

Abstract
The SARS-CoV-2 nucleocapsid protein (N) is responsible for the viral genome packaging and virion assembly. Being highly abundant in the host cell, N interacts with numerous human proteins and undergoes multisite phosphorylation in vivo. When phosphorylated within its Ser/Arg-rich region, a tract highly prone to mutations as exemplified in the Omicron and Delta variants, N recruits human 14-3-3 proteins, potentially hijacking their functions. Here, we show that in addition to phosphorylated Ser197, an alternative, less conserved phosphosite at Thr205, absent in SARS-CoV N, binds 14-3-3 with micromolar affinity and is in fact the preferred binding site. Fluorescence anisotropy reveals a distinctive pT205/pS197 binding selectivity towards the seven human 14-3-3 isoforms. While explaining the structural basis for the discovered selectivity towards SARS-CoV-2 N phosphopeptides, our crystal structures enable prediction of N interactions with 14-3-3, suggesting a link between the strength of this interaction and replicative fitness of emerging coronavirus variants.
Introduction
The current pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), exposed a number of weaknesses in our approach to tackle viral outbreaks. The lack of understanding host-virus interactions at the molecular level slowed the development of novel antiviral therapies and understanding the causes of virus pathogenicity, resulting in lack of preparedness and, consequently, in major health, economic and social impacts for the society. One of the most abundant virus proteins, the 46-kDa nucleoprotein (N), is common for single-stranded RNA viruses, including the positive-sense coronaviruses. It is responsible for the replication, packaging and storage of viral genomic RNA 1,2. N is one of the most abundant coronavirus proteins in the infected cells (reaching ∼1 % of a total number of cell proteins) 3–5. In SARS-CoV-2, N has been proposed to be the major factor of the enhanced pathogenicity 6. Sharing 89.1% sequence identity with SARS-CoV N, SARS-CoV-2 N consists of the structured N-terminal RNA-binding domain 7, the structured C-terminal dimerization domain 8, disordered terminal tails and the long unstructured interdomain linker (residues 176-247) (Fig. 1a) 9,10.

SARS-CoV-2 N site-specifically phosphorylated at Ser197 interacts with human 14-3-3γ. a. Schematic representation of the SARS-CoV-2 N domain structure. NTD and CTD, interdomain linker, Ser/Arg-rich region are labeled, Ser197 and Thr205 phosphorylation sites are marked by red circles, and blue rectangles indicate peptide fragments prone to amyloid aggregation 22. b. Local alignment of the SR-regions of SARS-CoV-2 and SARS-CoV N proteins is shown, with the phosphosites of interest marked by red font and potential 14-3-3-binding sites by bold font. Note that SARS-CoV N has only the first potential 14-3-3-binding site. c. Phosphorylation of SARS-CoV-2 N variants analyzed by a Phos-tag gel electrophoresis. d. The same samples as in c were analyzed by SDS-PAGE. e,f. The interaction of human 14-3-3γ with singly phosphorylated SARS-CoV-2 N.S197TAG (e) or the unphosphorylated SARS-CoV-2 N counterpart (f) analyzed by SEC when 14-3-3 was added to N in excess at 220 mM NaCl in the samples and running buffer, with SDS-PAGE of all fractions along the elution profiles. M - molecular mass markers (indicated in kDa to the left). Positions of proteins are indicated by arrows on the right. Note that fractions 4-6 (dashed rectangles) contained the 14-3-3:N complex only in the case of S197TAG.
The latter linker domain contains a Ser/Arg-rich region (residues ∼176-210) 11–13 that becomes heavily phosphorylated upon host infection 4,14–16 and is also a hotspot of viral mutations 17–19. This interdomain linker contributes to higher-order oligomerization of N molecules 20, to the N-M interactions 21, and to liquid-liquid phase separation with RNA 11,17 via the tentative amyloid-like aggregation of specific peptide stretches 179-184, 217-222 and 243-248 22. Phosphorylation of several Ser/Thr residues in the SR-rich region controls the flexibility, electrostatics, and functions of SARS-CoV-2 N 17,23,24. Apart from affecting properties of N itself, phosphorylation can also regulate N interaction with other proteins such as 14-3-3 25,26 -the universal and abundant phosphopeptide-binding protein-protein interaction hub represented by seven specific isoforms in human (β, γ, ε, ζ, η, σ, τ) 27,28. In line with this, SARS-CoV N undergoes phosphorylation-dependent association with human 14-3-3 in the cytoplasm of the transfected cells, which is proposed to control nucleocytoplasmic shuttling of N and hijack 14-3-3 functions via sequestration 25,26.
In previous work, we have shown that upon phosphorylation of the SR-rich region, SARS-CoV-2 N is specifically recognized by human 14-3-3 proteins, to form a 2:2 complex 26. The use of truncated N variants narrowed down the binding site to phospho-Ser197 26. This site is conserved between SARS-CoV and SARS-CoV-2 N proteins (Fig. 1a), as well as several animal coronaviruses, and represents a nearly canonical 14-3-3-binding motif SRNpS197TP 26. Nonetheless, other phosphosites within SARS-CoV-2 N promoting 14-3-3 binding were also proposed 26. Despite biochemical advances on SARS-CoV-2 N interaction with 14-3-3 proteins, the structural basis for such interaction remained elusive.
Production of phosphorylated N is challenged by the dense distribution of phosphorylatable residues and associated heterogeneity of poly-phosphorylated N forms (Fig. 1b) 26,29. Here, we first used genetic code expansion to verify phosphoserine exclusively at position Ser197 in SARS-CoV-2 N is sufficient to enable binding to 14-3-3. Intriguingly, when poly-phosphorylated by a kinase, we found that the naturally occuring SARS-CoV-2 N variant S197L still displayed a strong phosphorylation-dependent interaction with 14-3-3. We therefore identified an adjacent phosphosite at position Thr205, that also contributes to 14-3-3 binding and is absent in SARS-CoV N. Using fluorescence anisotropy, we determined micromolar dissociation constants for the interaction of both phospho-motifs with all seven human isoforms of 14-3-3, which revealed the characteristic affinity profiles reflecting the affinity hierarchy of the seven 14-3-3 isoforms 28 and a remarkable selectivity of 14-3-3 towards the SARS-CoV-2 N phosphopeptides. Crystal structures of the corresponding 14-3-3 complexes helped rationalize the site-selective binding of the SARS-CoV-2 N phosphopeptides to 14-3-3. This also captured for the first time residues 193-210 of the functional SR-rich region, providing a structural framework for the analysis of the existing and emerging SARS-CoV-2 mutations.
Results
Alternative 14-3-3-binding sites within SARS-CoV-2 N
The key 14-3-3-binding site of SARS-CoV-2 N was proposed to center at phospho-Ser197, which is shared by SARS-CoV and SARS-CoV-2 N proteins (Fig. 1b) 26. However, direct evidence for its role was lacking and whether other phosphosites contributed to 14-3-3 binding remained unknown. To address this, we first sought to obtain N protein containing the Ser197 phosphosite only. Due to the strong propensity of this protein to undergo multisite phosphorylation 26, it was not possible to make monophosphorylated N enzymatically. Therefore, we used a genetic code expansion approach to site-specifically install phosphoserine at a genetically programmable TAG stop codon 30. Incorporation of phosphoserine into this protein, N.S197TAG, was confirmed by the discrete upward shift on a Phos-tag gel electrophoresis (Fig. 1c, d). In contrast, SARS-CoV-2 N phosphorylated during co-expression with protein kinase A (PKA) produced multiple upward shifted bands corresponding to multisite phosphorylation (Fig. 1c, d), in agreement with earlier observations 26.
