

Abstract
Delivery of gene editing components such as the Cas nuclease and single guide RNAs (sgRNAs) into plant cells is commonly accomplished by agrobacterium-mediated transformation. Although Arabidopsis is easy to transform, generation of biallelic edited plants requires screening a large number of plants in subsequent generations. Here, we describe optimization of the Tobacco rattle virus (TRV) for in planta delivery of sgRNAs fused to a tRNAIleu that induces efficient multiplex somatic and biallelic heritable editing in Arabidopsis. Inclusion of tRNAIleu enhances the systemic movement of TRV and the mutant phenotype is visible in the initial TRV::sgRNA-tRNAIleu infected Arabidopsis, which allows for the uncovering of lethal phenotypes. Mutant progeny are recovered in the next generation (M1) at frequencies ranging from 30-60%, with 100% mutant recovery in the following (M2) generation. TRV::tRNAIleu system described here allows generation of biallelic edited plants in a single generation and is amenable for large-scale high throughput CRISPR screens.
Introduction
Developing newer, more efficient gene editing methods in plants is critical to performing complex gene function studies and for the advancement of plant biotechnology. The advent and rapid deployment of CRISPR/Cas-based technology has allowed for the generation of mutant genotypes with highly specific mutations. CRISPR has been readily employed in plant biotechnology and adapted to many model and crop plant species1,2,3. So far, most methods for highly efficient, CRISPR-based gene editing rely on traditional transgenic approaches to deliver the Cas nucleases and single guide RNA (sgRNA) components. This is a major limitation since transgenes are left behind in the genome of transformed plants. This can lead to unknown off-target effects that can be difficult to mitigate.
As one of the first plant genomes to be sequenced, the model plant Arabidopsis thaliana has a plethora of genetic resources and decades of readily available experimental evidence from gene-function studies. However, one major limiting factor when using Arabidopsis for high throughput gene-function studies is the generation of stable, homozygous knockouts. Current, high-throughput, heritable editing technologies for testing gene function have not been adapted in Arabidopsis, as it relies on traditional agrobacterium-mediated transformation. While agrobacterium-mediated plant transformation in Arabidopsis can be a robust gene editing tool4, it suffers from several drawbacks. Namely, it requires several subsequent, self-crossing generations of large screening populations to generate biallelic homozygous mutant lines. Further, it leaves behind transgenes which could have unknown, off-target effects. Given the large-scale data that are being generated with omics-based approaches, it is increasingly important to be able to quickly evaluate the function of genes of interest to guide future research.
One option to deliver CRISPR editing components without the use of transgenics is to use viral vectors to deliver sgRNA targeted to a specific gene of interest in plants already expressing Cas nuclease5,6. Since the viral nucleic acid genome is not integrated into the host genome and is not transmissible to subsequent generations, progeny seeds of infected plants are virus and transgene free. Some advancements have been made in this area by engineering Tobacco rattle virus (TRV)7,8, Potato virus X (PVX)9, and Barley stripe mosaic virus (BSMV)10 viral vectors for delivery of sgRNAs targeted to genes of interest. A major limitation of using viral vectors is that many viruses are unable to enter meristematic tissue. As a result, gene editing is largely restricted from germline cells; hence, the gene editing is not heritable. Another major limitation of using viral vectors for gene editing is that each virus has a limited host range, so different viruses need to be developed and optimized for different plants.
Recently, we reported a novel method for delivery of sgRNAs that induces high efficiency somatic and heritable editing in Nicotiana benthamiana using a modified TRV vector system8. TRV is a single stranded positive sense RNA virus with a bipartite genome11. RNA1 of TRV encodes replicase proteins (134 kDa and a translational readthrough 194 kDa) from the genomic RNA, a 29 kDa movement protein (MP) from subgenomic RNA, and a 16 kDa cysteine-rich RNA silencing suppressor protein from a separate subgenomic RNA (Fig. 1a). RNA2 of TRV encodes the coat protein (CP) from the genomic RNA and two non-structural proteins (29.4 kDa and 32.8 kDa) from separate subgenomic RNAs (Fig. 1a). We engineered the TRV RNA2 by replacing the two non-structural proteins with multiple cloning sites (MCS) (Fig. 1a) or a gateway cloning cassette to facilitate virus-induced gene silencing (VIGS) to knockdown expression of target genes of interest in plants12,13. TRV has a wide host range and can infect many dicotyledonous plants. Additionally, TRV-VIGS vectors have been widely used by the scientific community for gene function studies in more than 25 plant species including Arabidopsis11,14,15.
