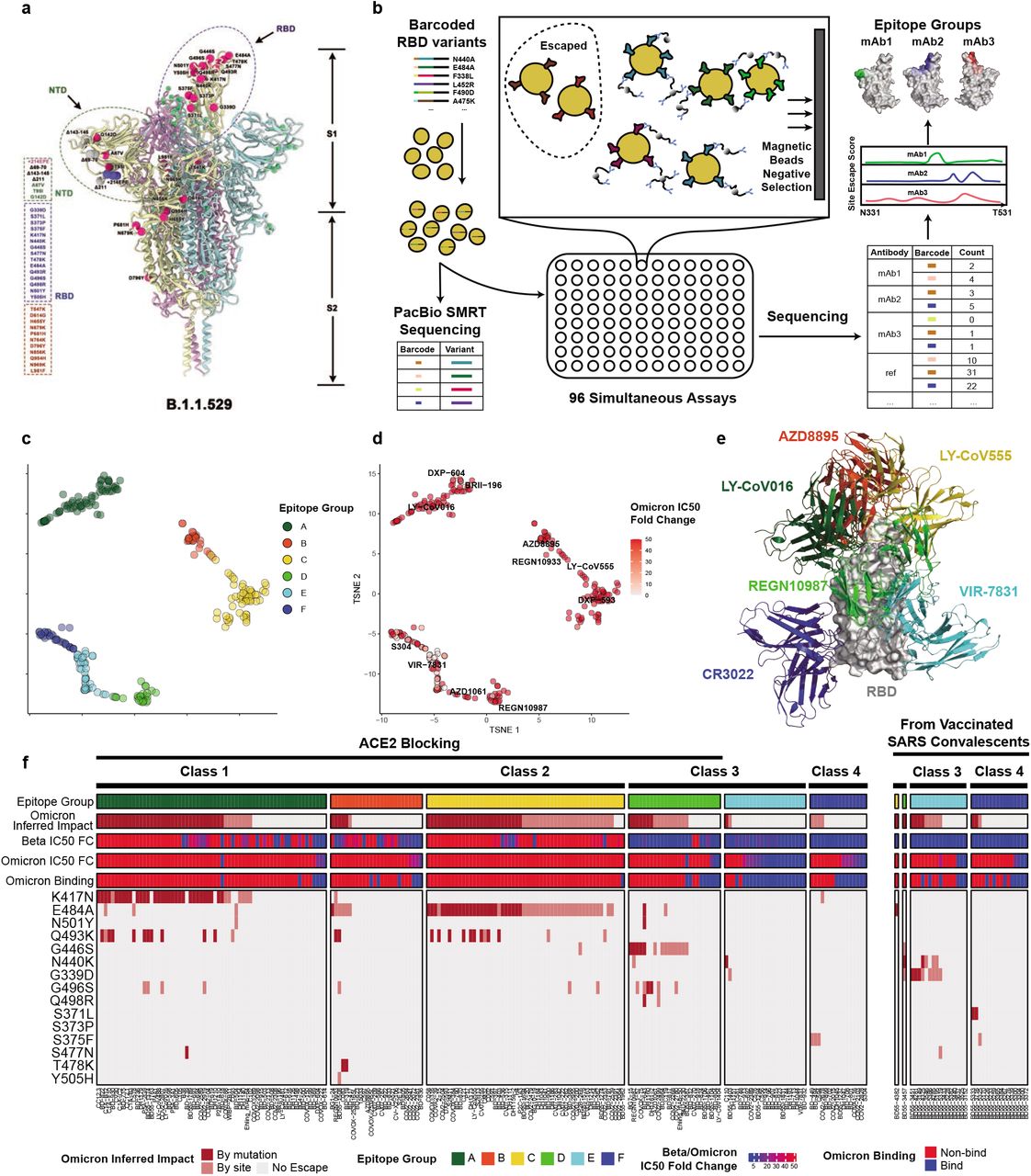
a) Illustration of B.1.1.529’s mutation location on the spike protein. b) Schematic of MACS-based high-throughput yeast display mutation screening. c) t-SNE embedding and unsupervised clustering of SARS-CoV-2 human NAbs based on each antibody escaping mutation profile. A total of 6 epitope groups (Group A-F) could be defined. d) B.1.1.529 pseudovirus neutralizing IC50 of each NAb. e) Representative NAb structures of each epitope group. f) The impact of each RBD mutation of B.1.1.529 on NAbs’ binding. Each NAb’s binding to B.1.1.529 RBD was validated through ELISA and BLI.
The S protein is the target of essentially all NAbs found in the convalescent sera or elicited by vaccines. Most of the N-terminal domain (NTD) neutralizing antibodies target an antigenic “supersite” in NTD, involving the N3 (residues 141 to 156) and N5 (residues 246 to 260) loops 15,16, and are thus very prone to NTD mutations. Omicron carries the Δ143-145 mutation, which would alter the N3 loop and most likely result in immune escape of most anti-NTD NAbs (Extended Data Fig. 1). Compared to NTD targeting NAbs, RBD targeting NAbs are particularly abundant and potent, and display diverse epitopes. Evaluating how Omicron affects the neutralization capability of anti-RBD NAbs of diverse classes and epitopes is urgently needed.

NTD-binding NAbs are shown together in different colors in complex with one NTD, and most of them contact with residues 142-145, which indicates their high probability to be escaped by G142D and Δ143-145 of Omicron. Missense mutations and deletions of Omicron NTD are colored blue and red, respectively.