Co-elution of human 14-3-3γ with SARS-CoV-2 N.S197TAG phosphoprotein, but not with the unphosphorylated SARS-CoV-2 N counterpart, during size-exclusion chromatography confirmed the formation of the phosphorylation-dependent complex (Fig. 1e, f), indicating that the Ser197 phosphosite plays a role in recruiting 14-3-3.
We then asked if the S197L mutation associated with the variant of the coronavirus identified in 2020 in Europe and Brazil 31–34, which blocks phosphorylation at this position, would preclude SARS-CoV-2 N from forming complexes with 14-3-3γ. The S197L mutant was co-expressed with PKA for phosphorylation and then purified similar to the wild-type protein. As a control we produced unphosphorylated N.S197L protein in the absence of PKA, and validated the differences in phosphorylation status of the two by Phos-tag and SDS-PAGE (Fig. 1c and d). Unexpectedly, the phosphorylated S197L mutant showed a very efficient interaction with 14-3-3γ, as confirmed by SEC with SDS-PAGE analysis of the fractions (Fig. 2a, b) as well as MALS analysis of the molecular mass of the complex obtained (150.8 kDa, Mw/Mn = 1.000; Mw expected for the 2:2 complex is 148.3 kDa) (Fig. 2c). Interestingly, at lower than 220 mM NaCl, we could detect nonspecific binding of the unphosphorylated S197L mutant of SARS-CoV-2 N to 14-3-3γ (data not shown). This could be completely blocked by increasing salt concentration to 300 mM, at which the specific interaction of the PKA-phosphorylated S197L mutant was still detectable (Fig. 2d-f).
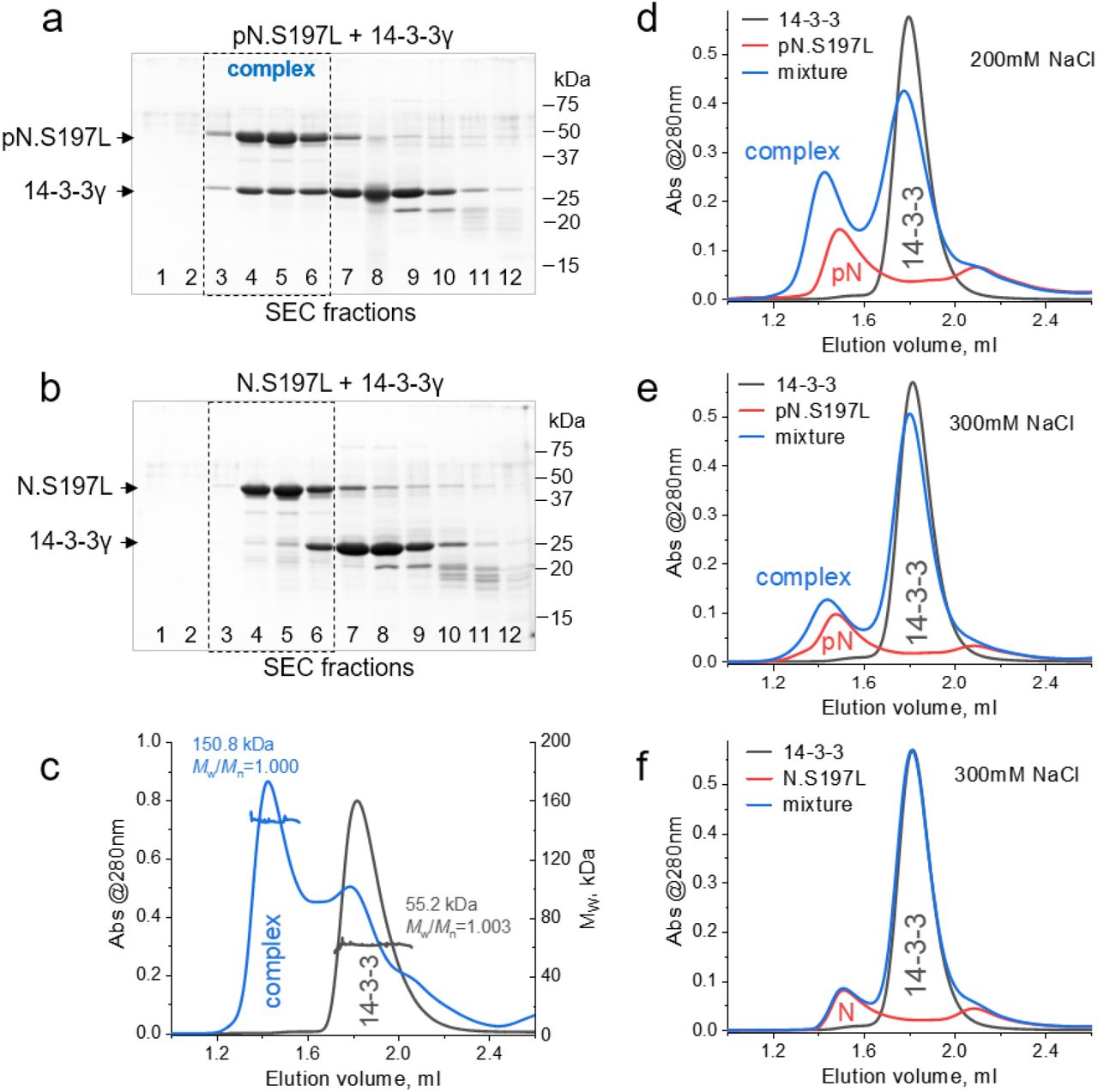
The S197L mutant of SARS-CoV-2 N interacts with human 14-3-3γ in a phosphorylation-dependent manner. a,b. The interaction of human 14-3-3γ with PKA-phosphorylated (a) or unphosphorylated (b) SARS-CoV-2 N.S197L mutant analyzed by SEC and SDS-PAGE of all fractions along the elution profiles. 14-3-3γ was pre-incubated in excess with N in 220 mM NaCl, and SEC was run in the same buffer. Molecular mass markers are indicated in kDa on the right. Positions of proteins are indicated by arrows on the left. Note that fractions 3-6 (dashed rectangles) contained the 14-3-3:N complex only in the case of pN.S197L. c. SEC-MALS profiles for 14-3-3γ or its mixture with phosphorylated N.S197L protein obtained using a Superdex 200 Increase 5/150 column at 220 mM NaCl in the premixed samples and running buffer. Average MW values and polydispersity indexes (Mw/Mn) for the peaks of interest are indicated. d-f. Effect of salt on the interaction. SEC profiles of 14-3-3γ, phosphorylated N.S197L or their mixture, obtained at 200 mM NaCl (d) or 300 mM NaCl (e) in the premixed samples and running buffer. Note that the complex is detectable even at 300 mM NaCl. f. 300 mM NaCl in the premixed samples and SEC buffer fully blocks nonspecific interaction of 14-3-3γ with unphosphorylated N.S197L.
The ability of N.S197L to efficiently interact with 14-3-3γ in a phosphorylation-dependent manner indicated that other site(s) besides Ser197 contribute to 14-3-3 binding. In particular, the functional SR-rich region of N contains another potential 14-3-3-binding motif around Thr205 (SRGTSP), which is phosphorylated in vivo 4,14. It has a sequence similar to the Ser197 site (SRNSTP), and since other phosphosites of the SR-rich region have more divergent sequences from the consensus 14-3-3-binding sites [RXX(pS/pT)XP/G] 35, we focused on the interaction of 14-3-3 with these two representative phosphopeptide fragments, SSRNpS197TPGSS and SSRGpT205SPARM. Interestingly, this Thr205 site is not conserved in SARS-CoV N, which has only the Ser197 site (Fig. 1b) 26. SARS-CoV N still showed a pronounced phosphorylation-dependent interaction with human 14-3-3γ (Supplementary Fig. 1).
