

Abstract
Snakebite envenoming affects more than 250,000 people annually in sub-Saharan Africa. Envenoming by Dispholidus typus (boomslang) results in venom induced consumption coagulopathy, whereby highly abundant prothrombin-activating snake venom metalloproteinases (SVMPs) consume clotting factors and deplete fibrinogen. The only available treatment for D. typus envenoming is the monovalent SAIMR Boomslang antivenom. Treatment options are urgently required because this antivenom is often difficult to source and, at $6,000/vial, typically unaffordable for most snakebite patients. We therefore investigated the in vitro and in vivo preclinical efficacy of four SVMP inhibitors to neutralise the effects of D. typus venom; the matrix metalloproteinase inhibitors marimastat and prinomastat, and the metal chelators dimercaprol and DMPS. The venom of D. typus exhibited an SVMP-driven procoagulant phenotype in vitro. Marimastat and prinomastat demonstrated equipotent inhibition of the SVMP-mediated procoagulant activity of the venom in vitro, whereas dimercaprol and DMPS showed considerably lower potency. However, when tested in preclinical murine models of envenomation, DMPS and marimastat demonstrated partial protection against venom lethality, demonstrated by prolonged survival times of experimental animals, whereas dimercaprol and prinomastat failed to confer any protection at the doses tested. The results presented here demonstrate that DMPS and marimastat show potential as novel small molecule-based therapeutics for D. typus snakebite envenomation. These two drugs have been previously shown to be effective against Echis ocellatus venom induced consumption coagulopathy (VICC) in preclinical models, and thus we conclude that marimastat and DMPS may be valuable early intervention therapeutics to broadly treat VICC following snakebite envenoming in sub-Saharan Africa.
1. Introduction
More than 250,000 cases of snakebite envenoming are estimated to occur annually in sub-Saharan Africa1, disproportionately affecting those in rural, impoverished communities without adequate access to healthcare2,3. Venom-induced consumption coagulopathy (VICC) is a common manifestation of snakebite envenoming, during which procoagulant venom toxins consume clotting factors resulting in the ensuing depletion of fibrinogen and, ultimately, coagulopathy4. Several clotting factors are the target for procoagulant snake venom toxins, and these include Factor X, Factor V, fibrinogen and prothrombin4. While infrequent, envenomings by the rear fanged African colubrid Dispholidus typus (boomslang) are characterised by causing VICC5–7. The venom of this species is known to potently activate prothrombin8, resulting in the liberation of thrombin, and the subsequent downstream consumption of fibrinogen and fibrin, causing dysregulation of coagulation8–10. The activation of prothrombin is likely the result of snake venom metalloproteinases (SVMPs)9,10, which are the dominant toxin type present in the venom and account for almost 75% of the proteinaceous toxins11,12. Other minor toxin families identified in the D. typus venom proteome include three-finger toxins, phospholipases A2 (PLA2s), cysteine-rich secretory proteins (CRISPs), snake venom serine proteases and C-type lectin-like toxins (of which each constitute <10% of the venom proteome)11, though their contribution to envenoming pathology remains unclear.
Dispholidus typus is broadly distributed throughout much of sub-Saharan Africa (sSA) and whilst incidences of envenoming are fortunately rare, the rapid and severe VICC consequences pose considerable clinical challenges. This is because the only specific treatment for D. typus envenoming is the monospecific F(ab’)2 antivenom “SAIMR Boomslang” (South African Vaccine Producers Pty Ltd), which has limited availability outside of the Southern Africa Economic Community, and costs as much as US$6050 per vial13. Given that D. typus exhibits a broad geographical distribution throughout much of sub-Saharan Africa (sSA), the only specific for treatment for envenomings caused by this species is largely unobtainable for snakebite victims who either cannot afford, or do not have access to, the antivenom9, and thus investigating novel treatments is a research priority.
More generally, it is well recognised that conventional polyclonal antibody-based antivenoms have several shortcomings, despite being life-saving therapeutics. In addition to often being unaffordable to many snakebite victims, they are associated with high rates of adverse reactions14,15, and have poor dose efficacy, with only ~10-20% of the active immunoglobulins recognising and binding to venom toxins16. Logistically, antivenoms are poorly suited for the rural locations in which they are typically required; for example many antivenoms rely on cold chain transport and storage and must be administered intravenously by trained staff in healthcare facilities17. Indeed, up to 75% of deaths from snakebite are estimated to occur before patients are able to reach healthcare facilities, thus there is a compelling need to identify novel snakebite treatments that could be administered in the community soon after a bite18.