B.1.1.529 escapes NAbs of diverse epitopes
RBD-directed SARS-CoV-2 NAbs can be assigned into different classes or binding sites based on structural analyses by cryo-EM or high-resolution crystallography 3–5,17; however, structural data only indicates the contacting amino acids, but does not infer the escaping mutations for a specific antibody. Recent advances in deep antigen mutation screening using FACS (fluorescence-activated cell sorting)-based yeast display platform has allowed the quick mapping of all single amino acid mutations in the RBD that affect the binding of SARS-CoV-2 RBD NAbs 1,18. The method has proven highly effective in predicting NAB drug efficacy toward mutations 2. However, to study how human humoral immunity may react to highly mutated variants like B.1.1.529 requires mutation profiling of a large collection of NAbs targeting different regions of RBD, and FACS-based yeast display mutation screening is limited by low experimental throughput. Here we further developed a MACS (magnetic-activated cell sorting) -based screening method which increases the throughput near 100-fold and could obtain comparable data quality like FACS (Fig 1b; Extended Data Fig 2). Using this method, we quickly characterized the RBD escaping mutation profile for a total of 247 NAbs (Supplymentary Files 1-6). Half of the NAbs were part of the antibodies identified by us using single-cell VDJ sequencing of antigen-specific memory B cells from SARS-CoV-2 convalescents, SARS-CoV-2 vaccinees, and SARS-CoV convalescents who recently received SARS-CoV-2 vaccines. The other half of NAbs were identified by groups worldwide and usually have antibody-antigen structures 3,5,6,12,17,19–42 (Supplementary Files 7).

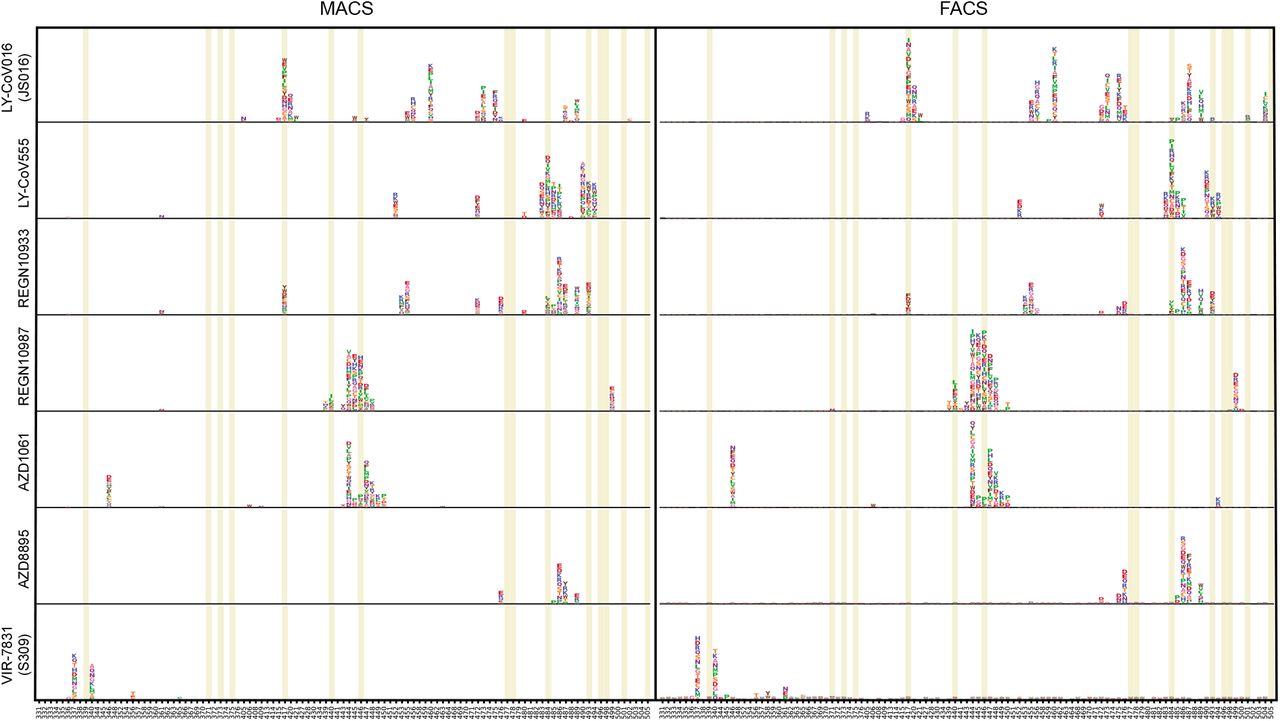
Deep mutational scanning maps with MACS-based (left) and FACS-based assays (right) of therapeutic neutralizing antibodies that have received emergency use authorization from the US FDA. Sites mutated in Omicron variant are highlighted.
The high-throughput screening capability allowed us to classify these NAbs into six Epitope Groups (A-F) using unsupervised clustering without dependence on structural studies, and the grouping is highly concordant with the knowledge-based structural classifications 3–5,17 (Fig. 1c and 1e). In particular, Group A-D NAbs largely correspond to the RBS A-D NAbs described by Yuan et al. 4, and their epitopes include RBD residues involved in binding to ACE2. Group A and B NAbs, represented by CB6/LY-CoV016 and AZD8895 respectively, usually can only bind to the ‘up’ RBD, whereas most of the Group C and D members, such as LY-CoV555 and REGN10987, bind to RBDs regardless of their ‘up’ and ‘down’ conformations. Group E and F NAbs target the S309/VIR-7831 site and CR3022 site, respectively, and neutralize SARS-CoV-2 using other mechanisms than directly interfering with ACE2 binding.