The results obtained using SARS-CoV-2 N.S197L, S197TAG and SARS-CoV N suggested that the pS197 and pT205 are alternative (sufficient but not necessary) 14-3-3-binding sites within the SR-rich region.
Ser197 and Thr205 phosphopeptides display nonparallel affinity profiles towards human 14-3-3 isoforms
Synthetic phosphopeptides corresponding to the Ser197 and Thr205 fragments, each additionally bearing a tryptophan residue for more accurate quantitation, were labeled by fluorescein 5-isothiocyanate (FITC) and used to determine binding affinities against all seven human 14-3-3 isoforms by fluorescence anisotropy. For this, homotypic MBP-tagged full-length 14-3-3 constructs were used to obtain highly pure protein in an efficient manner 28. SEC-MALS analysis of the MBP-14-3-3ζ protein confirms that the MBP fusion does not disrupt the functional dimeric state of 14-3-3 (Supplementary Fig. 2).
We first made sure that peptide binding to 14-3-3 was saturable and specific, and that the MBP tag does not change the KD value determined (Fig. 3a). The same FITC-labeled phosphopeptide failed to bind to noncognate protein lysozyme; likewise, free FITC did not show significant binding to MBP-14-3-3 (Fig. 3a). This is consistent with previous observations showing that FITC-labeled phosphopeptides and their unlabeled counterparts bind similarly to 14-3-3 28,36. In addition, the FITC-peptide complexes with 14-3-3 could be disrupted by titration with either the corresponding unlabeled peptides or the canonical phosphopeptide from unrelated protein HSPB6 (pB6) 37 (Supplementary Fig. 3). These data demonstrate the chosen N peptides bind specifically to the common peptide-binding grooves of 14-3-3.

Affinity and selectivity of SARS-CoV-2 N phosphopeptide binding to the human 14-3-3 protein family. a. Fluorescence anisotropy titration data for 14-3-3γ or MBP-14-3-3γ binding to the FITC-labeled SARS-CoV-2 N pT205 peptide. The corresponding apparent KD values (mean ± SD, n=3) are shown. Titration of the same peptide by lysozyme and titration of free FITC by MBP-14-3-3γ are shown as controls. b, c. Titration of FITC-labeled SARS-CoV-2 N phosphopeptides pT205 (b) or pS197 (c) by the seven human 14-3-3 isoforms. The data (mean ± SD, n=3) are color-coded with respect to the 14-3-3 isoforms; fitting curves and the KD values at equilibrium are collected in Supplementary Fig. 4 and Table 1. In a-c, background anisotropy was subtracted from all values. d. Affinity profiles of the SARS-CoV-2 N phosphopeptides versus 14-3-3 isoforms shown as heatmap gradients according to the KD scale on the right. The source KD values are found in Table 1. e, f. Affinity profiles presented as -logKD values (e) or selectivity coefficients (f) are correlated with the proteome-wide affinity hierarchy of the 14-3-3 isoforms represented as average ΔΔG values for the individual isoforms (indicated by asterisks) 28. Coefficient of determination R2 and Pearson’s r coefficient characterizing the linear correlations are indicated.
Both SARS-CoV-2 N phosphopeptides showed saturable interaction with all seven human 14-3-3 isoforms, enabling quantitative analysis of the dissociation constants (Fig. 3b and c, Supplementary Fig. 4). The average binding affinities of 14-3-3 to the SARS-CoV-2 N phosphopeptides were somewhat lower than to the pB6 peptide, yet were in the same micromolar range (Table 1 and Supplementary Fig. 3). For each SARS-CoV-2 N peptide, we detected a remarkable peptide-affinity trend following the general peptide-affinity hierarchy of 14-3-3 isoforms reported recently (Fig. 3d and e) 28. For the pT205 peptide, the binding affinity to 14-3-3 decreased from gamma (γ) > eta (η) > beta (β) > epsilon (ε) > zeta (ζ) > tau (τ) > sigma (σ); the order for the pS197 peptide was: β ∼ γ ∼ η > ζ ∼ τ ∼ ε > σ (Fig. 3d). As a result, transformation of the KD values into -logKD enabled to detect that both trends correlate well with the interactome-wide affinity hierarchy of the 14-3-3 isoforms (Pearson’s r=0.87 for pS197 and r=0.93 for pT205, Fig. 3e) 28. To facilitate calculations of how custom peptide binding to 14-3-3 isoforms correlates with their general affinity hierarchy based on the set of seven experimentally determined (or predicted) KD values for individual 14-3-3 isoforms, we created a simple Excel-based tool (Supplementary Data file 1).
Apparent KD values and selectivity coefficients for the interaction of seven 14-3-3 isoforms with two SARS-CoV-2 N phosphopeptides as determined by fluorescence anisotropy.
The affinity profiles of 14-3-3 for the two SARS-CoV-2 N peptides were significantly shifted and nonparallel to each other. Due to the shift, the affinity of the weakest pT205 peptide binder, 14-3-3σ (KD ∼33 μM), was still 2 times higher than for the strongest pS197 binders, 14-3-3β/γ/η (KD ∼70 μM). Overall, the 14-3-3 family covered affinities to SARS-CoV-2 phosphopeptides differing by an order of magnitude [∼10-fold KD ratio between the weakest (14-3-3σ/pS197) and the strongest (14-3-3γ/pT205) pair]. The average affinity of human 14-3-3 proteins to the studied SARS-CoV-2 phosphopeptides (KD ∼50 μM) is roughly in the same order of magnitude as the average affinity to twenty three unrelated peptides analyzed previously (KD ∼45 μM) 28. This suggests viable competition of the SARS-CoV-2 N phosphopeptides with the host cell phosphopeptides for the 14-3-3 binding, especially with the more weakly bound portions of the interactome.
Selectivity trend of 14-3-3 isoforms to SARS-CoV-2 N phosphopeptides
Curiously, the affinities to all 14-3-3 isoforms were consistently higher for the pT205 peptide than for the pSer197 phosphopeptide (4.3-fold ratio of the average KD values) (Fig. 3f). Moreover, we found a remarkable selectivity trend reflecting a linear correlation of the KD ratio for the 14-3-3 isoforms with their general affinity hierarchy (R2=0.84, Pearson’s r=0.93; Fig. 3f) 28. In other words, not only does 14-3-3γ turn out to be the strongest binder, it is also the most selective isoform with respect to the SARS-CoV-2 phosphopeptides pS197 and pT205 (the KD; 197/KD; 205 ratio of ∼7), while 14-3-3σ is the weakest and simultaneously the least selective (Fig. 3f, Table 1). Yet, even for 14-3-3σ the affinity to the pT205 peptide was ∼3 times higher than for the pS197 peptide. This observation was totally unexpected given the very similar sequences of both peptides, implying that the corresponding protein-peptide complexes feature different interfaces and chemical contacts. To understand the molecular basis of this phenomena, we crystallized 14-3-3σ complexes with both phosphopeptides and determined structures by X-ray crystallography at high resolution (Table 2).
X-ray data collection and refinement statistics.