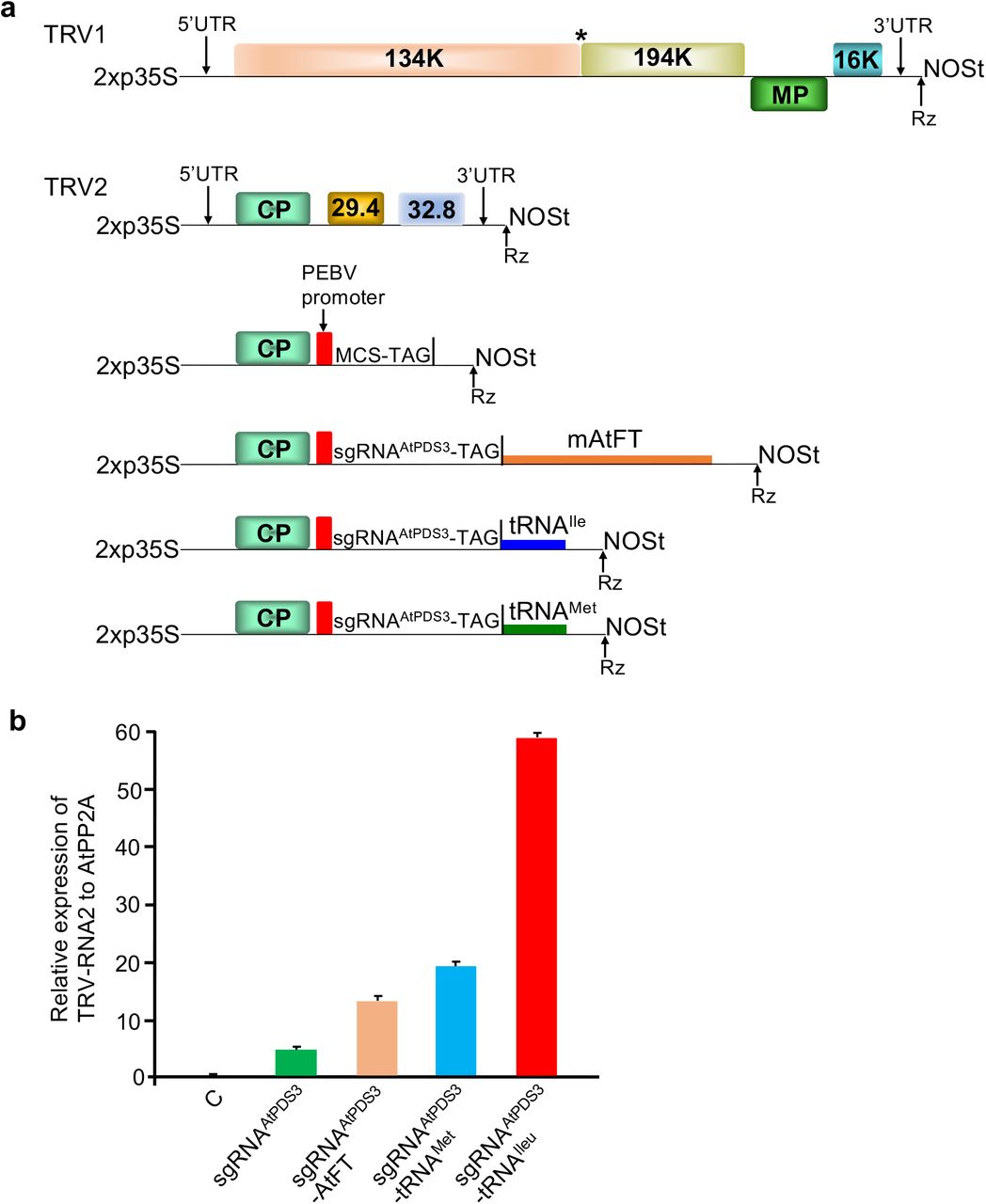
a Schematic representation of TRV1, TRV2 and various TRV2 derivative vectors. b Comparison of systemic movement of TRV with various mobile sequences. Relative levels of TRV RNA2 were measured by quantitative real-time PCR using AtPP2A as a reference. Data from three replicates were combined and values are shown as mean +SD
A major advantage of TRV vectors is that they induce very mild symptoms on the infected plants16,17 and can transiently invade the meristematic region18. To express gene editing components such as sgRNAs, we modified the TRV RNA2 vectors used in VIGS by including a subgenomic promoter from another tobravirus, Pea early browning virus (PEBV) (Fig. 1a)7. Expression of sgRNAs using TRV with the pPEBV promoter (pPEBV) induced moderate editing of targeted genes in the infiltrated and systemic leaves of N. benthamiana plants7. However, the efficiency of heritable editing in the progeny plants was only 0.2%7. To overcome this, we recently reported a modified TRV system in which sgRNAs are expressed as fusion with mobile RNA sequences such as Arabidopsis Flowering Locus T (AtFT) and transfer RNAs (tRNAs) (Ellison et al., 2020). In N. benthamiana, TRV with sgRNAs augmented with mobile RNA sequences induced very high efficiency somatic editing in the infiltrated plants and 65-100% heritable editing efficiency in the next generation (M1)8. Recently, the expression of sgRNAs fused to AtFT sequence using PVX has also been shown to induce heritable editing with an efficiency of 22% in N. benthamiana9.
N. benthamiana serves as an excellent model species for viral studies due to the wide range of plant viruses able to infect it and ability to robustly express heterologous proteins19,20. However, it lacks the depth of genetic resources that are available for Arabidopsis. Hence, we sought to optimize our recently reported TRV system in N. benthamiana for use in Arabidopsis. While our studies were being conducted, another study showed that TRV with sgRNA flanked by self-splicing tRNA21 could induce low efficiency heritable epigene editing in the Arabidopsis M1 generation but not in the original infiltrated plants22. Furthermore, the geminivirus, Cotton leaf crumple virus (CLCrV) expressing a sgRNA has been shown to induce low efficiency heritable editing, but only in the second generation (M2) of Arabidopsis plants23. Here, we report a simple, optimized, agrobacterium-based flooding method combined with sgRNAs fused to the tRNA isoleucine (sgRNA-tRNAIleu) that induces highly efficient multiplex somatic and heritable editing in Arabidopsis. The sgRNA-tRNAIleu is easily deliverable by TRV-based viral vector (TRV::sgRNA-tRNAIleu). We show that the mutant phenotype was visible in the initial TRV::sgRNA-tRNAIleu infected plants and hence the system allows uncovering of lethal phenotypes. Furthermore, we observed 30-60% heritable editing efficiency in the M1 generation and 100% heritable editing efficiency in the M2 generation.
Results
Optimization of efficient delivery of sgRNA using TRV into Arabidopsis plants expressing SpCas9
We first tested TRV-based delivery of the sgRNA by using our previously established method for gene expression knockdown in Arabidopsis via VIGS24. We recently demonstrated that in N. benthamiana, TRV2 with a sgRNA fused to tRNAIleu moves more efficiently and generates high efficiency heritable editing compared to TRV with sgRNA alone8. Therefore, we aimed to adapt and optimize this system in Arabidopsis. We infiltrated TRV1 and TRV2 containing a sgRNA designed to target the Arabidopsis thaliana PHYTOENE DESATURASE 3 (AtPDS3) gene fused to tRNAIleu (TRV2::sgRNAAtPDS3-tRNAIleu) (Fig. 1a) into 2 week old Arabidopsis Col-0 plants expressing SpCas9 (Col-0::SpCas9) (Supplementary Fig. 1a) by following the method described in24. About 3% of the infiltrated plants (1/36) showed visible photobleached spots or streaks on the leaves (Supplementary Fig. 2a). Since PDS in plants is required for the biosynthesis of photoprotective carotenoids, loss of function in AtPDS3 will result in a photobleached phenotype.

a Plants used for syringe infiltration of agrobacterium harboring TRV vectors. b Plate with seedlings in which leaves are pricked with agrobacterium harboring TRV vectors (black arrows). c Plate with seedlings flooded with agrobacterium harboring TRV vectors.