Inferred from the escaping mutation profiles, various single mutations of Omicron could impair NAbs of different epitope groups (Fig 1f). Specifically, NAbs in Group A-D, whose epitope overlaps with ACE2-binding motif, are largely escaped by single mutations of K417N, G446S, E484A, Q493K, and G496S. Also, a subset of NAbs of Group E and are escaped by single mutations of G339D, N440K, S371L, S375F. However, due to the extensive mutations accumulated on Omicron’s RBD, studying NAb’s response to Omicron only in the single mutation context is insufficient. Indeed, B.1.1.529 pseudovirus neutralization and Spike ELISA binding assay showed that single mutation tolerating NAbs could also be escaped by Omicron due to multiple synergetic mutations on their epitopes (Fig 1d and 1f). In total, over 85% of the tested human NAbs are escaped, suggesting that Omicron could cause significant humoral immune evasion.
ACE2-blocking NAbs are more vulnerable to B.1.1.529
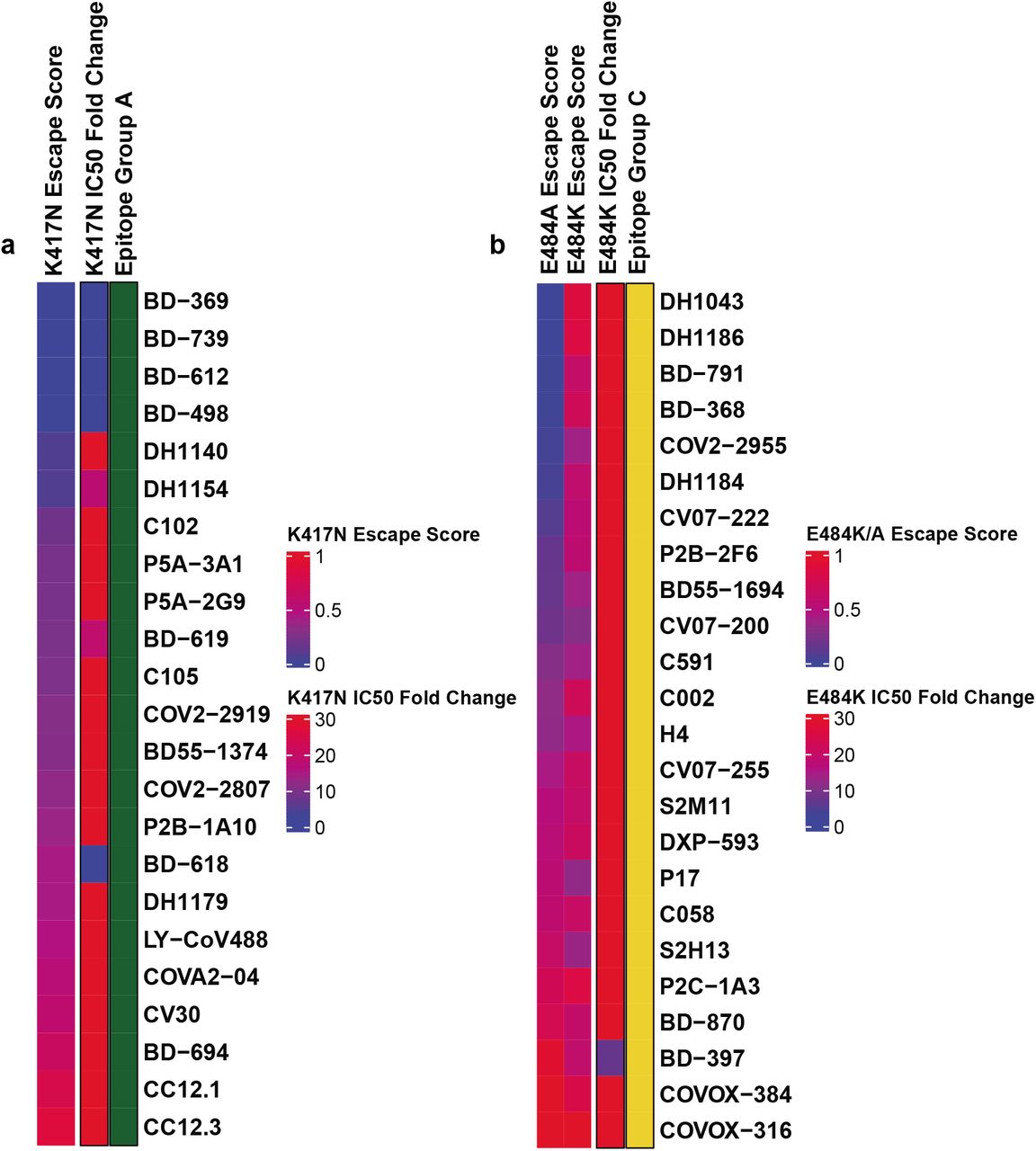
It is crucial to analyze how each group of NAbs react to Omicron to instruct the development of NAb drugs and vaccines. Group A NAbs mainly contains the VH3-53/VH3-66 germline gene-encoded antibodies, which are abundantly present in our current collection of SARS-CoV-2 neutralizing antibodies 19,23,24,28,43–45, including several antibodies that have obtained emergency use authorization (CB6/LY-CoV016) 21 or are currently being studied in clinical trials (P2C-1F11/BRII-196, BD-604/DXP-604) 20,46 (Fig. 2a, e). Group A NAbs often exhibit less somatic mutations and shorter CDR3 length compared to other groups (Extended Data 5a, b). The epitopes of these antibodies extensively overlap with the binding site of ACE2 and are often evaded by RBD mutations on K417, D420, F456, A475, L455 sites (Fig 2i, Extended Data Fig 3a). Most NAbs in Group A were already escaped by B.1.351 (Beta) strain, specifically K417N (Extended Data Fig 7a), due to a critical salt bridge interaction between Lys417 and a negatively charged residue in the antibody (Fig. 2i, m). The NAbs that survived Beta strain, such as BRII-196 and DXP-604, are insensitive to the K417N single site change but could also be heavily affected by the combination of K417N and other RBD mutations located on their epitopes, like S477N, Q493R, G496S, Q498R, N501Y, and Y505H of Omicron, causing lost or reduction of neutralization (Fig 2i; Extended Data Fig 4a).