Crystallography explains the selectivity of 14-3-3 for SARS-CoV-2 N phosphopeptides
X-ray structures of 14-3-3σ with both peptides (Table 2) had one 14-3-3 dimer in the asymmetric unit, where each protomer was occupied by the target phosphopeptide. Eight out of ten pS197 phosphopeptide residues and all ten residues for the pT205 phosphopeptide are clearly defined in electron density, revealing remarkable details of the protein/peptide interaction (Fig. 5a, b). The central six residues including the phosphosite are canonically placed facing the conserved residues of the amphipathic 14-3-3 groove, forming a developed network of hydrogen bonds (Fig. 4a, b and Supplementary Fig. 5). Residues at +1 and -1 positions with respect to the phosphosite are H-bonded to the side chains of the conserved 14-3-3σ residues Asn175 and Asn226, while hydroxyl groups of both Thr198 and Ser206 of the peptides (position +1) are identically involved in H-bond formation with Asn175 and the conserved Lys122 residue of 14-3-3σ (Fig. 4a, b and Supplementary Fig. 5). An additional H-bond in the case of the pS197 peptide between the side chain of its Asn196 residue and Asn226 of 14-3-3σ is replaced by the additional hydrophobic contact in the case of the pT205 peptide, formed between the methyl group of phospho-Thr205 residue and the side chain of 14-3-3σ’s Val178 (Fig. 4). In contrast to the nearly equivalent six-residue segments of both peptides (Cα RMSD=0.7 L), crystal structures revealed significant differences in their C-terminal regions. In the case of the pS197 peptide, we could not detect any stabilizing in-trans interactions after Pro199. Lack of defined electron density for the last two Ser residues of this peptide indicate their flexibility and hence inability to form stabilizing interactions (Fig. 4a). On the contrary, the pT205 peptide was additionally stabilized in-trans by i) the hydrophobic contacts of its Ala208 with Val46 of 14-3-3 and Met210 with Phe118 and Ile168 of 14-3-3, ii) the H-bonding interaction between the peptide’s carbonyl of Ala208 with the side chain of Ser45 of 14-3-3σ and iii) the salt bridge between the peptide’s Arg209 with Glu14 of 14-3-3σ (Fig. 5b). Interestingly, Glu14 is absolutely conserved in all human isoforms of 14-3-3 (Supplementary Fig. 6). Therefore, the salt bridge interaction involving Arg209 is likely shared by all 14-3-3 isoforms and, together with factors i)-ii), confer the consistently higher affinity of the pT205 peptide versus pS197 peptide to 14-3-3 proteins, as observed biochemically (Fig. 3).

Crystal structures reveal interfaces between 14-3-3σ and two alternative 14-3-3-binding sites within SARS-CoV-2 N. a,b. Molecular contacts formed between a 14-3-3σ subunit (gray cartoon) and the pS197 phosphopeptide (magenta sticks) (a) or the pT205 phosphopeptide (orange sticks) (b). Hydrogen bonds are indicated by yellow dashed lines, hydrophobic interactions are indicated by large semi-transparent spheres, water molecules mediating 14-3-3-peptide contacts are in aquamarine. 2Fo-Fc electron density maps for the phosphopeptides contoured at 1σ are shown as semi-transparent blue surfaces in the inserts. Sequences of the phosphopeptides are shown above the inserts. Note that two C-terminal serines in the pS197 peptide structure and a tryptophan residue, attached to each peptide from the N terminus, are not visible due to disorder. c. A model showing that the two phosphopeptides (magenta and orange sticks) of the same N chain cannot occupy both grooves of the 14-3-3σ dimer (shown as semi-transparent cartoon) simultaneously. The pT205 peptide is depicted either bound in the bottom 14-3-3 groove (semi-transparent sticks) or as a continuation of the pS197 peptide bound in the top 14-3-3 groove. Distances between two phospho-groups in one continuous peptide (black dashed line) or as separate 14-3-3-bound entities (red dashed line) are indicated.

Possible effects on 14-3-3 binding of the SARS-CoV-2 N mutations improving fitness of the virus. a. Sequence of the N region (193-210) covered by crystal structures of the 14-3-3/SARS-CoV-2 N peptide complexes aligned with the canonical 14-3-3-binding motifs 35. The suboptimal 14-3-3-binding sites within N are shown by boxes. Colors correspond to Fig. 4. b-d. Structural models showing the expected interaction of the mutated N peptides with 14-3-3 based on the crystal structures determined in the present study. The mutated residues are shown in green, the polar contacts are shown by black dashed lines, the hydrophobic interactions are indicated by semi-transparent spheres.
The studied peptides of the same SARS-CoV-2 N polypeptide chain cannot simultaneously occupy both grooves present in the dimer of 14-3-3, as the pS197 and pT205 phosphosites are only 7 residues apart (the interphosphate distance is 20 L). For bidentate binding to 14-3-3, at least 15 residues in the extended conformation 38 are needed to sufficiently separate the phosphosites (e.g. 22 in the 5N6N structure 39) and cover the required ∼35 L distance between the two binding sites of the 14-3-3 dimer (Fig. 4c). While within the same SARS-CoV-2 N molecule pS197 and pT205 are obviously alternative 14-3-3-binding sites, we cannot exclude a mixed pS197/pT205 binding within the heterotetrameric SARS-CoV-2 N/14-3-3 complex. Our data suggest that binding of two pT205 motifs to the 14-3-3 dimer would be thermodynamically preferred, though mutations in the SR-rich region might affect this selectivity.
Structure-based prediction of effects of the N mutations on 14-3-3 binding
The conformation of the 14-3-3-bound peptides allowed us to rationalize the effect of N mutations present in different coronavirus variants (Table 3) by predicting their influence on 14-3-3 binding. First of all, due to the phosphorylation-dependent interaction with 14-3-3 proteins, the S197L and T205I mutations would simply block the interactions at the corresponding site (while leaving the possibility of interaction using the alternative one). Interestingly, the S197L mutation was reported to have a statistically mild outcome 32. The T205I mutation, present in multiple variants of SARS-CoV-2, has either mild outcome or co-occurs with multiple mutations in other SARS-CoV-2 genes (https://viralzone.expasy.org/9556), making predictions about its physiological effect challenging. A similar situation applies to the P199L mutation, which should decrease the affinity to 14-3-3 at the pS197 site because of the importance of the canonical Pro residue at this position 35 (Table 3, Fig. 5a). Nevertheless, the P199L-mutated N would still retain uncompromised interactions with 14-3-3 using the pT205 site. The S194L mutation associated with severe COVID-19 32 can lead to site-specific strengthening of the interaction with 14-3-3 due to the additional hydrophobic contacts between Leu194 and a leucine residue of 14-3-3 facing this position in the crystal structure (Fig. 5b). The additional stabilization within the 14-3-3 complex is predicted also for the widespread combined R203K/G204R mutation present in at least several variants-of-concern (e.g., alpha, gamma, omicron) 40. As R203K/G204R mutation is expected to enhance phosphorylation of N 40, based on structural observations we anticipate this would alter 14-3-3 interactions. While the R203K mutation is expected to slightly decrease the in-cis contact of the side chain with the phosphate group, the Arg204 side chain is placed at an appropriate distance for making a salt bridge with the conserved Asp225 of 14-3-3 (Fig. 5c). The S202R mutation has the most profound effect on the efficiency of N to package RNA and produce new viral particles 18 and has at least two predictable effects. First, the S202R mutation is expected to enhance phosphorylation of Thr205, at least by basophilic protein kinases (by forming a canonical sequence RRXT instead of just RXT 41). Second, the S202R mutation converts the..SRGpTSP.. 14-3-3-binding site from suboptimal to canonical (..RRGpTSP..) 35, and thereby is expected to enhance 14-3-3 binding (Fig. 5d). The effect of the R203M mutation associated with the Delta variant is difficult to predict at the level of 14-3-3 binding, because this mutation would first of all interfere with Thr205 phosphorylation in the SMGT205SP context, at least by basophilic protein kinases.
Coupling between the effect of mutations in N of SARS-CoV-2 variants and binding to 14-3-3 proteins. Bold font indicates prediction of the most pronounced stabilizing effect on 14-3-3 binding. Underlined italics indicate that the mutation blocks 14-3-3 binding at the corresponding site.