To this end, small molecule-based drugs (i.e. ‘toxin inhibitors’) have received considerable interest as novel snakebite therapeutics, both as potential individual treatments or in combination with existing antivenoms18–20. Small molecule drugs have a number of potentially advantageous characteristics over antivenoms, including improved affordability and stability, oral delivery format, higher tolerability20, and improved tissue penetration21,22. Previously, Ainsworth et al demonstrated the in vitro inhibitory effect of the metal chelator EDTA against SVMP-mediated prothrombin degradation caused by D. typus venom, suggesting that small molecule inhibitors may be effective therapeutics for D. typus envenoming9. In the same study, Ainsworth et al demonstrated in a murine preclinical model that EDTA was protective against the lethal effects of Echis ocellatus venom, an African viper which, similar to D. typus venom, contains a high abundance of SVMP toxins9, including prothrombin activators, and causes VICC in envenomed victims23,24. Other small molecule drugs with SVMP-inhibiting potential include other metal chelators, such as dimercaprol and DMPS (2,3-dimercapto-1-propanesulfonic acid)25–27, and the mimetic matrix metalloprotease inhibitors marimastat, batimastat and prinomastat22,24,28. Marimastat and batimastat were found to effectively inhibit SVMP activity and reduce haemorrhagic pathologies in murine models of E. ocellatus envenoming24, and inhibit the procoagulant effects of several viper venoms in vitro24,25. Similarly, prinomastat (AG-3340) showed inhibitory activity against the haemorrhagic effects of both purified SVMPs and the crude venom of E. ocellatus28. The metal chelators dimercaprol and DMPS have also been shown to inhibit SVMP activity of E. ocellatus venom in vitro, with DMPS also demonstrating in vivo preclinical neutralisation of venom lethality and haemorrhage26. While D. typus venom is dominated by SVMPs, PLA2 toxins are also thought to contribute to the coagulopathy induced by this venom29. The small molecule drug varespladib has been extensively investigated as an inhibitor of venom PLA2 toxins found in a wide geographical range of snake species, with such studies showing potent neutralisation of PLA2 activity25,27,30 and associated anticoagulant, haemorrhagic, myotoxic and neurotoxic pathologies31–35. Thus, small molecule treatments for snakebite have the potential to overcome the current species-specific and restrictive geographical utility of current antivenoms.
Despite these promising recent research outcomes, further investigation is required to explore the inhibitory breadth and potency of small molecule toxin inhibitors due to the ubiquitous variability in snake venom composition and therefore, also, the variant toxin specificities of these inhibitory small molecule drugs. In particular, despite overarching similarities in venom composition and ensuing snakebite pathology between Echis spp. and D. typus9, the SVMPs of D. typus have evolved their prothrombin activating ability independently of those found in the venom of Echis spp.10. Consequently, building on previous principles demonstrated for Echis spp.24–26,36, in this study four small molecule drugs were investigated in vitro and in vivo to assess their inhibitory potential against the venom of D. typus. To do so, we applied in vitro metalloproteinase and coagulation bioassays on crude and nanofractionated venom, and in vivo murine models of envenoming to assess neutralisation of venom lethality.
2. Methods
2.1 Venoms
Lyophilised D. typus venom (Product code L1403, origin South Africa, purity >99%) was sourced from Latoxan (Portes les Valence, France) and stored at 4 °C to ensure long-term stability. Prior to use, venom was resuspended in PBS (pH 7.4, Gibco) at 1 mg/mL for in vitro experiments and 5 mg/mL for in vivo experiments.
2.2 Drug preparations for in vitro studies against crude venom
The small molecule SVMP inhibitors tested were; dimercaprol (2,3-dimercapto-1-propanol, ≥98 % iodometric, Cat no: 64046, Sigma), DMPS (2,3-dimercapto-1-propane-sulfonic acid sodium salt monohydrate, 98%, Cat no: H56578, Alfa Aesar), marimastat ((2S,3R)-N4-[(1S)-2,2-Dimethyl-1-[(methylamino)carbonyl]propyl]-N1,2-dihydroxy-3-(2-methylpropyl)butanediamide, >98%, Cat no: 2631, Tocris Bioscience), prinomastat hydrochloride (Cat no: HY-12170A, >98%, MedChemExpress). Varespladib (2-[[3-(2-Amino-2-oxoacetyl)-2-ethyl-1-(phenylmethyl)-1H-indol-4-yl]oxy]-acetic acid, Cat no: SML1100, >98% HPLC, Sigma) was used as a small molecule drug control. All drugs were reconstituted in dimethyl sulfoxide (DMSO) (Sigma) to 10 mM stocks and stored at −20 °C. Daughter plates were created at 1 mM concentrations in 384-well format to allow the creation of assay-ready plates using a VIAFLO 384 electronic pipette (Integra). Both daughter plates and assay-ready plates were stored at −20 °C, with the latter used within a month of creation. For the SVMP assay 0.91 μL of each drug was plated (final reaction volume of 91 μL), while 0.5 μL was plated for the coagulation assay (final reaction volume of 50 μL). For marimastat, prinomastat and varespladib, dose response curves were created at a final concentration range of 10 μM to 4.8 pM using a two-fold dilution (50 μL drug into 50 μL of DMSO), with each concentration tested in duplicate. For DMPS and dimercarpol, dose response curves were created at a final concentration range of 160 μM to 76.3 pM using a two-fold dilution (50 μL drug into 50 μL of DMSO), with each concentration tested in duplicate.
2.3 In vitro neutralisation of coagulopathic crude venom activity
To assess the inhibitory potency of the selected drugs against coagulopathic venom activity we used a previously described absorbance-based plasma clotting assay37. Citrated bovine plasma (VWR) was defrosted and centrifuged at 858 x g for 5 minutes to remove precipitates before use. Thereafter, 100 ng of venom in 10 μL PBS was added to each well in the 384-well assay-ready plate (containing 0.5 μL of 1 mM of inhibitor) using a VIAFLO 384, the plate was then briefly spun down in a Platefuge (Benchmark Scientific) and incubated at 37 °C for 25 minutes, followed by a further five minutes acclimatisation at room temperate. Next, 20 μL of 20 mM CaCl2 was added using a MultiDrop 384 Reagent Dispenser (ThermoFisher Scientific), followed by the immediate addition of 20 μL citrated bovine plasma. The plate was then immediately read for kinetic absorbance at 595 nm for 116 minutes using a FLUOstar Omega platereader (BMG Labtech).