(a)-(f) correspond to aggregated site escape scores of epitope group A-F, respectively. Escape hotspots of each epitope group are annotated by arrows.

(a)-(f) correspond to aggregated footprints of epitope group A-F, respectively. Publicly availble structures of neutralizing antibodies in complex with SARS-CoV-2 RBD are gathered from PDB and classified into groups. Different colors distinguish epitope groups, and the darkness reflects group-specific site popularity to appear on the complex interface. Common interface residues of each group are marked by arrows.

a-d VDJ combination of antibodies’ heavy chain variable region and their mutation rate for group A-D, respectively. For each group, the upper semicircle represents IGHV gene distribution and the lower semicircles represent IGHJ gene distribution.
e-h Escape maps of represetative RBD NAbs for group A-D respectively. For each site, the height of a letter indicates the detected mutation escape score of its corresponding residue. Site mutated in Omicron (B.1.1.529) are highlighted.
i-l Heatmaps of site escape scores for RBD NAbs of epitope group A-D, respectively. ACE2 interface residues are annotated with red blocks, and mutated sites in Omicron are marked red. Annotates on the right side of heatmaps represent pseudovirus neutralizing IC50 fold change for Omicron and Beta in comparison with D614G.
m-p Typical structures in complex with RBD of group A-D antibodies. Residues involved in important contacts or related to Omicron mutations are marked.
The VH1-58 gene-encoded NAbs are enriched in Group B (Fig. 2b). These NAbs such as AZD8895 38, REGN10933 44, and BD-836 47 bind to the left shoulder of RBD, often focusing on the far tip (Fig 2n). These NAbs are very sensitive to the change of F486, N487, and G476 (Fig 2j, Extended Data Fig 3b). Fortunately, F486 and a few other major targeting sites of these NAbs are critically involved in ACE2-binding, and therefore they are generally harder to be escaped. NAbs that focus on the far tip of the left RBD shoulder, such as AZD8895 and BD-836, could survive Beta (fig 2f); however, Omicron could significantly reduce Group B NAbs’ binding affinity to RBD, potentially through S477N/T478K/E484A on their epitope (Extended Data Fig 4b) 48, resulting in the loss of neutralization. Group B NAbs are mostly escaped by Omicron and should not be chosen for drug development against Omicron.
Group C NAbs are frequently encoded by VH1-2 and VH1-69 (Fig. 2c). The majority of NAbs in this group could bind to both “up” and “down” RBDs, resulting in higher neutralization potency compared to other groups (Extended Data Fig 5c). Several highly potent antibodies are found in Group C, including BD-368-2/DXP-593 46, C002 3, and LY-CoV555 49. They bind to the right shoulder of RBD (Fig 2o), and are mostly prone to the change of Glu484 (Extended Data Fig 3c, 4c), such as the E484K mutation found in Beta (Fig 2g). The E484A mutation seen in Omicron elicited a similar escaping effect, although the change to Ala is slightly subtler, and could be tolerated by certain antibodies in this group (Extended Data Fig 7b). All Group C NAbs tested are escaped by Omicron (Fig 2k).

a) The length of H chain complementarity-determining region 3(HCDR3) amino acid sequence. Scatters show the individual HCDR3 length of antibody, vertical bars show the mean value, and error bars show mean ± sd. b) The V segment amino acid(a.a.) mutation rate. Mutation count divided by the a.a. length of V segment is defined as the mutation rate of V segment. Scatters show the individual H chain V segment mutation rate of antibody, vertical bars show the mean value, and error bars show mean ± sd.
c-e) IC50 of antibodies in the D614G(c), Beta(d), and Omicron(e) mutant pseudovirus neutralizing assay. Scatters show the individual IC50 of antibody, diamonds show the IC50 geometric mean(GM) of each epitope, and error bars show GM ± sd in the log10 scale. Dotted lines show the limit of detection, which is from 0.0005 ug/ml to 10 ug/ml. IC50 GMs are also noted on the figure.

The concentrations of RBD are shown in different colors. Dissociation constant(KD), associationconstant(Ka) and dissociation rate constant(Kd) are noted on the figure. The assays without binding are marked as “Escaped”.