a Phenotype in systemic leaves of Arabidopsis plant that was syringe infiltrated with agrobacterium harboring TRV1 and TRV2::sgRNAAtPDS3-tRNAIleu. White photobleached region on the leaves (asterisks) is due to editing and loss of AtPDS3 function. b Phenotype in systemic leaves of transplanted Arabidopsis plants that were treated by flooding of agrobacterium harboring TRV1 and TRV2::sgRNAAtPDS3-tRNAIleu. White photobleached region on the leaves (asterisks) is due to editing and loss of AtPDS3 function.
Next, we tested two other methods to evaluate if somatic editing efficiency could be improved. In the first method, we grew plants on MS media for ten days and transferred the seedlings to a sterile petri plate with a mixture of agrobacterium harboring TRV1 and TRV2::sgRNAAtPDS3-tRNAIleu constructs. Then, needles were used to deliver multiple pricks to the leaves of individual plants (Supplementary Fig. 1b) (see Methods section for details). Twenty-four hours post-pricking, plants were transplanted into soil. In the second method, seeds were grown on MS plates for ten days, then flooded with a mixture of agrobacterium harboring TRV1 and TRV2::sgRNAAtPDS3-tRNAIleu (Supplementary Fig. 1c) (see Methods section for details). Three to four days post-flooding, plants were transplanted into soil. Only about 8% (3/36) of the pricked plants showed visible photobleached spots or streaks on the leaves. However, with the agrobacterium flooding method, about 22% of the plants (8/36) showed the photobleaching phenotype (Supplementary Fig. 2b). To assess that the observed phenotype is due to mutations in the AtPDS3 gene, we PCR-amplified the region flanking the target site using DNA extracted from the entire leaves showing the photobleaching spots or streaks. Sanger sequencing of the PCR products followed by Synthego ICE (Inference of CRISPR Edits) analysis (https://ice.synthego.com/#/)25 confirmed the mutations in the AtPDS3 gene (Supplementary Fig. 3). Together, these results indicated that the agro-flooding method is more efficient for the delivery of sgRNAs into Arabidopsis expressing SpCas9 to induce editing.

The indel mutation types and frequencies identified from Sanger sequencing of amplicons spanning the AtPDS3 target site in leaves of three independent plants that were treated with agro-flooding and showed a photobleaching phenotype.
TRV with sgRNAAtPDS3 fused to tRNAIleu exhibits better systemic movement in Arabidopsis compared to sgRNAAtPDS3 fused to other mobile elements
Since the agro-flooding approach was more efficient, we followed this method in the rest of our experiments described in this manuscript. We recently reported that in addition to sgRNAs fused to tRNAIleu, sgRNAs fused to AtFT without the start codon (mAtFT) and tRNA methionine (tRNAMet) also facilitate efficient movement of TRV in N. benthamiana8. Therefore, we tested systemic movement efficiency of TRV with sgRNAAtPDS3 fused to mAtFT (sgRNAAtPDS3-mAtFT) and tRNAMet (sgRNAAtPDS3-tRNAMet) along with sgRNAAtPDS3-tRNAIleu (Fig. 1a). Ten day old Col-0 or Col-0::SpCas9 Arabidopsis seedlings on MS plates were flooded with agrobacterium harboring TRV1 and TRV2 with various sgRNAAtPDS3 mobile RNA fusions. After three to four days, seedlings were transplanted into soil. Three weeks after transplanting, the newly developed leaves were used for qRT-PCR analysis using primers that anneal to the 3’ end of TRV RNA2. A significantly higher amount of TRV was present in systemic leaves of plants infected with TRV2 with sgRNAAtPDS3-tRNAIleu compared to TRV2 with sgRNAAtPDS3 fused to other mobile RNA sequences, or when compared to sgRNAAtPDS3 with no mobile RNA fusion as a control (Fig. 1b). Systemic movement of TRV2 with sgRNAAtPDS3-tRNAMet was better than that of sgRNAAtPDS3-mAtFT or sgRNAAtPDS3 alone (Fig. 1b). These results indicate that the sgRNA augmented with tRNAIleu moves better in Arabidopsis compared to other tested sequences.
Efficient heritable editing of AtPDS3 using TRV with sgRNAAtPDS3 fused to tRNAIleu
Next, we set out to systematically characterize somatic editing efficiency of the AtPDS3 gene using TRV2 with sgRNAAtPDS3-tRNAIleu. Ten day old Col-0::SpCas9 seedlings on MS plates were flooded with agrobacterium containing TRV1 and TRV2::sgRNAAtPDS3-tRNAIleu (Fig. 2a). Three days later, seedlings were transplanted into soil. About 15-20 days later, we observed the white photobleaching spots or streaks on the leaves (Fig. 2b). To determine the editing efficiency, we collected leaf tissue from completely photobleached regions, regions with overlapping photobleached and green (referred to as mosaic regions) and the green regions. DNA extracted from these samples was used to PCR amplify the region flanking the AtPDS3 target site and the products were Sanger sequenced and subjected to Synthego ICE analysis. We observed a very high percentage of indels ranging from 60% to 99% at the AtPDS3 target site in tissue collected from the completely photobleached regions (Fig. 2c and Fig. 3). In the mosaic regions, the editing efficiency varied from 20% to 70%, and editing efficiency was about 0% to 26% in the green regions (Fig. 2c and Fig. 3). During later stages of development, we observed complete photobleaching of some of the rosette and cauline leaves (Fig. 2d). In addition, some of the siliques and seed pods were completely photobleached (Fig. 2d and Supplementary Fig. 4). Together, these results indicate that sgRNAAtPDS3 fused to tRNAIleu induces mutations in the AtPDS3 gene leading to the photobleached phenotype in Arabidopsis.


a Schematic of TRV RNA2 vector with sgRNAAtPDS3 fused to tRNAIleu. b Phenotype of Arabidopsis plants infected with TRV1+TRV2::sgRNAAtPDS3-tRNAIleu. White photobleached regions on the leaves is indicative of loss of AtPDS3 function. c Editing efficiency in green (G), overlapping region between green and white (M) and white (W) region of leaves from 5 independent TRV1+TRV2::sgRNAAtPDS3-tRNAIleu infected Arabidopsis plants. Indel frequency was assessed by Sanger sequencing of amplicons spanning the AtPDS3 target site followed by Synthego ICE analysis. d White photobleaching phenotype in some cauline leaves, stem and flowers due to editing in AtPDS3. Right panel is an enlarged version of part of the left panel. e White photobleaching phenotype of siliques. Right panel is an enlarged version of part of the left panel.