Assays were performed in triplicate and each assay contained technical duplicates at each dose. Positive control values were generated using DMSO + venom, and negative control values were generated using DMSO in the absence of venom. All compounds were analysed for their ability to return clotting to normal at the timepoint at which the positive and negative absorbance values were furthest apart. For this, the raw values were normalised to show percentage of normal clotting, e.g. a value of 100% meant the compound returned clotting to that of the negative control. These percentage values were plotted and fitted with a nonlinear regression curve for the normalised response (variable slope) using to calculate the IC50 data and 95% confidence intervals for each compound using GraphPad Prism 9.0 (GraphPad Software, San Diego, USA). Multiple comparisons one-way ANOVA test was used to compare IC50 values generated for each replicate plate, using GraphPad Prism 9.0.
2.4 Venom nanofractionation
To further explore the inhibitory specificity of the selected drugs, we fractionated D. typus into toxin constituents and repeated the plasma bioassay. Venom nanofractionation29,38 was performed on a UPLC system (‘s Hertogenbosch, The Netherlands) controlled by Shimadzu Lab Solutions software. Venom solution was prepared by dissolving lyophilised D. typus venom into water (purified by Milli-Q Plus system, Millipore) to a concentration of 5.0 mg/mL and stored at −80 °C until use. For each analysis, 50 μL venom solution (1.0 mg/mL) was injected by a Shimadzu SIL-30AC autosampler after diluting the stock venom solutions (5.0 ± 0.1 mg/mL) in Milli-Q water. A Waters XBridge reversed-phase C18 column (4.6 × 100 mm column with a 5 μm particle size and a 300 Å pore size) was used for gradient separation at 30 °C. Mobile phase A was composed of 98% water, 2% acetonitrile (ACN) (Biosolve) and 0.1% formic acid (FA) (Biosolve), while mobile phase B was composed of 98% ACN, 2% water and 0.1% FA. The total solvent flow rate was maintained at 0.5 mL/min and the gradients were run as follows: linear increase of eluent B from 0 to 50% in 20 min followed by a linear increase to 90% B in 4 min, then isocratic elution at 90% for 5 min, subsequently the eluent B was decreased from 90% to 0% in 1 min followed by an equilibration of 10 min at 0% B. The column effluent was split as two parts (9:1), with the smaller fraction (10%) sent to a Shimadzu SPD-M20A prominence diode array detector. The larger fraction (90%) was directed to a FractioMate nanofractionator (SPARK-Holland & VU) and fractions collected onto transparent 384-well plates (F-bottom, rounded square well, polystyrene, without lid, clear, non-sterile; Greiner Bio One). The nanofractionator was controlled by FractioMator software (Spark-Holland) to collect fractions continuously at a resolution of 6 s/well. After collection, the well plates with venom fractions were dried overnight in a Christ Rotational Vacuum Concentrator (RVC 2-33 CD plus, Zalm en Kipp, Breukelen, The Netherlands), to remove any solvent remaining in the wells. The vacuum concentrator was equipped with a cooling trap maintained at −80 °C during operation. The dried plates were then stored at −20 °C until bioassaying.
2.5 In vitro neutralisation of coagulopathic venom toxin fractions
The small molecule inhibitors marimastat ((2S,3R)-N4-[(1S)-2,2-Dimethyl-1-[(methylamino)carbonyl] propyl]-N1,2-dihydroxy-3-(2-methylpropyl) butanedia-mide), prinomastat hydrochloride (AG-3340 hydrochloride), dimercaprol (2,3-Dimercapto-1-propanol), DMPS (2,3-dimercapto-1-propane-sulfonic acid sodium salt monohydrate) and varespladib (A-001) were purchased from Sigma-Aldrich. Bovine plasma (Sodium Citrated, Sterile Filtered, Product Code: S0260) was purchased from Biowest. For assay preparation, the CaCl2 (Biosolve), which was used to de-citrate plasma to initiate coagulation in the coagulation assay, was dissolved in Milli-Q water to 20 mM. The inhibitors were dissolved in DMSO (≥ 99.9%, Sigma-Aldrich) to a concentration of 10 mM and stored at −20 °C. The plasma was defrosted and then centrifuged at 2000 rpm (805 × g) for 4 min in a 5810 R centrifuge (Eppendorf) to remove possible particulate matter. The inhibitor stock solutions were diluted in PBS buffer to the described concentrations, then 10 μL of each diluted inhibitor solution was pipetted to all wells of plate wells containing freeze-dried nanofractionated venom fractions by a VWR Multichannel Electronic Pipet, followed by centrifuging the plate for 1 min at 805 x g. Next, a pre-incubation step for 30 min at room temperature was performed. Final concentrations of inhibitor solutions used for the coagulation bioassay were 20, 4, 0.8, 0.16 and/or 0.032 μM (with corresponding DMSO final concentrations of 0.02%, 0.004%, 0.0008%, 0.00016% and 0.000032%, respectively). After this incubation step, the HTS coagulation assay was performed as described by Still et al 37. A Multidrop 384 Reagent Dispenser (Thermo Fisher Scientific) was used to dispense 20 μL of CaCl2 solution onto all wells of the 384-well plates, followed by 20 μL plasma after rinsing of the Multidrop with deionized water between dispensing. Kinetic absorbance measurements were conducted immediately for 100 min at 595 nm and 25 °C using a Varioskan Flash Multimode Reader (Thermo Fisher Scientific). Venom-only analyses were performed as control experiments, for which 10 μL PBS instead of inhibitor solution was added to all wells of the vacuum-centrifuge-dried nanofractionated well plates. Each nanofractionation analysis was performed in at least duplicate.