(a) K417N escape scores and corresponding K417N pseudovirus neutralizing IC50 fold change compared to D614G pseudovirus of antibodies within epitope group A.
(b) E484K/E484A escape scores and corresponding E484K pseudovirus neutralizing IC50 fold change compared to D614G pseudovirus of antibodies within epitope group C.
Group D NAbs consist of diverse IGHV gene-encoded antibodies (Fig. 2d). Prominent members in this group include REGN10987 44 and AZD1061 38 (Fig 2h). They further rotate down from the RBD right shoulder towards the S309 site when compared to Group C (Fig 2p). As a loop formed by residues 440-449 in RBD is critical for the targeting of this group of NAbs, they are sensitive to the changes of Asn440, Lys444, Gly446, and Asn448 (Extended Data Fig 3d, 4d). Most NAbs of Group D remain active against Beta; however, G446S would substantially affect their neutralization capability against Omicron (Fig 2I). Also, for those NAbs that could tolerate G446S single mutation, the N440K/G446S combination may significantly reduce their binding affinity, resulting in that most Group D NAbs are escaped by Omicron.
SARS cross-reactive NAbs are less affected by B.1.1.529
Group E and F NAbs are rarer when compared to the other four groups. The archetypical member of each group was originally isolated from a SARS-CoV convalescent, and displays SARS-CoV-2 cross-neutralizing activity. There is no clear VDJ convergent effect compared to Group A, B, and C (Fig 3a, b), and the mutation rate and CDR3 length are larger than other groups. NAbs in Group E and F rarely compete with ACE2; thus, their average IC50 is higher than NAbs in Group A-D (Extended Data 5c, d, e). High-resolution structures of Group E and F NAbs are limited; thus, their epitope distribution and neutralization mechanism are not well-understood.

a-b VDJ combination of antibodies’ heavy chain variable region and their mutation rate for group E-F, respectively. For each group, the upper semicircle represents IGHV gene distribution and the lower semicircles represent IGHJ gene distribution.
c-d Escape maps of represetative RBD NAbs for group E-F respectively. For each site, the height of a letter indicates the detected mutation escape score of its corresponding residue. Site mutated in Omicron (B.1.1.529) are highlighted.
e-f Heatmaps of site escape scores for RBD NAbs of epitope group E-F, respectively. ACE2 interface residues are annotated with red blocks, and mutated sites in Omicron are marked red. Annotates on the right side of heatmaps represent pseudovirus neutralizing IC50 fold change for Omicron and Beta in comparison with D614G.
g-j Typical structures in complex with RBD of group E-F antibodies. Residues involved in important contacts or related to Omicron mutations are marked.
NAbs in Group E, such as VIR7831/S309, may recognize a mixed protein/carbohydrate epitope, involving the N-linked glycan on Asn343 6. Inferred from the escaping mutation profiles (Fig 3c), Group E NAbs are often sensitive to changes of R346, T345, and G339 (Extended Data Fig 3e, 4e). The G339D mutation would affect a subset of NAbs’ neutralization performance. Also, part of Group E NAbs’ epitope would extend to the 440-449 loop, making them sensitive to N440K and G446S on Omicron (Fig 3e).
Group F NAbs such as CR3022 and S304 target a cryptic site in RBD that is generally not exposed (Fig 3h, i), therefore their neutralizing activities are generally weaker 7. Group F NAbs are often sensitive to changes of K378, T376, and F374 (Extended Data Fig 3f, 4f). A loop involving RBD residues 371-375 lies in the ridge between the E and F sites; therefore, a subset of Group F NAbs, including some Group E NAbs, could be affected by the S371L/S373P/S375F mutations if their epitopes extend to this region (Fig 3d, f). Interestingly, a part of Group F NAbs is highly sensitive to G504 and V503, similar to the epitopes of S2. (Fig 3j), suggesting that they can compete with ACE2. Indeed, several NAbs, such as BD55-5300 and BD55-3372, exhibited high neutralization potency compared to other NAbs in Group F. However, These antibodies’ neutralization capability might be undermined by N501Y and Y505H of Omicron.
B.1.1.529 escapes the majority of NAb drugs
As for NAb drugs, consistent with their escaping mutation profiles, LY-CoV016/LY-CoV555 cocktail, REGN-10933/REGN-109876 cocktail, and AZD1061 are escaped by Omicron (Fig 4a). The binding affinity of AZD8895 and BRII-196 toward Omicron RBD is significantly reduced, potently due to multiple mutations accumulating on their epitopes, such that AZD8895 and BRII-196 failed to neutralize Omicron. VIR7831 retains strong RBD binding capability, although Gly339 is part of its epitope, the G339D mutation in Omicron does not appear to affect VIR7831’s binding; however, VIR7831’s IC50 is slightly reduced to 181 ng/mL. DXP-604’s binding affinity against Omicron RBD is largely reduced compared to wildtype RBD; nevertheless, it can still neutralize Omincron at an IC50 of 280 ng/mL, a nearly 30-fold reduction compared to wildtype (Fig 4b). Additionally, several NAbs in Group E and F have shown high potency against Omicron and broad pan-sarbecovirus neutralization ability, promising for NAb drug development (Fig 4c). Many more NAbs identified from vaccinated SARS convalescents are waiting to be characterized.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a, Neutralization of SARS-CoV-2 variants of concern (pseudotyped VSV) by 9 clinical antibody drugs. Data are collected from three technical replicates, and error bars show mean±s.d. b, IC50 values calculated from data in Fig. 4a. c, SARS-CoV-2 variants pseudovirus IC50 and sarbecovirus RBD binding affinity (mea-sured by ELISA OD450) of selected potent Omicron-neutralizing antibodies.