The identified indel mutation types and indel frequencies derived from Sanger sequencing of amplicons spanning the AtPDS3 target site followed by Synthego ICE analysis in green (G), overlapping region between green and white (M) and white (W) region of leaves from 5 independent TRV1+TRV2::sgRNAAtPDS3-tRNAIleu infected Arabidopsis plants shown in Fig. 2c.
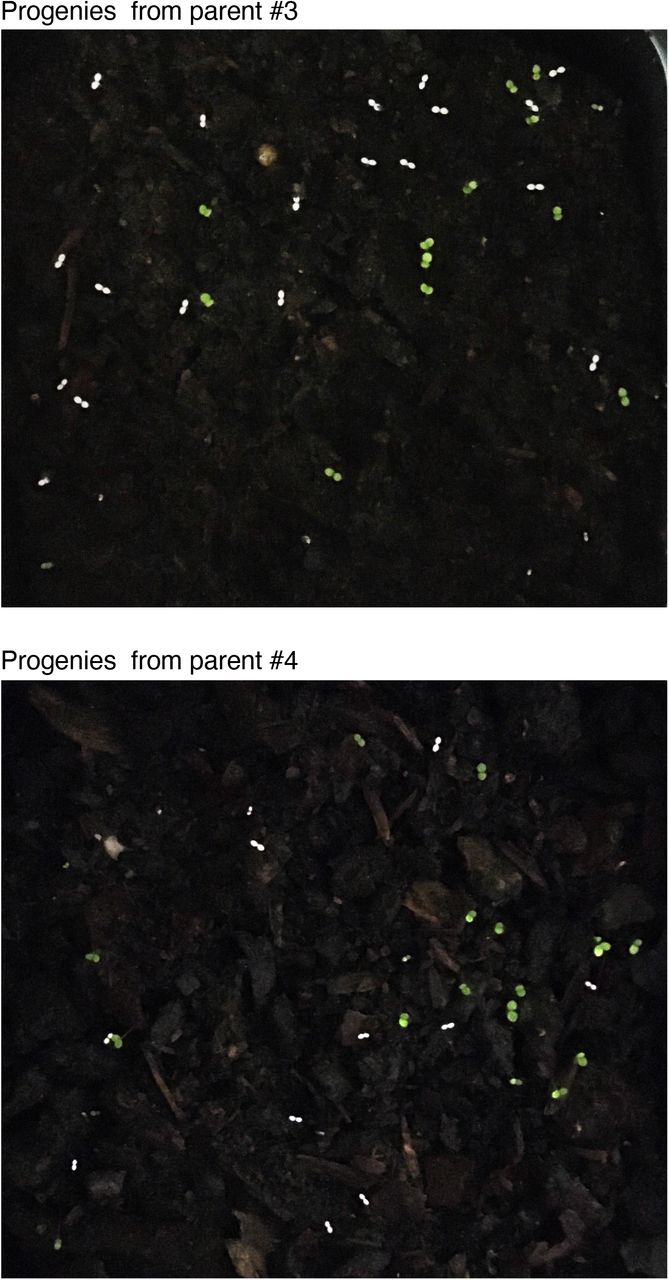
Next, we assessed whether TRV2::sgRNAAtPDS3-tRNAIleu infected plants that show somatic editing also possess AtPDS3 germline editing that is transmissible to the next generation. To accomplish this, we collected seeds from the aforementioned parent plants infected with TRV1 and TRV2::sgRNAAtPDS3-tRNAIleu (Fig. 2) in three batches between principal growth stage 8 to 5 days after principal growth stage 6.926 that we refer to here as early, middle and late stage developed seeds. We observed very few photobleached seedlings collected from late stage developed seeds compared to early and middle stage developed seeds. From the middle stage developed seeds, 30-60% of progeny seedlings were completely photobleached, indicating biallelic mutations in AtPDS3 (Fig. 4a and Supplementary Fig. 5). The next generation sequencing (NGS) of the AtPDS3 PCR products flanking the target site (amplicons) of 8 white seedlings from parent 1 and parent 2 showed indel frequencies ranging from 86-93% and 95-99%, respectively (Fig. 4b and Supplementary Table 1). The NGS of amplicons of 8 green seedlings from each parent indicated indel frequency of 57-62% in these plants (Fig. 4b and Supplementary Table 2). We also performed Sanger sequencing of the region flanking the AtPDS3 target region of 8 white progeny seedlings from two additional parents. Our Synthego ICE analysis indicated indel frequencies of 88-99% for progenies from parent 3 and 93-99% for progenies from parent 4 at the target site in AtPDS3 (Supplementary Fig. 6). Together, these results indicate that TRV2::sgRNAAtPDS3-tRNAIleu induces high efficiency somatic and heritable editing in Arabidopsis.

M1 progenies from two parental plants infected with TRV1+TRV2::sgRNAAtPDS3-tRNAIleu showing a photobleaching phenotype.