The resulting coagulation chromatograms were plotted as described by Slagboom et al29, with each chromatogram reconstructed to display ‘very fast coagulation’, ‘slightly/medium increased coagulation’ and ‘anticoagulation’. To plot the very fast coagulation chromatogram, the average slope of the first five minutes in the assay was plotted, and for the slightly/medium coagulation chromatogram the average slope of the first 20 minutes was plotted. For anticoagulant chromatogram the final (end-point) read at 100 minutes was plotted. Clotting velocities were all plotted against the venom nanofractionation time, producing positive peaks for procoagulant compounds and negative peaks for anticoagulant compounds.
2.6 In vitro neutralisation of venom SVMP activity
The SVMP activity of crude D. typus venom in the presence of inhibitors or vehicle control (DMSO), was measured using a quenched fluorogenic substrate (ES010, R&D Biosystems), in line with principles previously described26. The substrate was suspended in reaction buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5) and used at a final concentration of 10 μM (supplied as a 6.2 mM stock). Reactions consisted of 1 μg of venom (1 μg in 15 μL PBS) co-incubated with 0.91 μL of 1 mM of inhibitor. The 384 well plate (Greiner) was briefly spun down in a Platefuge (Benchmark Scientific) and incubated at 37 °C for 25 minutes, with an additional 5 minutes acclimatisation at room temperate, before the final addition of the freshly diluted fluorogenic substrate (75 μL of 12.1 μM). The plate was immediately run on a CLARIOstar platereader (BMG Labtech) at an excitation wavelength of 320-10 nm and emission wavelength of 420-10 nm with 10 flashes per well at 25 °C for 100 cycles (each cycle time 79 seconds). The assay was performed independently in technical duplicate. The end-reads were calculated for each sample at the time where all fluorescence curves had typically reached a plateau (maximum fluorescence). SVMP activity was calculated for each test condition as a percentage of the mean of the DMSO only wells (100% activity), with a baseline of the marimastat 10 μM controls representing 0% activity. IC50 values were calculated from the percentage inhibition values by fitting a nonlinear regression curve for the normalised response (variable slope) for each compound using GraphPad Prism 9.0 (GraphPad Software, San Diego, USA). The best-fit IC50 values for each replicate were compared to identify significant differences between the IC50 values for each drug using one-way multiple comparisons ANOVA analysis in GraphPad Prism 9.0.
2.7 In vivo neutralisation of venom lethality
2.7.1 Animal ethics
All animal experiments were performed using protocols approved by the Animal Welfare and Ethical Review Boards of the Liverpool School of Tropical Medicine and the University of Liverpool, under project licence (P58464F90) approved by the UK Home Office in accordance with the UK Animal (Scientific Procedures) Act 1986.
2.7.2 Animal maintenance
Male CD1 mice (18-20g) were sourced from Charles River (UK) and acclimatised for a minimum of one week before experimentation. Mice were grouped in cages of five, with room conditions of approximately 22 °C at 40-50% humidity, with 12/12 hour light cycles, and given ad lib access to CRM irradiated food (Special Diet Services, UK) and reverse osmosis water in an automatic water system. Mice were housed in specific-pathogen free facilities in Techniplast GM500 cages containing Lignocell bedding (JRS, Germany), Sizzlenest zigzag fibres as nesting material (RAJA), and supplied with environmental enrichment materials.