Discussion
The high-throughput yeast screening method provides a laboratory means for quickly examining the evolution outcome under a particular NAb, however, the current throughput using FACS is limited and can’t be used to evaluate a large NAb library. By virtue of MACS, we are able to increase the throughput by two orders of magnitude. In doing so, we were able to gain statistical confidence for the survival proportion of anti-RBD NAbs in each epitope group against Omicron. In addition to mutations in the RBD, changes in other regions of the S protein are also concerning. The Δ69-70 mutation in the N-terminal domain (NTD) may enhance viral infectivity 50. The P681H mutation, located in the furin cleavage site, could promote furin cleavage and therefore accelerate cell fusion, similar to P681R 51.
To date, a large number of SARS-CoV-2 NAbs have been identified from convalescents and vaccinees. The most potent NAbs are frequently found in Groups A-D as we described above, and tend to directly interfere with the binding of ACE2. Nevertheless, the neutralizing powers of these NAbs are often abrogated by RBD mutations in the evolutionary arms race between SARS-CoV-2 and human humoral immunity. Indeed, we showed that Omicron would escape from the majority of SARS-CoV-2 NAbs in this collection. Some superpotent NAbs in Group A may still function due to their extremely high affinity for RBD (such as DXP604), albeit at reduced efficacy. A subset of Group B NAbs (including AZD8895) is also more difficult to be evaded by single amino acid changes, as they target a small epitope involving several RBD residues that are essential for ACE2 binding; however, Omicron significantly reduced the binding affinity of those NAbs, possibly through multiple synergetic mutations, eventually causing lost of neutralization. On the other hand, Groups E and F NAbs are less affected by Omicron, likely because they are not abundant in population, hence exerting less evolutionary pressure for RBD to mutate in the corresponding epitope groups. These NAbs target conserved RBD regions in Sarbecovirus and therefore are ideal targets for future development of pan-Sarbecovirus NAb drugs.
Our study also offers important instructions for vaccine design. Vaccines are the most important tools for eventually vanquishing the SARS-CoV-2 pandemic. Based on our findings, we hypothesize that a vaccination strategy that helps to enrich Groups E and F NAbs would be promising to achieve broad protection. Along this line, Tan et al. recently showed that high-level and broad-spectrum NAbs are present in the SARS-CoV convalescents who have received an mRNA vaccine based on the SARS-CoV-2 Spike 52. We suspect that Groups E and F NAbs existed in SARS-CoV convalescents and might have been boosted upon SARS-CoV-2 vaccination. Because Groups E and F NAbs are the only effective NAbs against Omicron, the vaccines preferentially stimulating these antibodies are highly desired. Future studies are required to investigate a potential serial immunization scheme involving different inactivated Sarbecoviruses or their RBDs is capable of broadly protecting from future SARS-CoV-2 VOCs and even future Sarbecoviruses.
Author contributions
Y.C. and X.S.X designed the study. Y.C. and F.S coordinated the characterizations of the NAbs. J.W., F.J., H.L., H.S. performed and analyzed the yeast display mutation screening experiments. T.X., W.J., X.Y., P.W., H.L. performed the pseudovirus neutralization assays. W.H., Q.L., T.L., Y.Y., Q.C., S.L., Y.W. prepared the VSV-based SARS-CoV-2 pseudovirus. A.Y., Y.W., S.Y., R.A., W.S. performed and analyzed the antigen-specific single B cell VDJ sequencing. X.N., R.A. performed the antibody BLI studies. Z.C., S.D., P.L., L.W., Z.Z., X.W., J.X. performed the antibody structural analyses. P.W., Y.W., J.W, H.S, H.L. performed ELISA experiment. X.H. and R.J. coordinated the blood samples of vaccinated SARS convalescents. Y.C., X.W., J.X., X.S.X wrote the manuscript with inputs from all authors.
Declaration of interests
X.S.X. and Y.C. are inventors on the patent application of DXP-604 and BD series antibodies. X.S.X. and Y.C. are founders of Singlomics Biopharmaceuticals Inc. Other authors declare no competing interests.
Methods
Antigen-specific B cell sorting and sequencing
PBMCs were separated from whole blood samples based on the detailed protocol as described previously 12. Briefly, blood samples were first diluted with 2% FBS (Gibco) in PBS (Invitrogen) and subjected to Ficoll (Cytiva) gradient centrifugation. After red blood cell lysis and washing steps, PBMCs were resuspended with 2% FBS in PBS for downstream B cell isolation or 10% DMSO (Sigma-Aldrich) in FBS for further preservation.