Discussion
Since the beginning of the pandemic SARS-CoV-2 acquired mutations that improve the invasion, replication, evasion of the immune system, or transmissivity and become widespread. The benefits of mutations in the S-protein can often be rationalized via their direct effect on viral entry, which have now been structurally characterized 42,43. However, the effects of mutations in N remain largely unknown. Curiously, the majority of the widespread N mutations beneficial for the virus are mapped to a very small segment, residues 194-205 (Fig. 1a, Table 3) 18,19,33. In fact, all variants-of-concern defined by the World Health Organization feature at least one mutation in the 199-205 subregion 18. This coincides with the maximum Shannon entropy in the SARS-CoV-2 genome centered at residue 203 and surrounding positions within N 19. However, this mutational hotspot SR-rich region of N is disordered, making it challenging to study the consequences of relevant mutations at the structural level. The fact that N appears to have many functional roles, coupled with the lack of appropriate model systems for testing the effect of mutations on N functionality in vivo, further challenges our ability to deconvolute the underlying effects.
Despite these challenges, recent work has shown that the viral-like particles can be used to rapidly assess evolved SARS-CoV-2 variants 18. According to this study, the large effect of the S202R and R203M mutations within the mutational hotspot of SARS-CoV-2 N can be explained by the vastly improved RNA packaging mechanisms and production of higher titers of infectious virions (166-and 51-fold, respectively) 18. The molecular and structural consequences of these and other widespread mutations of N have yet to be elucidated.
We questioned how post-translational modifications and these mutations might affect N interactions with host cell factors. In previous work, we showed that SARS-CoV-2 N phosphorylated at more than 20 sites, including several ones within the SR-rich region (176-210) that are phosphorylated in vivo 4,14, can be specifically recognized by human 14-3-3 proteins, presumably, using the conserved motif phosphorylated at Ser197 26.
In the present work, we first used a unique SARS-CoV-2 N protein variant containing a single phosphoserine at position Ser197 to verify the role of this site in recruiting 14-3-3 proteins (Fig. 1). Consistent with this, we show that SARS-CoV N, which shares the pS197 site with SARS-CoV-2 N, also interacts with 14-3-3 in a phosphorylation-dependent manner (Supplementary Fig. 1). Surprisingly, the S197L variant of SARS-CoV-2 N found in the natural variant circulating in Europe in 2020 and associated with a generally mild outcome 32, also displayed a hallmark phosphorylation-dependent interaction with 14-3-3 (Fig. 2). This indicated that N had other 14-3-3-binding phosphorylation sites besides pS197. Since the SR-rich tract, the key determinant of 14-3-3 binding 26, also contains another similar site around Thr205, we compared the interaction of both phosphorylated fragments, pS197 and pT205, with all human isoforms of the 14-3-3 protein.
Using high-throughput fluorescence anisotropy data (Fig. 3), we found that both SARS-CoV-2 N phosphopeptides display the characteristic peptide-affinity profiles correlating well (r=0.87 and 0.93) with the general affinity hierarchy of the human 14-3-3 isoforms 28. The affinity profiles for the two SARS-CoV-2 N fragments to 14-3-3 proteins were significantly shifted and nonparallel to each other, indicating selectivity toward the 14-3-3 isoforms (Fig. 3). Unexpectedly, the pT205 fragment interacted with all seven 14-3-3 proteins with a consistently higher affinity than the pS197 fragment. Moreover, the interaction of the pT205 peptide with the weakest isoform, 14-3-3σ, was still stronger than the interaction of the pS197 peptide with the strongest isoform, 14-3-3γ (Fig. 3). Most importantly, we identified the remarkable selectivity profile for the 14-3-3 family, a phenomenon that, to the best of our knowledge, has not been previously recognized with respect to the 14-3-3 proteins. This selectivity trend also correlated well (R2=0.84; r=0.93) with the general peptide-affinity hierarchy of the 14-3-3 isoforms 28. According to these results, not only the strongest peptide binder, 14-3-3γ, binds the pT205 peptide stronger, but that its selectivity to this peptide over the pS197 peptide is more pronounced than in the case of generally the weakest isoform, 14-3-3σ (Fig. 3f, Table 1).
To explain the established binding preferences of 14-3-3 proteins toward one of the apparently similar phosphopeptides, we determined crystal structures of 14-3-3σ with both peptides (Fig. 4 and Table 1). The X-ray structures enabled tracing of residues 193-200 and 201-210, respectively, and conferred the following detailed analysis. The central portions of the peptides adopt very similar conformations forming roughly equivalent contacts with 14-3-3. By contrast, specific stabilizing interactions beyond the central region were observed only in the case of the pT205 peptide. The pS197 and pT205 fragments are mutually exclusive, because their simultaneous placement within the 14-3-3 dimer is not possible due to steric reasons (Fig. 4c).
The crystal structures of the 14-3-3-bound SARS-CoV-2 N fragments 193-200 and 201-210 provide a valuable structural framework for analyzing the effect of the widespread mutations and testing new hypotheses. Collectively, our analyses based on structural, thermodynamic and bioinformatic data support the hypothesis 19 that beneficial SARS-CoV-2 N mutations positively affect 14-3-3 binding (Fig. 5).
Last but not least, the 14-3-3-binding region, 193-210, is immediately flanked by the regions 179-184 and 217-222 (Fig. 1a) that were proposed to drive amyloid aggregation of N in the course of liquid-liquid phase separation in the presence of RNA 22. Because this process plays a role in viral replication 17, one can expect the influence of N phosphorylation and 14-3-3 binding as a next level of regulation of these processes and also a basis for the effect of the widespread mutations. While this warrants further investigation, we note that interaction with host signaling proteins such as 14-3-3, mediated by structurally disordered segments containing phosphorylation sites, is emerging as a cornerstone in virus-host interactions 25,26,28,44–48.
Methods
Peptides and chemicals
SARS-CoV-2 N peptides phosphorylated at Ser197 (WSSRNpSTPGSS) or Thr205 (WSSRGpTSPARM) were synthesized by Severn Biotech (UK). The phosphopeptide of human HSPB6 (pB6; WLRRApSAPLPGLK) was obtained as previously described 37. Expected and experimentally determined masses of the peptides are listed in Supplementary Table 1. FITC was from Sigma-Aldrich (cat no. F7250). All common chemicals were of the highest purity and quality available, and all solutions were made with milliQ grade water.
FITC labeling of the SARS-CoV-2 N phosphopeptides
SARS-CoV-2 N peptide labeling was achieved in 0.1 M carbonate buffer, pH 9.0, by mixing a 5 mg/ml solution of the peptide with 1/10 volume of the FITC stock solution (9 mg/ml) in DMSO. The pB6 phosphopeptide 37 was labeled according to the same scheme and was used for controls. The mixtures were wrapped with aluminum foil and incubated overnight at room temperature, then the reaction was quenched by the addition of Tris to a final concentration of 1 M. The FITC-labeled peptides were separated from the unreacted label by size-exclusion chromatography on a 2 ml Sephadex G10 column equilibrated and run using a 20 mM Tris-HCl buffer, pH 7.5, 150 mM NaCl at a 0.5 ml/min flow rate. The runs were operated by the Prostar 335 system (Varian Inc., Australia) following the full-spectrum absorbance. The resulting spectrum, and absorbance at 280 (A280) and 493 nm (A493) in particular, was used to estimate the phosphopeptide concentration (Cpeptide), taking into account FITC absorbance at 280 nm using the formula 49:
 where εpeptide, 280 is the extinction coefficient of the peptide at 280 nm (for all peptides in the study 5500 M-1 cm-1). FITC concentration was determined from the FITC-specific absorbance at 493 nm using the extinction coefficient of 68,000 M-1 cm-1. The completion of the labeling reaction was confirmed by LC-MS on a Bruker Impact II mass-spectrometer (Supplementary Table 1 and Supplementary Fig. 7).
where εpeptide, 280 is the extinction coefficient of the peptide at 280 nm (for all peptides in the study 5500 M-1 cm-1). FITC concentration was determined from the FITC-specific absorbance at 493 nm using the extinction coefficient of 68,000 M-1 cm-1. The completion of the labeling reaction was confirmed by LC-MS on a Bruker Impact II mass-spectrometer (Supplementary Table 1 and Supplementary Fig. 7).