2.7.3 Co-incubation model of preclinical efficacy
The median murine lethal dose (LD50) for D. typus venom administered by intravenous injection was previously determined as 22.29 μg per mouse9. To determine the efficacy of small molecule inhibitors against D. typus venom, a refined version of the WHO recommended antivenom ED50 neutralisation experiments was used, in which ~4 x LD50 doses of venom (90 μg) were pre-incubated with each small molecule inhibitor. Drug stocks were freshly prepared to allow for a ratio of 1:1.33 venom to inhibitor as previously defined by marimastat in vivo testing against other snake venoms36. Drugs tested in vivo were dimercaprol (2,3-dimercapto-1-propanol ≥98 % iodometric, Cat no: 64046, Sigma-Aldrich), marimastat (>98% HPLC, Cat no: M2699, Sigma-Aldrich), and prinomastat hydrochloride (≥95% HPLC, Cat no: PZ0198, Merck), all resuspended at 1 mg/mL in water, and DMPS (2,3-dimercapto-1-propanesulfonic acid sodium salt monohydrate, 95%, Cat no: H56578, Alfa Aesar) resuspended at 1mg/mL in PBS. Groups of five mice received experimental doses that consisted of either: (a) venom only (4 x LD50 dose) or (b) venom (4 x LD50 dose) with drug (118 μg) or (c) drug only (118 μg) to assess drug safety. The control group was the venom only group, against which all drug treatments were compared. Each experimental group comprised 5 animals as this was previously determined to be the minimum number of animals required to produce statistically significant results39. No randomisation was used to allocate experimental groups - mice were randomly allocated into cages of five prior to the experiment, and each cage formed one treatment group. No criteria for including or excluding animals was applied, and all data points were included in analyses. A total of 45 mice were used. All experimental doses were prepared to a volume of 200 μL in PBS and incubated at 37 °C for 30 mins prior to intravenous injection via the tail vein. Animals were monitored for humane endpoints (loss of righting reflex, seizure, external haemorrhage) for six hours, and any animals showing such signs were immediately euthanised by rising concentrations of carbon dioxide. All observations were performed by mixed gender experimenters who were blinded to the drug group allocation. Time of death, number of deaths and number of survivors were recorded, where deaths and times of death represent implementation of humane endpoint-dictated euthanasia. Kaplan-Meier survival plots were generated using GraphPad Prism 9.0 (GraphPad Software, San Diego, USA) and log-rank (Mantel-Cox) tests were used to statistically compare the survival times between groups treated with and without drug.
3. Results
3.1 Small molecule drugs have varying effects on the procoagulant activity of crude D. typus venom
The addition of D. typus venom to recalcified bovine plasma in the coagulation assay resulted in earlier stimulation of clotting compared to the no venom control (natural clotting), highlighting the procoagulant nature of this venom (Figure 1A). The effects of the small molecules against the procoagulant activity of crude D. typus venom are shown in Figure 1. Weak inhibitory effects were observed for the metal chelators dimercaprol and DMPS at micromolar concentrations. As shown in Figure 1B, concentrations of 160 μM showed strong inhibitory activity for both drugs, but this inhibitory effect rapidly decreased at lower concentrations, with no effect observed at concentrations of 10 μM and lower. IC50 values were determined to be 77.7 μM for dimercaprol and 120 μM for DMPS (Figure 1C), although due to the small number of data points between 0 and 100% inhibition these values must be interpreted with caution and 95% confidence intervals were unable to be calculated. Contrastingly, the peptidomimetic matrix metalloproteinase inhibitors marimastat and prinomastat potently neutralised the procoagulant effects of D. typus venom (Figure 1B). The IC50 values for marimastat and prinomastat were determined to be 34.2 nM (95% CI 24.2 to 48.5 nM) and 75.6 nM (95% CI 58.6 to 97.7 nM) respectively (Figure 1C), demonstrating that marimastat was significantly more potent in this assay than prinomastat (p = 0.01). The PLA2 inhibitor varespladib (control non-SVMP inhibiting drug used throughout) had no neutralising effect on venom-induced coagulation at any of the tested drug concentrations (Supplemental File S1), a result in line with our expectations of procoagulant venom activity being mediated by SVMP toxins.

Inhibitory activity is expressed as a percentage of normal clotting, where 100% inhibition represents return of clotting to normal plasma clotting levels. A) Clotting as indicated by the increase in OD595 in the presence of crude D. typus venom (black circles) compared to normal clotting in the absence of venom (grey squares). Data points represent the mean of twelve individual values recorded over three independent technical replicates, and error bars represent standard deviation. B) Inhibitory activity of marimastat (teal circles), prinomastat (dark purple circles), dimercaprol (light purple circles) and DMPS (pink circles) over a two-fold serial dilution curve, from which IC50 values were calculated. Data points represent the mean of six individual values recorded over three independent technical replicates, and error bars represent standard deviation. C) Calculated IC50 values for marimastat, prinomastat, dimercaprol and DMPS. Data points represent the best fit IC50 value and error bars represent 95% confidence intervals (not calculated for dimercaprol and DMPS).
3.2 Inhibitory effects of small molecule drugs on nanofractionated D. typus venom coagulotoxins
To further characterise the coagulopathic activity of D. typus venom, and to better explore the specificity of the various small molecule inhibitors against specific toxins, we repeated the coagulation assay experiments using nanofractionated venom. As previously described, this method uses fractionated venom as the basis for measurements of the velocity of clotting in different wells in comparison to control wells, with procoagulant toxins producing positive peaks in the resulting bioassay chromatogram and anticoagulant toxins producing negative peaks37. In the venom-only analysis, broad positive peaks (18.4-22.0 min) were observed for both the ‘very fast coagulation’ chromatograms and the ‘slightly/medium increased coagulation’ chromatograms, indicative of an overall procoagulant effect of the venom. Detected bioactivities of D. typus venom were correlated with previously generated LC-MS and proteomics data29, and a candidate toxin mass of 23 kDa was identified for the pro-coagulant activity, which is within the range of SVMPs. The inhibitory effects of the matrix metalloproteinase inhibitors marimastat and prinomastat on nanofractionated D. typus venom toxins are depicted in Figure 2A and 2B respectively. The peaks in the very fast and slight/medium increased coagulation chromatograms decreased with increasing concentrations of both marimastat and prinomastat, indicative of a dose-dependent restoration of normal clotting velocity. All coagulopathic activities were inhibited at 0.8 μM marimastat and 0.16 μM prinomastat for very fast coagulation chromatograms, and at 4 μM for both inhibitory molecules for slightly/medium increased coagulation activity. In terms of the metal chelators dimercaprol and DMPS, their inhibitory effects on nanofractionated D. typus venom toxins are depicted in Figure 2C and 2D, respectively. By increasing the concentration range of dimercaprol, the procoagulation activity of D. typus venom was inhibited, with very fast coagulation activity fully inhibited at 4 μM, and slightly/medium increased coagulation activity at 20 μM. However, no substantial inhibition of procoagulant venom activity was observed with DMPS at any tested concentration up to 20 μM. These results reflect the considerable potency differences observed between the peptidomimetic inhibitors and the metal chelators in the crude venom plasma bioassays.