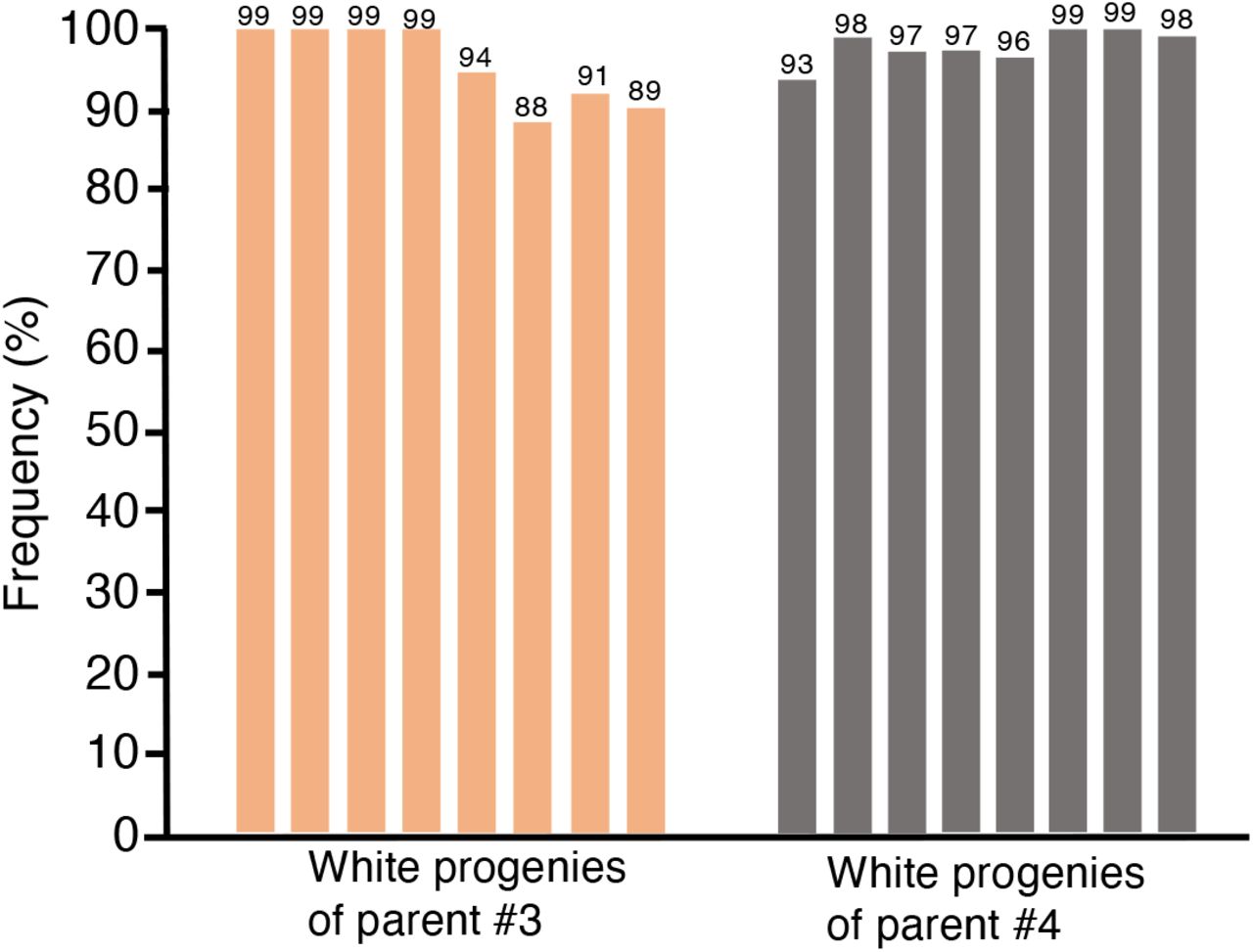
Indel mutation frequencies assessed by Sanger sequencing of amplicons of AtPDS3 target site followed by Synthego ICE analyses of M1 progenies of parental plants 3 and 4 infected with TRV1+TRV2::sgRNAAtPDS3-tRNAIleu. Numbers shown on top of each bar indicate the percentage of indels.

a Phenotype of M1 progenies of seeds collected from two parental plants infected with TRV1+TRV2::sgRNAAtPDS3-tRNAIleu. White photobleached seedlings are indicative of biallelic editing and loss of AtPDS3 function. b Indel mutation frequencies in selected 8 white and 8 green progenies from each parent shown in (a) assessed by NGS of amplicons spanning the AtPDS3 target site.
Efficient multiplex editing of AtCHLI1 and AtCHLI2 using TRV with sgRNA fused to tRNAIleu
To assess the efficiency of multiplex editing using this system, we targeted two genes that encode two isoforms of the I subunit of magnesium-chelatase (CHLI), AtCHLI1 (At4g18480) and AtCHLI2 (At5g45930). Along with CHLH and CHLD, CHLI is involved in catalyzing the first committed step toward chlorophyll synthesis. Knockout in both AtCHLI1 and AtCHLI2 genes results in albino sterile plants27. To target both AtCHLI1 and AtCHLI2 genes, we cloned two sgRNAs which target these genes in tandem with a 23-base pair (bp) spacer and placed them downstream of the PEBV promoter in TRV2 (TRV2::sgRNAAtCHLI1-tRNAIleu-S-sgRNAAtCHLI2-tRNAIleu) (Fig. 5a). Recently, we showed that the sgRNAs arranged in tandem with a 23 bp spacer works efficiently for multiplexing in N. benthamiana8.

a Schematic of TRV RNA2 vector with sgRNAAtCHLI1 and sgRNAAtCHLI2 fused to tRNAIleu with a 23 bp spacer. b Phenotype of Arabidopsis plants infected with TRV1+TRV2::sgRNAAtCHLI1-tRNAIleu-23bp-sgRNAAtCHLI2-tRNAIleu. White albino and yellow leaves are indicative of editing and loss of function of both AtCHLI2 and AtCHLI2. c Indel mutation frequencies in AtCHLI1 and AtCHLI2 from six independent plants showing albino and yellow leaves phenotype.
We performed a flooding treatment of ten day old Col-0::SpCas9 Arabidopsis seedlings with agrobacterium harboring TRV1 and TRV2::sgRNAAtCHLI1-tRNAIleu-S-sgRNAAtCHLI2-tRNAIleu. Starting from 15-20 days post-transplanting after TRV treatment, plants exhibited a yellow or white leaf color phenotype (Fig. 5b and Supplementary Fig. 7). The NGS amplicon sequencing of PCR products flanking AtCHLI1 and AtCHLI2 generated from plant tissue detected indel frequencies of 79-94% in the AtCHLI1 gene and 45-66% in the AtCHLI2 gene (Fig. 5C and Supplementary Tables 3 and 4). Together, these results indicate that TRV augmented with tRNAIleu can be used for multiplex gene editing in Arabidopsis. We were unable to estimate the heritable transmission of the knockout phenotype in the next generation because the parent plants showing phenotypes produced less seeds and the progeny resulting from these seeds were green or some were dead. We reasoned that since knocking out of both AtCHLI1 and AtCHLI2 genes results in sterility, the seeds that were produced carried wildtype genes.