The labeling efficiency was 74-76% for all phosphopeptides. Labeled peptides were stored in aliquots at -80 °C.
Plasmids and mutagenesis
Cloning of the SARS-CoV-2 N sequence (Uniprot ID P0DTC9) was described in previous work 26. The protein contained an N-terminal His6-tag cleavable by rhinovirus 3C protease, which left extra residues GPA on the N terminus of the protein. The wild-type SARS-CoV-2 N plasmid was used as template to PCR amplify the S197L mutant by the megaprimer method 50 using the S197L reverse mutagenic primer (5’-CTGGAGTTAAATTTCTTGAACTG-3’, the mutated codon underlined). For site-specific phosphoserine incorporation into position 197 of SARS-CoV-2 N using amber codon suppression 30, we created the S197TAG mutant, which was obtained by the megaprimer method using the reverse mutagenic primer (5’-CTGGAGTCTAATTTCTTGAACTG-3’, the mutated codon underlined) and DNA template being the SARS-CoV-2 N plasmid containing a C-terminal His6-tag cleavable by 3C protease. The resulting PCR product containing the S197TAG substitution and carrying the C-terminal cleavable His6-tag was treated with NdeI/HindIII restriction endonucleases and then cloned into a pRBC_14-3-3γ/His6SUMO_mBAD43-204 vector 30 pretreated with the same endonucleases. The C-terminal His6-tag enabled the use of the phosphoserine-incorporating E.coli system [BL21 ΔserB(DE3) cells transformed by the pKW2-EFSep plasmid (chloramphenicol resistance) and the target N plasmid] having the Release Factor 1 (responsible for translation termination at TAG codons) 51. Despite contamination by the truncated product due to the preliminary translation termination at the TAG197 codon, only the C-terminally His-tagged full-length target protein with incorporated phosphoserine could be purified by immobilized metal-affinity chromatography (IMAC). After cleavage by 3C protease, such protein would retain extra residues GLEVLFQ on its C terminus. The correctness of the obtained constructs and the presence of the desired mutations were confirmed by DNA sequencing in Evrogen, Moscow (see exemplary chromatograms for S197L and S197TAG in Supplementary Fig. 8).
The SARS-CoV N-containing pGEX plasmid (ampicillin resistance) was kindly provided by Prof. Fulvio Reggiori (University of Groningen, The Netherlands).
Untagged full-length human 14-3-3γ (Uniprot ID P61981) cloned into a pET21 vector (ampicillin resistance) 52, the surface entropy reduction mutants 53 of the C-terminally truncated human 14-3-3σ (residues 1-231), i.e. Clu1 (159KKE161 → 159AAA161) and Clu3 (75EEK77 → 75AAA77), each containing an N-terminal 3C protease cleavable His6-tag and cloned into the pET28 vector (kanamycin resistance) have been described previously 37. Homotypic constructs of the full-length human 14-3-3 protein isoforms beta (β), gamma (γ), eta (η), tau (τ), zeta (ζ), epsilon (ε), and sigma (σ), fused to the C terminus of the maltose-binding protein (MBP) and described earlier 28, were kindly shared by the laboratory of Prof. Gilles Travé (Université de Strasbourg, Illkirch, France). Parameters of proteins used are provided in Supplementary Table 2.
Protein expression and purification
Proteins were expressed in E.coli BL21(DE3) by the addition of IPTG up to 0.5 mM for 20 h at 25 °C (N proteins), or for 4-6 h at 37 °C (14-3-3 proteins). To ensure phosphorylation of SARS-CoV-2 N, its S197L mutant, or SARS-CoV N in E.coli cells, target proteins were co-expressed with the catalytically active subunit of mouse protein kinase A (PKA) encoded in a separate pACYC-PKA plasmid (chloramphenicol resistance), essentially as described earlier 26. The site-specifically phosphorylated SARS-CoV-2 N protein Ser197TAG was expressed in BL21 ΔserB(DE3) cells tailored to co-translational phosphoserine incorporation 51.
All N proteins, and Clu1 and Clu3 mutants of 14-3-3σ used for crystallization, were purified by subtractive immobilized metal affinity and size-exclusion chromatography, where two IMAC steps were separated by 3C cleavage of the His6-tag 37. To remove large amounts of contaminating nucleic acids non-specifically bound to N protein, we used a continuous washing step (50 column volumes) by 3 M NaCl before elution with imidazole during IMAC1. Most proteins were obtained at amounts exceeding 10 mg per 1 liter of bacterial culture. While the yield of the SARS-CoV-2 N.Ser197TAG phosphoprotein was particularly low (∼1 mg per 1 liter of bacterial culture) and the full-length protein was expressed alongside with the truncated side-product 1-196, the resulting protein was indeed phosphorylated according to Phos-tag SDS-PAGE 54. All purified N preparations used in this study showed an A260/A280 ratio of 0.57-0.6 and were free from nucleic acids. To minimize protein degradation and dephosphorylation, SARS-CoV-2 N.S197TAG was purified in the presence of inhibitors of proteases (EDTA-free Pierce™ Protease Inhibitor Mini Tablets, cat. no. A32955) and phosphatases (orthovanadate, glycerophosphate and NaF).
SARS-CoV N was expressed as a GST-fusion and purified by a combination of a GSTtrap step 1, 3C proteolysis to cut off GST, a GSTtrap step 2 and heparin-affinity chromatography.
Untagged 14-3-3γ was expressed in E.coli BL21(DE3) cells by IPTG induction (final concentration of 1 mM) and purified by ammonium sulfate fractionation, anion-exchange and size-exclusion chromatography as described earlier 26. Expression of the MBP-14-3-3 constructs was the same, but these proteins were purified using a combination of MBPTrap column and size-exclusion chromatography 28. Protein concentrations were determined spectrophotometrically at 280 nm on a NP80 nanophotometer (Implen, Germany) using extinction coefficients listed in Supplementary Table 2.
Analytical size-exclusion chromatography
The interaction of N proteins and 14-3-3γ was analyzed using a Superdex 200 Increase 5/150 column (GE Healthcare) operated at a 0.45 ml/min flow rate on a Prostar 335 chromatographic system (Varian, Australia). When required, the eluted fractions were collected in eppendorf tubes and their protein content was analyzed by SDS-PAGE 55. In specific cases, a Superdex 200 Increase 10/300 column and a 0.8 ml/min flow rate was used. The buffer, used to equilibrate the columns, contained 20 mM Tris-HCl pH 7.6, 3 mM NaN3 and 200-300 mM NaCl. We noticed that free unphosphorylated SARS-CoV-2 N.S197L absorbed onto the column when equilibrated with <220 mM NaCl. Therefore, the reported experiments contained at least this much salt in the SEC buffer. Experiments were done at least three times with qualitatively very similar results.
Multi-angle light scattering (MALS)
To determine absolute masses of MBP-14-3-3ζ, 14-3-3γ and its complex with phosphorylated SARS-CoV-2 N.S197L, we used SEC coupled to MALS. To do this, a chromatographic system was connected to the miniDAWN detector (Wyatt Technology) calibrated relative to the scattering from toluene. The concentration signal was obtained from SEC traces recorded at 280 nm and weight extinction coefficients 1.377 for MBP-14-3-3ζ, 1.13 for 14-3-3γ, and 1.02 for the 14-3-3γ/pN.S197L complex. The Mw distributions were calculated in Astra 8.0 software (Wyatt Technology) using dn/dc=0.185. Protein content in the peaks was additionally analyzed by SDS-PAGE 55.