The negative peaks indicate anticoagulant activity where velocity is lower than the assay solution in control wells without venom toxins, and the positive peaks indicate pro-coagulant activity where velocity is higher than that in control wells without venom toxins. The top superimposed chromatograms are characteristic profiles of the UV trace at 220, 254 and 280 nm. PBS indicates venom only samples where PBS was used as a control for the inhibitors. Traces with different colours indicate different concentrations (final) of inhibitors in the assay.
A very weak signal (peak centre at 19.6 min) was also detected in terms of anticoagulant venom activity. A previous study observed a much clearer negative anticoagulant peak with D. typus venom, though the venom was applied at a five-fold higher concentration (5.0 mg/mL venom) than that used in this study (1.0 mg/mL venom)29. While varespladib has previously been demonstrated to be a potent inhibitor of anticoagulant venom activities induced by PLA2 toxins33,40–42, in this study it produced no inhibitory effects on venom-induced coagulation, whether procoagulant or anticoagulant, at the maximal drug dose tested (20 μM) (Supplemental File S2). However, due to the weak anticoagulant venom activity observed in these experiments, the assay window for measuring such inhibition is limited.
3.3 Small molecule drugs inhibit crude D. typus venom SVMP activity
To determine the specific inhibitory effects of the selected small molecule drugs on SVMP toxin activity, we performed IC50 screens of the five different toxin inhibitors in a previously defined kinetic enzymatic assay SVMP using crude D. typus venom. As previously observed43, D. typus venom demonstrated strong SVMP-specific activity in this assay. The inhibitory effects of the matrix metalloproteinase inhibitors marimastat and prinomastat and the metal chelators DMPS and dimercaprol against the venom SVMP activity of D. typus are displayed in Figure 3. The peptidomimetic inhibitors marimastat and prinomastat demonstrated nanomolar IC50 values of 14.5 (95% CI 13.9 to 15.2 nM) and 25.9 nM (95% CI 22.6 to 29.7 nM) respectively, and complete inhibition of venom activity at 156 nM for marimastat and 2.5 μM for prinomastat (Figure 3A). Inhibition of SVMP activity by dimercaprol and DMPS as measured by IC50 values was significantly lower than that observed for marimastat and prinomastat (p < 0.02 for all comparisons). Dimercaprol and DMPS demonstrated highly comparable inhibition of D. typus SVMP activity with complete inhibition obtained at 40 μM and IC50 values of 6.68 (95% CI 6.36 to 7.02 μM) and 6.61 μM (95% CI 6.38 to 6.87 μM), respectively (Figure 3B), with no significant differences between the two IC50 values. As anticipated, the control drug used in this study, the PLA2 inhibitor varespladib, showed no inhibitory activity at any of the concentrations tested (maximum concentration 10 μM) (Supplemental File S3).
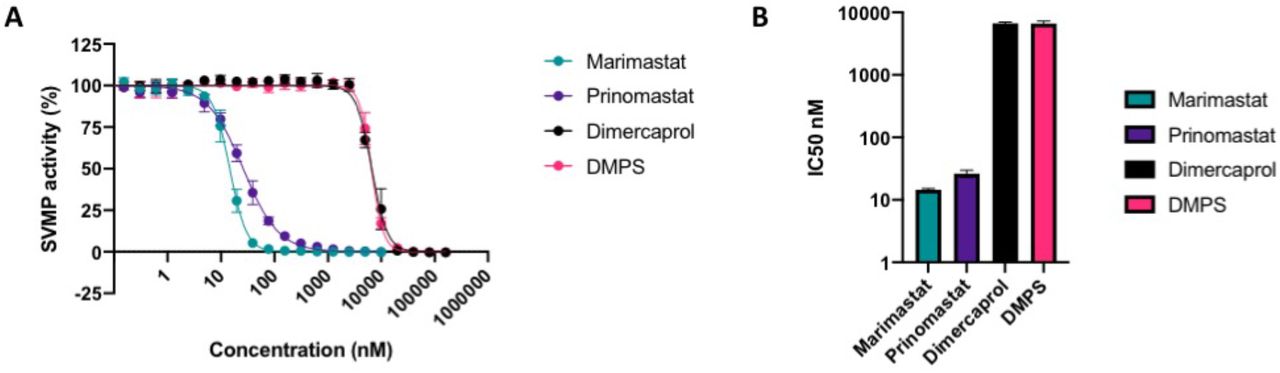
SVMP activity of D. typus crude venom in the presence of marimastat (teal circles), prinomastat (dark purple circles), dimercaprol (light purple circle) and DMPS (pink circles) over a two-fold serial dilution curve ranging from which IC50 values were calculated. Data points represent the percentage of crude venom SVMP activity generated from the mean of four individual values recorded over two independent technical replicates, and error bars represent standard deviation. B) IC50 values of SVMP inhibition for marimastat, prinomastat, dimercaprol and DMPS.