Starting with freshly isolated or thawed PBMCs, B cells were enriched by positive selection using a CD19+ B cell isolation kit according to the manufacturer’s instructions (STEMCELL). The enriched B cells were stained in FACS buffer (1× PBS, 2% FBS, 1 mM EDTA) with the following anti-human antibodies and antigens: FITC anti-CD19 Antibody (Biolegend), FITC anti-CD20 Antibody (Biolegend), Brilliant Violet 421 anti-CD27 Antibody (Biolegend), PE/Cyanine7 anti-IgM, and fluorophore-labelled RBD (SARS-CoV-2 and SARS-CoV RBD, Sino Biological Inc.) and ovalbumin (Ova) for 30 min on ice. Cells were stained with 7-AAD for 10 minutes before sorting. Single CD19 or CD20+ CD27+ IgM-Ova-RBD-PE+ RBD-APC+B cells were sorted on an Astrios EQ (BeckMan Coulter) into PBS containing 30% FBS. Cells obtained after FACS were proceed to 5’-mRNA and V(D)J libraries preparation as previously described12, which were further submitted to illumine sequencing on a Hiseq 2500 platform, with the 26×91 pair-end reading mode.
V(D)J sequence data analysis
The raw FASTQ files were processed by Cell Ranger (version 6.1.1) pipeline using GRCh38 reference. Sequences were generated using “cellranger multi” or “cellranger vdj” with default parameters. Then we extracted the protein sequences and processed them by IMGT/DomainGapAlign (version 4.10.2) to obtain the annotations of V(D)J, regions of CDR and the mutation frequency53,54. Mutation count divided by the length of V gene peptide is defined as the amino acid mutation rate of V gene.
Recombinant antibody production
Paired immunoglobulin heavy and light chain genes obtained from 10X Genomics V(D)J sequencing and analysis were submitted to recombinant monoclonal antibody synthesis. Briefly, heavy and light genes were cloned into expression vectors respectively based on Gibson assembly, which were subsequently co-transfected into HEK293 cells. Then secreted monoclonal antibodies from cultured cell were purified by protein A affinity chromatography. The specificities of these antibodies were determined by ELISA binding analysis.
ELISA and neutralization assay
ELISA were conducted to evaluate antibody or plasma binding ability. Briefly, after coating with 0.03 μg/mL and 1 μg/mL RBD (Sino Biological Inc.) of different sarbecovirus and SARS-CoV-2 variants, blocking, and washing, 1μg/mL antibodies or serially diluted plasma samples were added to the plates. After incubation and wash, plates were incubated with diluted goat anti-human IgG (H+L)/HRP (JACKSON). Then plates were developed by addition of the TMB (Solarbio), then the developing reaction was stopped by adding H2SO4 and OD450 was measured by a ELISA microplate reader.
Neutralization assay was performed to evaluate neutralizing ability of antibody and plasma the detailed process was previously described by Cao et al.12. Briefly, serially diluted antibodies were first incubated with pseudotyped virus for 1h, and the mixture was then incubated with Huh-7 cells. After 24h incubation in an incubator at 37C°, cells were collected and lysed with luciferase substrate (PerkinElmer), then proceed to luminescence intensity measurement by a microplate reader. IC50 and NT50 were determined by a four-parameter non-linear regression model. Omicron pseudovirus contains the following mutations: A67V, H69del, V70del, T95I, G142D, V143del, Y144del, Y145del, N211del, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F.
Biolayer interferometry
Biolayer interferometry assays were conducted on Octet® R8 Protein Analysis System (Fortebio) following the manufacturer’s instruction. Briefly, after baseline calibration, Protein A biosensors (Fortebio) were immersed with antibodies to capture the antibody, then sensors were immersed in PBS with 0.05% Tween-20 to the baseline. After association with different concentrations of RBD or Spike of Sarbecovirus and SARS-CoV-2 variants (Omicron RBD: 40592-V08H85, Omicron Spike: 40589-V08H26), disassociation was conducted. Data were recorded using software Data Acquisition 11.1 (Fortebio) and analyzed using software Data Analysis HT 11.1 (Fortebio).
RBD Deep Mutational Scanning Library construction
The yeast-display RBD mutant libraries used here were constructed as described by Starr et al.,13 based on the spike receptor binding domain (RBD) from SARS-CoV-2 (NCBI GenBank: MN908947, residues N331-T531) with the modifications that instead of 16-neuclotide barcode, a unique 26-neuclotide barcode was appended to each RBD variant as an identifier in order to decrease sequencing cost by eliminating the use of PhiX. Briefly, three rounds of mutagenesis PCR were performed with designed and synthesized mutagenetic primer pools; in order to solid our conclusion, we constructed two RBD mutant libraries independently. RBD mutant libraries were then cloned into pETcon 2649 vector and the assembled products were electroporated into electrocompetent DH10B cells to enlarge plasmid yield. Plasmid extracted form E. coli were transformed into the EBY100 strain of Saccharomyces cerevisiae via the method described by Gietz and Schiestl55. Transformed yeast population were screened on SD-CAA selective plate and further cultured in SD-CAA liquid medium at a large scale. The resulted yeast libraries were flash frozen by liquid nitrogen and preserved at −80C°.
PacBio library preparation, sequencing and analysis
The correspondence of RBD gene sequence in mutant library and N26 barcode was obtained by PacBio sequencing. Firstly, the bacterially-extracted plasmid pools were digested by NotI restriction enzyme and purified by agarose gel electrophoresis, then proceed to SMRTbell ligation. Four RBD mutant libraries were sequenced in one SMRT cell on a PacBio Sequel ll platform. PacBio SMRT sequencing subreads were converted to HiFi ccs reads with pbccs, and then processed with a slightly modified version of the script previously described13 to generate the barcode-variant dictionary. To reduce noise, variants containing stop codons or supported by only one ccs read were removed from the dictionary and ignored during further analysis.