Efficient heritable multiplex editing of AtTRY and AtCPC genes using TRV with sgRNA fused to tRNAIleu
Since we were unable to assess the heritability of multiplex editing in AtCHLI1 and AtCHLI2, we targeted two MYB-like transcription factors TRIPTYCHON (AtTRY, At5g53200) and CAPRICE (AtCPC, At2g46410) using TRV2 augmented with tRNAIleu. AtTRY and AtCPC functions as negative regulators of trichome development in Arabidopsis, since try cpc double mutant plants display increased trichomes on leaves leading to a clustered leaf trichomes phenotype28,29. We cloned an sgRNA fused tRNAIleu that targets both AtTRY and AtCPC under the control of the PEBV promoter in TRV2 (TRV2::sgRNAAtTRY/AtCPC-tRNAIleu) (Fig. 6a). Agrobacterium harboring TRV1 and TRV2::sgRNAAtTRY/AtCPC-tRNAIleu was introduced into Col-0::SpCas9 Arabidopsis seedlings by the agro-flooding method as described above. Approximately 15-20 days post-transplanting of seedlings after agro-flooding we observed clustered trichomes on leaves (Fig. 6b and Supplementary Fig. 8). In addition to on the leaves, we also observed trichome clusters on the stems (Fig. 6b and Supplementary Fig. 8). To confirm that the observed clustered trichome phenotype is due to editing in the AtTRY and AtCPC genes, we PCR amplified the region flanking the TRY and CPC target sites using tissue from four independent plants and sequenced the amplicons by NGS. Our analysis indicated indel frequencies of 70-83% in the AtCPC gene and 71-76% in the AtTRY gene (Fig. 6c and Supplementary Tables 5 and 6). These results indicate that TRV with sgRNAAtTRY/AtCPC-tRNAIleu targets two genes simultaneously with high efficiency.


a Schematic of TRV RNA2 vector with single guide RNA targeting both AtTRY and AtCPC fused to tRNAIleu. b Phenotype of Arabidopsis plants infected with TRV1+TRV2::sgRNAAtTRY/AtCPC-tRNAIleu. Trichomes on the leaves and stems are due to editing and loss of function of both AtTRY and AtCPC. c Indel mutation frequencies in AtTRY and AtCPC from four independent plants showing leaf trichome phenotype. d Phenotype of M1 progenies of seeds collected from a parental plant infected with TRV1+TRV2::sgRNAAtTRY/AtCPC-tRNAIleu. Trichomes on leaves and stems is indicative of biallelic editing in both AtTRY and AtCPC. e Indel mutation frequencies in five M1 progenies of three independent parent plants assessed by NGS of amplicons spanning the AtCPC (top panel) and AtTRY (bottom panel) target sites. f Phenotype of M2 progenies of seeds collected from a M1 progeny. All progenies showed leaf trichome phenotype indicating highly heritable biallelic editing in both AtTRY and AtCPC.
To assess the heritability of the observed editing in AtTRY and AtCPC in TRV1 and TRV2::sgRNAAtTRY/AtCPC-tRNAIleu infected plants, we collected seeds from parental plants that showed the clustered trichome phenotype (Fig. 6b and Supplementary Fig. 8) and grew them in soil. About 35-45% of the progeny plants from each parental line exhibited the clustered leaf trichomes phenotype (Fig. 6d and Supplementary Fig. 9a). The NGS amplicon sequencing of PCR products flanking the target sites indicated indel frequencies ranging from 89-92%, 84-89% and 68-93% in AtCPC gene (Fig. 6e, top panel and Supplementary Table 7) and 79-82%, 65-86% and 69-88% in AtTRY gene (Fig. 6e, bottom panel and Supplementary Table 8) in progenies from parent 1, parent 2 and parent 3 respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a M1 progenies from seed collected from a parental plant infected with TRV1+TRV2::sgRNAAtTRY/AtCPC-tRNAIleu. b M2 progenies from seed collected from three independent M1 progenies.
To assess the transmission of knockout mutations in the 2nd generation, we collected and planted the seeds from M1 plants that showed the clustered trichomes phenotype. We observed that 100% of the M2 progenies displayed the clustered leaf trichomes phenotype (Fig. 6f and Supplementary Fig. 9b). Together, these results indicate that TRV augmented with tRNAIleu induces highly efficient multiplex heritable knockout mutations in Arabidopsis.
Discussion
Here, we describe an optimized TRV-based system for delivery of sgRNAs to induce highly efficient somatic and heritable editing in the model plant Arabidopsis. Our findings show that the fusion of tRNAIleu with sgRNA in TRV enhances systemic movement of the virus in Arabidopsis compared to sgRNA fused to mAtFT. This contrasts with our recent report which showed that both tRNAIleu and mAtFT promote better systemic movement of TRV in N. benthamiana8. Our findings are consistent with previous reports in Arabidopsis that the AtFT protein is the mobile form not the AtFT mRNA30,31. In wheat, systemic movement of BSMV with sgRNA fused to mutant wheat FT (BSMV-sg-mTaFT) was similar to the movement of BSMV expressing sgRNA alone (BSMV-sg)10. Although BSMV-sg-mTaFT induced similar somatic editing efficiency as BSMV-sg, BSMV-sg-mTaFT failed to induce heritable editing in wheat10. In rice, it was shown that mRNA of the FT ortholog, OsHd3a, was unable to move to the shoot apical meristem while the protein was fully mobile32. Due to the syntenic relationship between wheat and rice, this could be one reason why the BSMV-sg-mTaFT construct was unable to induce heritable editing.
Heritable editing in plants using viral vectors will depends on the ability of the virus to invade the meristematic region. Compared to most plant viruses, TRV can transiently invade the meristem early in the infection cycle18. Consistent with this, TRV has been used successfully to silence meristem genes like NFL (an ortholog of Arabidopsis LEAFY16) and floral homeotic genes such as DEFICIENS (an ortholog of Arabidopsis AP317) in N. benthamiana. Despite this, TRV expressing sgRNA alone could only induce very low frequency heritable editing in N. benthamiana7,8. However, the addition of tRNAIleu to TRV significantly enhanced heritable editing frequency in N. benthamiana8 and Arabidopsis (this report). We hypothesize that enhanced viral systemic movement facilitated by tRNAIleu leads to more virus invading into the meristem region which, in turn, promotes editing in a higher numbers of germline cells. Future systematic analysis of TRV and TRV derivatives with mobile sequences should provide insights into engineering principles that would facilitate better invasion of TRV and other viruses into the meristem region.