3.4 Marimastat and DMPS provide some protection against D. typus venom-induced lethality in vivo
Given that inhibition of SVMP and coagulotoxic activities in vitro have previously been demonstrated to translate into varying degrees of in vivo protection against systemic envenoming26,36 we next tested the capability of the four SVMP-inhibiting small molecule drugs to protect against D. typus venom-induced lethality in vivo. To do so, we used a modified version of the WHO-recommended protocol of murine venom neutralisation (ED50 assay). All five experimental animals treated intravenously with 4 x LD50 doses of D. typus venom (90 μg) succumbed to the lethal venom effects within the first hour of the experiment (mean 17 minutes, range 1 to 40 minutes), as shown in Figure 4. The intravenous co-delivery of preincubated drugs with D. typus venom revealed that both prinomastat and dimercaprol failed to protect against venom induced lethality at the single therapeutic dose tested (118 μg), with all experimental animals in these groups succumbing to venom lethality within the first 30 minutes (mean 7.8 minutes for both groups; prinomastat range 4 to 21 minutes; dimercaprol range 1 to 18 minutes), in a highly comparable manner to the venom only control. Contrastingly, DMPS and marimastat both showed a significant degree of protection against D. typus venom-induced lethality. Three of the five experimental animals dosed with DMPS were protected for the duration of the experiment, with two deaths occurring within the first hour, resulting in a mean survival time of 224 minutes compared to 17.2 minutes in the venom only group (log-rank test, p = 0.047). Of the animals dosed with marimastat, four of the five animals were protected for the duration of the experiment, with mean survival times of 316.2 minutes compared to 17.2 minutes in the venom only group (log-rank test, p = 0.002). The single non-surviving experimental animal in this drug group was euthanised at 141 minutes. No adverse effects were observed in experimental animals dosed with any of the drugs only controls and, consequently, all survived the duration of the experiment (Figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The data is shown in Kaplan-Meier survival graphs for experimental animals (n=5 per group, except prinomastat only group where n=1) treated with either: 90 μg (4 x LD50) of D. typus venom only (magenta), 118 μg of drug only (cyan) or 90 μg venom and 118 μg drug (black). Treatments were pre-incubated at 37 °C for 30 minutes prior to intravenous injection via the tail vein and animals were monitored for 6 hours. Data is shown for: A) DMPS, B) dimercaprol, C) marimastat, and D) prinomastat
4. Discussion
Conventional animal-derived antivenom, although a life-saving treatment, has numerous deficiencies that impair its utility in the treatment of snakebite. Although a seemingly effective antivenom for treating D. typus envenomation is available in South Africa, it is often difficult to source in other regions of the continent and can be catastrophically unaffordable for patients13, and this scenario encapsulates the challenges faced by snakebite victims world over. There is therefore an urgent need to develop alternative/supplementary therapeutics that are stable, effective, affordable and available in remote rural areas where medical access is limited. Small molecule inhibitors that can broadly neutralise a class of key toxins in snake venom following oral administration are possible solutions in this regard20 and Phase II clinical trials for small molecule inhibitors of snakebite are underway44. Rapid-onset pathologies such as VICC, together with tissue damage induced by SVMPs, are only partially neutralised by antibody based antivenoms45, which suffer from poor tissue distribution due to the inherent large size of antibodies46. By contrast, the drastically smaller size of the inhibitors tested in this study enables favourable properties of rapid and effective tissue penetration and potential for oral delivery, due to their pharmacokinetic and physicochemical properties34,35,47. Moreover, repurposing these small molecule inhibitors that are either licensed drugs or phase I-approved drug candidates could significantly shorten drug development times as safety profiles, pharmacokinetics, bioavailability and tolerance data on these molecules have already obtained47–49. Current evidence of the utility of small molecule inhibitors against snakebite indicates that they may be particularly effective as first line, early intervention, therapeutics and/or bridging therapies for initial and adjunct treatment in community settings, before patients are able to access antivenom in healthcare centres18,28,48.