MACS-based mutation escape profiling
ACE2 binding mutants were sorted based on magnetic beads to eliminate non-functional RBD variants. Briefly, the biotin binder beads (Thermo Fisher) were washed and prepared as the manufacturer’s instruction and incubated with biotinylated ACE2 protein (Sino Biological Inc.) at room temperature with mild rotation. The ACE2 bound beads were washed twice and resuspend with 0.1% BSA buffer (PBS supplemented with 0.1% bovine serum albumin), and ready for ACE2 positive selection. Transformed yeast library were inoculated into SD-CAA and grown at 30C° with shaking for 16-18h, then back-diluted into SG-CAA at 23C with shaking to induce RBD surface expression. Yeasts were collected and washed twice with 0.1% BSA buffer and incubated with aforementioned ACE2 bound beads at room temperature for 30min with mild rotating. Then, the bead-bound cells were washed, resuspend with SD-CAA media, and grown at 30C° with shaking. After overnight growth, the bead-unbound yeasts were separated with a magnet and cultured in a large scale. The above ACE2 positive selected yeast libraries were preserved at −80C° in aliquots as a seed bank for antibody escape mapping.
One aliquot of ACE2 positive selected RBD library was thawed and inoculated into SD-CAA, then grown at 30C° with shaking for 16-18h. 120 OD units were back-diluted into SG-CAA media and induced for RBD surface expression.
Two rounds of sequential negative selection to sort yeast cells that escape Protein A conjugated antibody binding were performed according to the manufacturer’s protocol. Briefly, Protein A magnetic beads (Thermo Fisher) were washed and resuspend in PBST (PBS with 0.02% Tween-20). Then beads were incubated with neutralizing antibody and rotated at room temperature for 30min. The antibody-conjugated beads were washed and resuspend in PBST. Induced yeast libraries were washed and incubated with antibody-conjugated beads for 30min at room temperature with agitation. The supernatant was separated and proceed to a second round of negative selection to ensure full depletion of antibody-binding yeast.
To eliminate yeast that did not express RBD, MYC-tag based RBD positive selection was conducted according to the manufacturer’s protocol. First, anti-c-Myc magnetic beads (Thermo Fisher) were washed and resuspend with 1X TBST, then the prepared beads were incubated for 30min with the antibody escaping yeasts after two rounds of negative selection. Yeasts bound by anti-c-Myc magnetic beads were wash with 1X TBST and grown overnight in SD-CAA to expand yeast population prior to plasmid extraction.
Overnight cultures of MACS sorted antibody-escaped and ACE2 preselected yeast populations were proceed to yeast plasmid extraction kit (Zymo Research). PCRs were performed to amplify the N26 barcode sequences as previously described13. The PCR products were purified with 0.9X Ampure XP beads (Beckman Coulter) and submitted to 75bp single-end Illumina Nextseq 500 sequencing.
Deep mutational scanning data processing
Raw single-end Illumina sequencing reads were trimmed and aligned to the reference barcode-variant dictionary generated as described above to get the count of each variant with dms_variants Python package (version 0.8.9). For libraries with N26 barcodes, we slightly modified the illuminabarcodeparser class of this package to tolerate one low sequencing quality base in the barcode region. The escape score of variant X is defined as F×(nX,ab / Nab) / (nX,ref / Nref), where nX,ab and nX,ref is the number of detected barcodes for variant X, Nab and Nref are the total number of barcodes in antibody-selected (ab) library and reference (ref) library respectively as described by Starr et al. 13. Different from FACS experiments, as we couldn’t measure the number of cells retained after MACS selection precisely, here F is considered as a scaling factor to transform raw escape fraction ratios to 0-1 range, and is calculated from the first and 99th percentiles of raw escape fraction ratios. Scores less than the first percentile or larger than the 99th percentile are considered to be outliers and set to zero or one, respectively. For each experiment, barcodes detected by <6 reads in the reference library were removed to reduce the impact of sampling noise, and variants with ACE2 binding below −2.35 or RBD expression below −1 were removed as previously described 13. Finally, we built global epistasis models with dms_variants package for each library to estimate single mutation escape scores, utilizing the Python scripts provided by Greaney et al. 18.
Antibody clustering
Antibody clustering and epitope group identification were performed based on the N×M escape score matrix, where N is the number of antibodies which pass the quality controlling filters, and M is the number of informative sites on SARS-CoV-2 RBD. Each entry of the matrix Anm refers to the total escape score of all kinds of mutations on site m of antibody n. The dissimilarity between two antibodies is defined based on the Pearson’s correlation coefficient of their escape score vectors, i. e. Dij=1-Corr(Ai,Aj). Sites with at least 6 escaped antibodies (site escape score >1) were considered informative and selected for dimensionality reduction and clustering. We utilized cmdscale R function to convert the cleaned escape matrix into an N×6 feature matrix by multidimensional scaling (MDS) with the dissimilarity metric described above, followed by unsupervised k-medoids clustering within this 6-dimensional antibody feature space. Finally, two-dimensional tSNE embeddings were generated with Rtsne package for visualization.
Acknowledgments
We thank Professor Jesse Bloom for his generous gift of the yeast SARS-CoV-2 RBD libraries. We thank Beijing BerryGenomics for the help on DNA sequencing. We thank Sino Biological Inc. for the technical assistance on mAbs and B.1.1.529 RBD expression. This project is financially supported by the Ministry of Science and Technology of China (CPL-1233).
References