Our findings in Arabidopsis (this report) and N. benthamiana8 show that the addition of tRNAIleu to sgRNAs in TRV enhances systemic movement of the virus and is capable of inducing efficient somatic and heritable editing in target genes. In a previous study, tRNAMet and tRNAGly were identified as mobile tRNAs in the phloem exudates of pumpkin but not tRNAIleu 33. When fused to a GUS transcript, tRNAMet and tRNAGly facilitate unidirectional movement of GUS from shoot to root, but not from root to shoot in Arabidopsis34. This could be one reason for reduced infection rate of BSMV-tRNAMet derivative in systemic leaves of wheat10. The addition of tRNAMet into BSMV may enhance downward movement into roots than into the systemic leaves. In the case of TRV, our results showed that the inclusion of tRNAIleu significantly enhances the accumulation of TRV in the systemic leaves of Arabidopsis compared to tRNAMet. Future studies are required to understand the mechanistic basis of how tRNAIleu facilitates better movement of TRV and possibly other viruses in plants.
Recently, TRV has been shown to induce a low percentage of heritable epigene editing in Arabidopsis22. The epigene editing phenotype was only observed in the M1 progenies but not in the initial TRV inoculated Arabidopsis plants. We reason that the low heritability could be because TRV2 with self-splicing tRNA21 flanking the sgRNAs was used in these studies22. Since the tRNA will be spliced to generate sgRNAs, the TRV RNA2 would also be spliced reducing the infectiousness of the virus. In this scenario, there will be a population of TRV RNA2 population in which the tRNA is not spliced, this non-spliced tRNA will fail to produce sgRNAs with precise 5’ and 3’ ends. In BSMV, the inclusion of self-splicing tRNA flanking sgRNA significantly reduced the systemic virus infectivity in wheat10. The self-splicing tRNA sequence is very similar to the tRNAGly, with the few base differences residing in the loop region of these tRNAs. TRV with sgRNA fused to tRNAGly did not enhance systemic movement of the virus and induced very low heritable gene editing in N. benthamiana8.
Very low efficiency, heritable editing observed only in the second (M2) generation in Arabidopsis has also been recently reported using the geminivirus CLCrV23. In that report, sgRNA fused to the N-terminus 102 bp of AtFT (AtFT1-102bp) was expressed using CLCrV. This study failed to observe mutant phenotypes in either the CLCrV-infected plants or in the M1 progenies, but a small number of mutant progenies in the M2 generation were recovered. Based on our findings in this paper and previous reports indicating AtFT mRNA is immobile in Arabidopsis30,31, we hypothesize that the very low efficiency of editing through CLCrV with AtFT1-102bp that was observed could be due to the virus being unable to efficiently invade the meristem.
The TRV system with tRNAIleu that we describe in this paper is the most efficient method for inducing somatic and heritable editing in Arabidopsis reported to date. Furthermore, we show that the system is suitable for multiplex editing with very high efficiency. It also facilitates uncovering lethal phenotypes because one could observe the phenotype in the original virus infected plants. Our findings indicate that biallelic mutants can be generated in a single generation. In the second generation, 100% of the M2 progenies exhibited the mutant phenotype. Introducing gene editing components into Arabidopsis through transformation is relatively easy compared to other plants; however, it still requires a significant amount of time to generate biallelic knockout mutants using CRISPR/Cas9. The optimized and simple agro-flooding method combined with TRV augmented with tRNAIleu described here is amenable for high throughput editing of candidate genes identified from different omics approaches.
Methods
Vector construction
The pYL192 (TRV113; Addgene ##148968), SPDK3876 (TRV2 with PEBV promoter8; Addgene #149275) have been described. TRV2-pPEBV-MCS-tRNAIleu (SPDK3888, Addgene #149276), TRV2-pPEBV-MCS-tRNAMet (SPDK3889, Addgene #149277), and TRV2-pPEBV-MCS-mAtFT (SPDK3895) have been described8. To generate sgRNAAtPDS3 vectors, oligonucleotides 9208 and 9209 and 8788 and 8789 were annealed and ligated into XbaI-SacI cut SPDK3888, SPDK3889, SPDK3895 and SPDK3876 resulting in SPDK3959 (TRV2-sgRNAAtPDS3-tRNAIleu), SPDK3952 (TRV2-sgRNAAtPDS3-tRNAMet), SPDK3969 (TRV2-sgRNAAtPDS3-tRNAmAtFT), and SPDK4151 (TRV2-sgRNAAtPDS3) vectors respectively. To generate sgRNAs targeting AtCHLI1 and AtCHLI2 with 23 bp spacer (SPDK4146), PCR products generated using 100272 and 100149 primers with SPDK3959 DNA template and 8787 and 100273 primers with SPDK3959 DNA template and Phusion DNA polymerase were digested with XbaI-EagI and EagI-SacI respectively and cloned into SPDK3888 cut with XbaI-SacI. The vector containing sgRNA targeting AtCPC and AtTRY was generated by cloning PCR product generated using 100279 and 8787 primers with SPDK3959 template was digested with XbaI-SacI and cloned into SPDK3888 cut with same enzymes. All primers used for cloning are listed in Supplementary Table 9.
Plant growth conditions
Col-0, Col-0::SpCas9 (line 13-19) or TRV infected plant progeny seeds were stratified at 4° C in water in the dark for 3 days before sowing in soil. Plants were grown in a growth chamber set at 26°/22°C day/night condition with a 12-h light/12-h dark photoperiod and a light intensity of 100 μE m-2 sec-1, 50% humidity. Col-0 and Col-0::SpCas9 seedlings from MS plates treated with agrobacterium with TRV were transplanted to soil and grown at the same conditions as plants started from the seeds.