Fluorescence anisotropy
Fluorescence anisotropy (FA) was measured at 25 °C on a Clariostar plus microplate reader (BMG Labtech, Offenburg, Germany) using a set of bandpass filters (482±16 nm and 530±40 nm), FITC labeled phosphopeptides (100 nM, on a 20 mM HEPES-NaOH buffer, pH 7.5, 150 mM NaCl, 0.02% Tween 20) and 384-well plates (Black Nunc™ 384-Shallow Well Standard Height Polypropylene Storage Microplates, Thermofisher scientific, cat. no. 267460). A series of concentrations of MBP-14-3-3 proteins in a 20 mM HEPES-NaOH buffer, pH 7.5, 150 mM NaCl, 0.01% Tween 20 (33 μl each) was diluted 2-fold by a 100 nM solution of either FITC-labeled peptide to obtain 66 μl samples containing 50 nM peptide, MBP-14-3-3 (from 0 to 110 μM) and 0.01% Tween 20. After 5 min incubation at room temperature the fluorescence anisotropy readout did not change with time, indicating equilibrium. Then, each sample was split into three 20 μl aliquots, the plates were centrifuged for 1 min at 25 °C at 250 g to remove air bubbles, and the FP data were recorded. The triplicate measurements were converted into mean ± standard deviation values, which were used for the data presentation and fitting. To determine KD values, the binding curves were approximated using the quadratic equation 56 in Origin 9.0 (OriginLab Corporation, Northampton, MA, USA). Selectivity coefficient reflecting the preferred 14-3-3 binding to the pT205 peptide than the pS197 peptide, which by definition is an equilibrium constant (Keq = 1/KD) for the corresponding displacement reaction, was determined as the ratio of KD for the pS197 peptide to KD for the pT205 peptide. To determine the correlation between selectivity or -logKD values for the 14-3-3/phosphopeptide pairs and average ΔΔG values characterizing the affinity hierarchy reported earlier 28, we used the standard Pearson’s correlation coefficient r and coefficient of determination R2.
Crystallization and X-ray data collection
Crystallization was performed using 14-3-3σ surface entropy reduction mutants Clu1 or Clu3 (storage buffer 20 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1 mM EDTA, 2 mM DTT, 2 mM MgCl2) mixed with either pS197 or pT205 phosphopeptide at a 1:5 protein:peptide molar ratio with the final protein concentration of 11.5 mg/ml. The best diffracting crystal for the 14-3-3σ Clu1/pT205 complex grew in condition H7 (0.2 M AmSO4, 100 mM BisTris pH 5.5, 25% PEG 3350) of JCSG+ screen (Molecular Dimensions, Sheffield, UK); the best diffracting crystal for the 14-3-3σ Clu3/pS197 complex grew in condition G2 (0.2 M NaBr, 100 mM BisTris propane pH 7.5, 20% PEG 3350) of PACT screen (Qiagen, Hilden, Germany). Crystals were mounted in nylon loops directly from crystallization drops and flash frozen in liquid nitrogen without additional cryoprotection.
Synchrotron diffraction data (Table 1) were collected at 100 K at the Diamond Light Source (Oxfordshire, UK) and processed using DIALS 57.
Structure determination and refinement
The structures were solved using MolRep 58 and an unliganded 14-3-3σ dimer (PBD 5LU2) as a search model. Peptide residues were manually built into difference electron density maps in Coot 59 and the overall structures were refined using Buster 2.10.4 60, which used NCS information (with pruning), TLS and all-atom individual isotropic B-factor restrained refinement. In the case of the 14-3-3σ Clu1/pT205 complex, the refinement was additionally restrained using the target structure of an unliganded 14-3-3σ dimer (PBD 3IQU, 1.05 L 61). The refined structures were validated using the comprehensive validation algorithm in Phenix 1.10 62. Structural illustrations were prepared using PyMOL 2.20 (Schrodinger, Inc.).
Data availability
The refined models and structure factors have been deposited in the Protein Data Bank under the accession codes 7QIK and 7QIP. All materials are available from the corresponding authors upon reasonable request.
Conflict of interests
The authors declare no conflicts of interest.
Author Contributions
NNS and AAA conceived studies on N interaction with 14-3-3; KVT, AAS, JLRS, NNS - expressed and purified proteins; KVT - performed fluorescence anisotropy experiments; JLRS - crystallized proteins; NNS - determined crystal structures; KVT, RBC, AAA, NNS - analyzed data; NNS and KVT made illustrations; NNS wrote the paper with input from RBC and AAA.
Supplementary information
Supplementary Tables
Monoisotopic masses of the synthetic peptides and their FITC-labeled derivatives used in the study.
Parameters of protein constructs used in this study.
Supplementary Figures
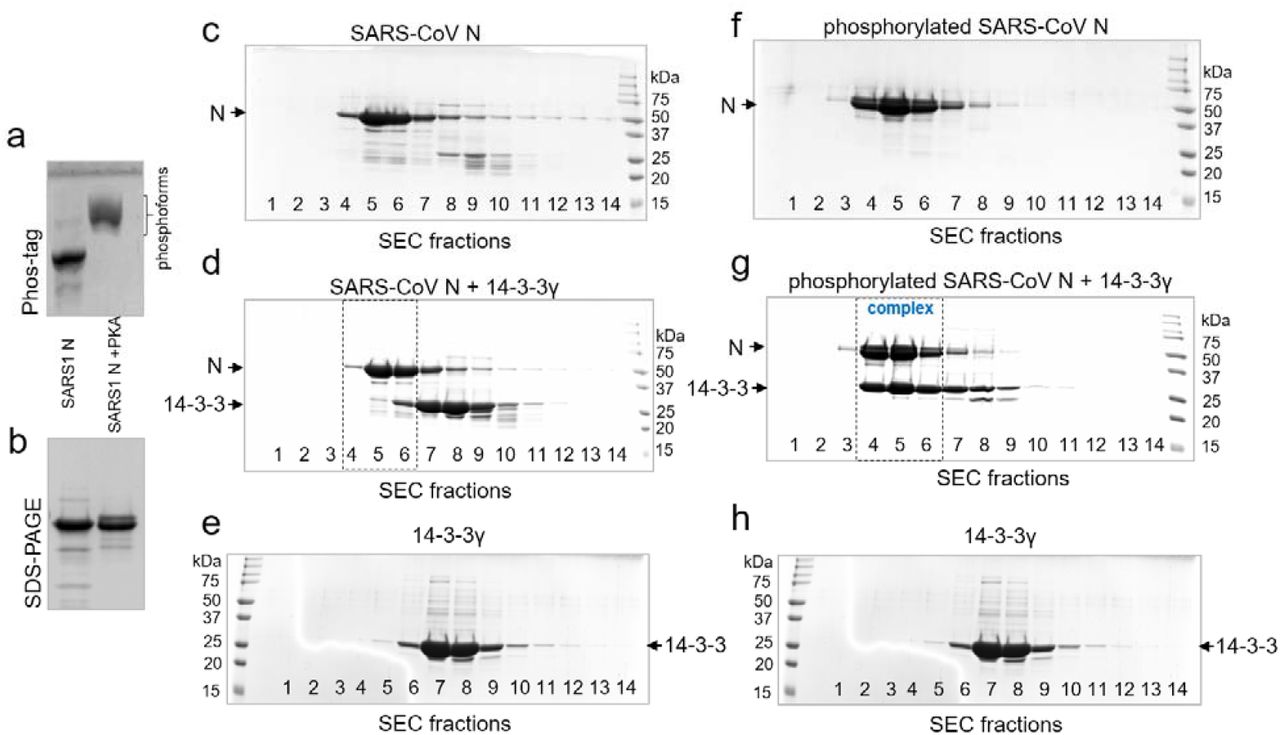
SARS-CoV N interacts with human 14-3-3γ in a phosphorylation-dependent manner. a. Phosphorylation of SARS-CoV N analyzed by a Phos-tag gel. b. The same samples as in a were analyzed by SDS-PAGE. c-e. The interaction of unphosphorylated SARS-CoV N with human 14-3-3γ analyzed by SEC at a 14-3-3 excess and 220 mM NaCl in the samples and running buffer, with SDS-PAGE of all fractions along the elution profiles. f-h. The interaction of PKA-phosphorylated SARS-CoV N with human 14-3-3γ analyzed by SEC at a 14-3-3 excess and 220 mM NaCl in the samples and running buffer, with SDS-PAGE of all fractions along the elution profiles. c and f correspond to SEC profiles of N or pN. d and g correspond to SEC profiles of 14-3-3 mixtures with either N or pN. e and h correspond to SEC profiles of 14-3-3. Molecular mass markers are indicated in kDa. Positions of proteins are indicated by arrows. Note that only phosphorylated SARS-CoV N forms a complex with 14-3-3γ (fractions 4-6 outlined by dashed rectangles).