In this study we assessed the ability of four small molecule inhibitors to neutralise D. typus venom toxin activities in vitro and in vivo. In line with previous findings8–11, our study demonstrates that D. typus venom toxicity is largely conferred by SVMP toxins, which are likely responsible for causing coagulopathy in vivo. Our data from in vitro assays of D. typus venom activity demonstrate that the matrix metalloproteinase inhibitors marimastat and prinomastat are potent inhibitors of the SVMP-mediated procoagulant effects of this venom, with both compounds demonstrating similar inhibitory activity in in vitro assays of plasma coagulation and SVMP activity. Indeed, marimastat and prinomastat showed nanomolar IC50 values in the crude venom plasma coagulation assay and crude venom SVMP assay, and both drugs showed inhibitory effects at low micromolar concentrations in the plasma coagulation assay with nanofractionated venom. However, and in contrast with in vitro SVMP-inhibiting prowess, in the in vivo assays, all animals in the prinomastat group succumbed to lethality from D. typus venom in murine models of envenomation, whilst an equivalent amount of marimastat conferred 80% protection. This was unexpected and hints at different levels of drug exposure and metabolism in this single dose intravenous-delivered model, as both matrix metalloproteinase inhibitors showed potent inhibitory activity in the in vitro assays and both have been shown in other studies to neutralise the in vivo lethal effects of E. ocellatus in murine models24,26. The metal chelators dimercaprol and DMPS demonstrated lower potency than marimastat and prinomastat in the in vitro studies of SVMP activity and plasma coagulation, whilst the PLA2 inhibitor varespladib, used as a non-SVMP inhibiting control, produced no inhibitory effects on venom. Of the two metal chelators, DMPS showed the weakest inhibitory activity in in vitro assays of venom bioactivity. Despite this reduced in vitro potency in comparison with the peptidomimetic matrix metalloproteinase inhibitors, DMPS conferred a degree of protection against venom lethality in murine models of envenomation, with significant increases in mean survival times and 60% of mice surviving until the end of the experimental time window. None of the experimental animals dosed with the other metal chelator, dimercaprol, survived the experiment, despite the comparable mechanism of action and in vitro inhibitory potency in the coagulation and SVMP assays. These results contrast with our previous preclinical study investigating E. ocellatus envenoming, which found that both dimercaprol and DMPS provided protection against lethal effects in this same intravenous murine model, though are consistent with DMPS exhibiting superior preclinical efficacy26.
The notable discrepancies between the in vitro and in vivo experiments described herein exemplifies the complexity associated with relying on in vitro potency-based screens as a means to predict the efficacy of small molecule drugs in in vivo experiments. While efficacy data gained from in vitro experiments is undoubtedly an essential prerequisite prior to preclinical efficacy testing, substantial differences in inhibitor potency at this step does not preclude preclinical efficacy, which ultimately is dictated by drug exposure. Equally, the preclinical model utilised here, consisting of the pre-incubation of drug with venom followed by codelivery intravenously, is largely detached from the clinical scenario of a snakebite. While this is the WHO-recommended method for preclinical assessment of antivenom efficacy, and thus is a logical starting point for assessing preclinical efficacy, this method does not reflect the biodistribution of venom during early envenomation, uses a non-clinically relevant route of venom injection, and does not take into account the pharmacokinetics/pharmacodynamics of the unbound test inhibitor50. Thus, the lack of efficacy observed with prinomastat (compared with marimastat) here, for example, may be the result of a lack of dose optimisation and thus sub-optimal exposure. Future work is required to better define the pharmacokinetic and pharmacodynamic profiles of small molecule drugs in preclinical models of snakebite envenoming to inform the design of optimised pre-clinical dosing regimens applicable for use in more biologically realistic models of envenoming (e.g. “challenge then treat models”)26,34,36,46,51.
In sub-Saharan Africa, VICC is only known to be commonly caused by Echis spp. and D. typus, and the venoms of these snakes have been shown to converge on similar SVMP-rich venom composition profiles9, suggesting that D. typus venom may be amenable to neutralisation by previously identified inhibitors of Echis venoms. This study investigated the ability of repurposed small molecule inhibitors to effectively neutralise D. typus venom activity in vitro and in vivo, and identified the SVMP inhibiting drugs DMPS and marimastat as two lead compounds that provide a significant degree of preclinical protection against the lethal effects of D. typus venom. Previous studies have demonstrated that both DMPS and marimastat also provide preclinical efficacy against E. ocellatus envenoming26,36, and this study therefore expands the range of snake species that victims of envenoming could potentially benefit from receiving an early intervention (e.g. oral) small molecule therapeutic. These findings therefore provide a strong rationale for the future clinical evaluation of the efficacy of such small molecule drugs in all cases of diagnostically indicated VICC following snakebite envenoming in sub-Saharan Africa.
Funding
This research was funded in whole, or in part, by the Wellcome Trust, grant numbers as detailed below. For the purpose of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. R.H.C. acknowledges funding support from the Director’s Catalyst Fund at LSTM [supported by Wellcome Institutional Strategic Support Fund 3 (204806/Z/16/Z) and LSTM Internal Funding]. N.R.C. acknowledges a UK Medical Research Council research grant (MR/S00016X/1) and a Sir Henry Dale Fellowship (200517/Z/16/Z) jointly funded by Wellcome and the Royal Society. N.R.C and J.K. acknowledge funding provided by a Wellcome project grant (221712/Z/20/Z). C.X. acknowledges funding support from the China Scholarship Council (CSC) fellowship (201706250035).
Author contributions
Conceptualization – JK, NRC
Methodology – SKM, RHC, CX
Investigation – SKM, RHC, CX, AW, SRH, RJE, JA, EC, RAH, NRC,
Data curation – SKM, RHC, CX, AW
Formal analysis – SKM, RHC, CX, AW
Original draft preparation – SKM, RHC, CX, NRC, JK
Editing - all authors
Acknowledgements
We thank the staff in the Biomedical Service Unit of the University of Liverpool for their support in the maintenance and care of the study mice.
References