Preparation of agrobacterium harboring TRV vectors
TRV1, TRV2 and various TRV2 derivative vectors were introduced into chemically competent agrobacterium strain GV3101. Transformants were confirmed by colony PCR. Agrobacterium harboring TRV vectors were grown in 5 ml of lysogeny broth (LB) with antibiotics for 16-18 h at 28° C. Agrobacterium cultures were centrifuged for 20 min at 3,500 relative centrifugal force (RCF). The LB medium was discarded and agrobacterium cells were resuspended in 5 ml of sterile water and centrifuged for 10 min 3,500 RPM. The resulting pellets were resuspended in sterile agro-infiltration buffer [10 mM MgCl2, 10 mM 2-(N-morpholino) ethanesulfonic acid, and 250 µM acetosyringone] to OD600 = 1.5 and incubated with slow shaking for 3 h at room temperature. The TRV1 and TRV2 or TRV2 derivatives were mixed at 1:1 ratio and used for various inoculation methods.
TRV inoculation
Leaf infiltration
Agrobacterium with TRV1 and TRV2 or TRV2 derivatives were infiltrated into 2 weeks old Col-0::SpCas9 plants using a 1mL needleless syringe as described in24. About 10 days post-infiltration, the infiltrated plants were monitored every day for any visible phenotypes.
Agro-pricking with a needle
Col-0 and Col-0::SpCas9 seeds were surface sterilized by placing in a 1.5 mL sterile tube and 1 ml 75% ethanol and 0.1% Triton X-100 (Sigma-Aldrich) was added and placed in Eppendorf Mixer 5432 for 4-5 minutes. Seeds were washed with 1 ml of 100% ethanol 2 times with 3 minute incubation each time in the mixer and seeds were dried overnight in the laminar hood. Sterilized seeds were placed on one-half strength MS medium containing Gamborg vitamins (Phyto Technologies Laboratories) solidified with 0.25% Phytoblend (Caisson Labs) in petri plates (100 mm × 25 mm). The MS plates with seeds were covered in aluminum foil and kept for three days at 4°C and transferred to a growth chamber set at 24°C with a light intensity of 100 μE m-2 sec-1 and a 12 h light/12 h dark photoperiod. Eight to ten days post-germination, the seedlings were transferred to a sterile petri plate and 10 ml of agrobacterium suspension with TRV1 and TRV2 or TRV2 derivatives was added. Leaves of each seedling were pricked using Monoject® Hypodermic Plastic Hub Needle 25g x 1”. The seedlings were incubated overnight at room temperature and next day they were planted on soil and grown in a growth chamber as described above.
Agro-flooding
Col-0 and Col-0::SpCas9 seeds were sterilized and set on MS plates as described above. Ten ml agrobacterium suspension with TRV1 and TRV2 or TRV2 derivatives was dispensed directly into the MS plates containing eight to ten days-old Arabidopsis seedlings. The plates were incubated for 4 days in the growth chamber at 24°C with a light intensity of 100 μE m-2 sec-1 and a 12 h light/12 h dark photoperiod. Seedlings were transplanted to soil and grown in a growth chamber as described above.
Quantitative real-time PCR analysis
Leaf tissue was collected from control and TRV infected plants and total RNA was extracted using TRIzol reagent (Life Technologies). Total RNA was treated with RNase free DNase I (New England Biolabs). First-strand cDNA was synthesized using SuperScript III reverse transcriptase (Life Technologies) and TRV-specific primer SP9239 or the control AtPP2A-R primer. RT–qPCR was performed using the Bio-Rad CFX96 qPCR instrument (Bio-Rad) with iTaq Universal SYBR Green Supermix (Bio-Rad), 2.5 μM each of TRV-specific primers (9238 and 9239) or AtPP2a-specific primers (AtPP2A-F and AtPP2A-R) and 5% diluted first-strand cDNA. Data presented are normalized for AtPP2A expression level.
Analysis of somatic and heritable editing efficiency in Arabidopsis
To assay editing in TRV infected plants or M1 progenies, Phire Plant Direct PCR Kit (Thermo Fisher Scientific) was used. A small leaf or a piece of leaf of about 2 mm in diameter was collected into a tube containing 20 μl of dilution buffer and the leaf sample was crushed using a 1 ml pipette tip. Centrifuged for 5 min at 14,000 rpm and the supernatant was transferred to a new tube. PCR reaction was performed using 2-5 μl of this genomic DNA as template and 25 pmol of primers in a 50 μl reaction. All primers used for genotyping are listed in Supplementary Table 9. The resulting amplicons were purified using Zymoclean Gel DNA Recovery Kit (Zymo Research). The amplicons were sent for Sanger sequencing (Eurofins Genomics) or Amplicon-Ez Next Generation Sequencing (NGS) (Genewiz/Azenta Life Sciences). Sanger trace sequencing file (.ab1 file) was used to assess indel percentage using Synthego ICE (Inference of CRISPR Edits) analysis pipeline (https://ice.synthego.com/) (Hsiau et al., 2019). The paired-end amplicon sequencing reads from Amplicon-EZ were de-multiplexed using ea-utils35. The demultiplexed reads were uploaded to Cas-Analyzer36 and compared against the targeted DNA sequence. In order to avoid the quantification of indels located far from the target sequence, we used a 40 bp window to either side of the target site for analysis. We also filtered out sequences with <5 reads to eliminate any sequencing errors from the analysis.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files, or from the corresponding author upon request.
Competing Interests
The authors declare no competing interests.
Author Contributions
U.N and S.P.D-K conceptualize the project and designed the experiments. U.N and J-Y.L performed the experiments. U.N and N.M analyzed the data and interpreted the results. D.F.V provided suggestions and discussed the results with S.P.D-K. Y.X. and J.S generated and characterized SpCas9 expressing Arabidopsis transgenic line. U.N., N.M., and S.P.D-K wrote the original draft of the manuscript. All authors reviewed and edited the manuscript.
Acknowledgements
We thank Professor Jen Sheen and Dr. Jian-Feng Li for providing SpCas9 expressing Arabidopsis line. We thank April DeMell for critical reading and editing the manuscript. This work was supported by Innovative Genomics Institute (to S.P.D-K and U.N), Agricultural Innovation through Gene Editing program grant no. 2020-67013-31544/project accession no. 1022332 from the USDA National Institute of Food and Agriculture (to S.P.D-K and U.N), and grant no. HR0011-17-2-0053 from the Defense Advanced Research Projects Agency (DARPA; to S.P.D-K and D.F.V).
References