SEC-MALS analysis of the MBP-14-3-3ζ fusion. a. SEC-MALS profile from the Superdex 200 Increase 10/300 column followed by absorbance at 280 nm showing the Mw distribution across the protein peak as calculated from MALS data. Polydispersity index (Mw/Mn) and the expected Mw for the protein dimer are indicated. Flow rate was 0.8 ml/min. b. SDS-PAGE analysis of the fractions collected from the column. Mass markers are indicated in kDa. Protein position and its apparent mass are marked by an arrow.

SARS-CoV-2 N phosphopeptides interact with the common peptide-binding 14-3-3 grooves. The FITC-labeled SARS-CoV-2 N pT205 phosphopeptide is outcompeted from its complex with human 14-3-3γ either by the same unlabeled phosphopeptide (a) or by the control unlabeled pB6 peptide (b) as monitored by fluorescence anisotropy. c. Titration of the FITC-labeled pB6 phosphopeptide by MBP-14-3-3ε, with the corresponding KD. In a-c, background anisotropy was subtracted from all values. The data are shown as mean ± standard deviation (n=3).
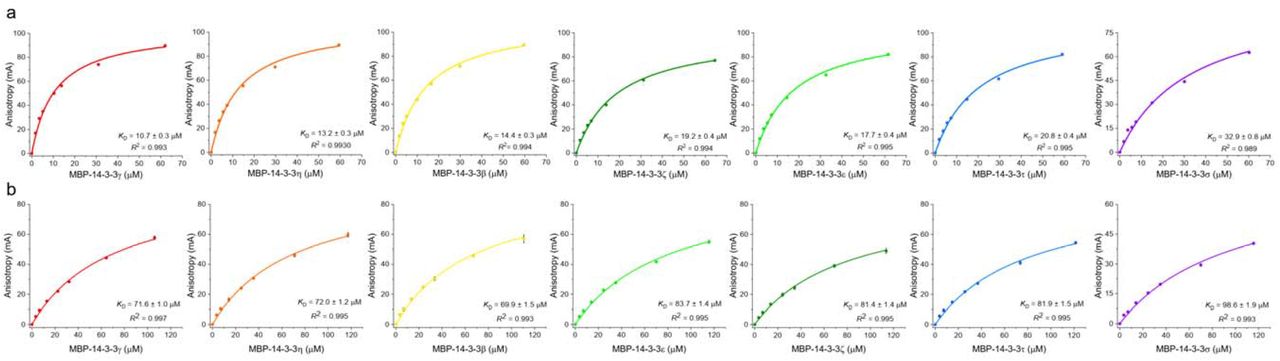
Titration of SARS-CoV-2 N phosphopeptides by the seven human 14-3-3 isoforms monitored by fluorescence anisotropy. a. Binding curves for the pT205 peptide. b. Binding curves for the pS197 peptide. Fitting of the curves was done in Origin 9.0 by standard quadratic binding equation 56. Apparent dissociation constants (KD) at equilibrium are shown on the panels along with the coefficient of determination R2. The data are shown as mean ± standard deviation (n=3). Fluorescence anisotropy data throughout titrations were corrected by subtracting the background anisotropy values.

The six core residues in the two SARS-CoV-2 N phosphopeptides form similar contacts with 14-3-3σ. a, b. Superimposition of the two crystal structures showing the similarity of the conformation of both peptides and molecular contacts formed, looking at two different angles. H-bonds are shown by yellow dashed lines, hydrophobic interaction between the methyl group unique to pT205 and the side chain of Val178 of 14-3-3σ is shown by large semi-transparent spheres, distances characterizing key polar contacts are indicated in □. Note that the hydrophobic contact made by the methyl group of pT205 is somewhat compensated in the pS197 peptide by an additional H-bond from the pS197 peptide’s Asn196 side chain and the side chain of Asn226 of 14-3-3σ, making the interaction of the core part of the peptides nearly equivalent in terms of the amount of chemical contacts.

The unique salt bridge-forming glutamate residue is highly conserved in the human 14-3-3 protein family. a. Crystal structure of 14-3-3σ subunit (grey cartoon) complexed with the pT205 phosphopeptide of SARS-CoV-2 N (orange sticks) showing the location of the unique salt bridge (red dashed lines). 2Fo-Fc electron density map for the analyzed region is contoured at 1σ. b. Close-up of the salt bridge between the side chains of Arg209 of the SARS-CoV-2 N peptide and Glu14 of 14-3-3σ showing characteristic distances in D. 2Fo-Fc electron density map for the analyzed region is contoured at 1σ. c. Local multiple sequence alignment of the human 14-3-3 isoforms in their N-terminal region, showing the absolute conservation of the glutamate partaking in the salt bridge to Arg209 of SARS-CoV-2 N. To the best of our knowledge, this glutamate is conserved in all 14-3-3 proteins. This implies that the ability to stabilize the binding of the pT205 phosphopeptide of SARS-CoV-2 N is shared by all isoforms with no exception.

MS-based validation of the peptides and their labeled derivatives. LC-MS spectra for the pS197 peptide (a) and its FITC-labeled derivative (b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sequencing results for the S197L and S197TAG mutants of SARS-CoV-2 N. The introduced changes are highlighted by red. On top, the nucleotide and amino acid sequences for the wild-type protein are shown for reference.
Acknowledgements
We thank Prof. Gilles Trave’s laboratory for providing the MBP-14-3-3 plasmids, Prof. Fulvio Reggiori for the SARS-CoV N plasmid, Dr. Olga Moroz for help with crystallization and Dr. Gergo Gogl for advice concerning fluorescence anisotropy. The authors are also thankful to Sam Smith for help with X-ray data collection. LC-MS and MALS were carried out at the Shared-Access Equipment Centre “Industrial Biotechnology” of the Federal Research Center “Fundamentals of Biotechnology” of the Russian Academy of Sciences. The study was supported by the Ministry of Science and Higher education of the Russian Federation in the framework of the Agreement no. 075-15-2021-1354 (07.10.2021) and the Wellcome Trust (206377 award to A.A.A.). R.B.C. acknowledges support of the Medical Research Foundation at the Oregon Health Sciences University (USA) and the Collins Medical Trust. Expression and purification of 14-3-3 proteins were partially supported by the Russian Science Foundation (no. 19-74-10031).
References




