

Abstract
The inferior colliculus (IC) is a midbrain hub critical for perceiving complex, time-varying sounds such as speech. In addition to processing ascending inputs from most auditory brainstem nuclei, the IC receives descending inputs from auditory cortex that control IC neuron feature selectivity, plasticity, and certain forms of perceptual learning. Although these corticofugal synapses primarily release the excitatory transmitter glutamate, many physiology studies show that auditory cortical activity has a net inhibitory effect on IC neuron spiking. Perplexingly, anatomy studies indicate that corticofugal axons primarily target glutamatergic IC neurons while only sparsely innervating IC GABA neurons, thereby suggesting that corticofugal inhibition of the IC occurs largely independently of feedforward activation of local GABA neurons. We shed light on this paradox using in vitro electrophysiology and optogenetics in acute IC slices from fluorescent reporter mice. We find that corticofugal synaptic strength is indeed stronger onto IC glutamate compared to GABA neurons. Nevertheless, two mechanisms enable reliable corticofugal activation of local GABA neurons. 1) Many IC GABA neurons fire tonically at rest, such that sparse and weak excitation suffices to significantly increase their spike rates. 2) Corticofugal activity triggers barrages of large amplitude, polysynaptic EPSPs in IC GABA neurons, owing to a dense intra-collicular connectivity from local axon collaterals of IC glutamate neurons. Consequently, repetitive activity in corticofugal fibers drives spikes in IC GABA neurons and generates substantial recurrent inhibition. Thus, descending signals trigger intra-collicular inhibition despite apparent constraints of monosynaptic connectivity between auditory cortex and IC GABA neurons.
Introduction
Feedback projections from the sensory neo-cortex to sub-cortical regions are ubiquitous in the mammalian brain. These corticofugal pathways enable “high-level”, cortical computations to rapidly control the nature of ascending sensory signals, thereby hypothetically supporting important “top-down” functions such as predictive coding, error propagation, or stream segregation (Briggs and Usrey, 2011; Stebbings et al., 2014; Usrey and Sherman, 2019; Asilador and Llano, 2020). Interestingly, corticofugal activity often has a net inhibitory effect upon spontaneous and sensory-evoked activity in sub-cortical circuits, consequently sharpening receptive fields and increasing the sparseness of neuronal representations along the ascending sensory hierarchy (Syka and Popelár, 1984; Zhang et al., 1997; Bledsoe et al., 2003; Boyd et al., 2012; Crandall et al., 2015; Vila et al., 2019; Born et al., 2021; but see Kirchgessner et al., 2021). Corticofugal projections originate almost exclusively from excitatory (glutamatergic) neurons in cortical layers 5 and 6 (Hattox and Nelson, 2007; Schofield, 2009; Slater et al., 2013; Asilador and Llano, 2020; Sherman and Usrey, 2021), the well-documented inhibitory effects suggest that corticofugal activity primarily recruits sub-cortical, feedforward inhibitory (GABAergic) interneurons rather than faithfully detonating spikes in projection neurons. However, the cellular and circuit-level mechanisms that enable corticofugal activity to reliably generate sub-cortical inhibition are poorly understood; this knowledge gap persists owing to the difficulty of quantifying corticofugal transmission in identified excitatory and inhibitory neurons.
In the central auditory system, the corticofugal projection from auditory cortex to the inferior colliculus (IC) is of particular importance as the IC relays the majority of ascending acoustic signals destined for forebrain circuits. Accordingly, corticofugal activity powerfully shapes how IC neurons respond to diverse sound features, often via inhibitory interactions: Stimulating the auditory cortex dampens or completely suppresses IC acoustic responses (Syka and Popelár, 1984; Bledsoe et al., 2003; Vila et al., 2019; Blackwell et al., 2020), a result further corroborated by intracellular data showing that auditory cortex stimulation can drive synaptic inhibition in individual IC neurons (Mitani et al., 1983; Qi et al., 2020). Conversely, silencing auditory cortex often potentiates spontaneous and sound-evoked activity in the IC (Nwabueze-Ogbo et al., 2002; Popelár et al., 2003; Popelář et al., 2016; but see Zhang and Suga, 1997), indicating that ongoing auditory cortical activity can have a net inhibitory effect on the moment-to-moment excitability of IC neurons. However, anatomical data show that IC GABA neurons receive surprisingly few synapses from auditory cortex; the majority of corticofugal axons instead targeting glutamatergic IC neurons (Nakamoto et al., 2013; Chen et al., 2018). Thus, rather than recruiting local inhibitory neurons to sharpen IC neuron tuning curves, auditory cortex may instead exert inhibitory control through long-range inhibitory pathways (Beneyto et al., 1998; Budinger et al., 2000), via local effects upstream of the IC (Kong et al., 2014), or through a newly discovered, monosynaptic GABAergic corticofugal projection (Bertero et al., 2021). Alternatively, circuit mechanisms beyond the orthodox feedforward inhibitory motif, whereby long-range excitation preferentially drives GABAergic interneurons (Pouille and Scanziani, 2001; Gabernet et al., 2005; Cruikshank et al., 2007; Boyd et al., 2012), may nevertheless enable the auditory cortex to generate local inhibition in the IC.
We used patch-clamp electrophysiology and optogenetics in brain slices from fluorescent GABA reporter mice to study corticofugal transmission onto identified IC neurons. We focused specifically on neurons in the dorso-medial “shell” IC because this sub-region receives the densest projection of corticofugal axons (Winer et al., 1998; Song et al., 2018; Oberle et al., 2022). Our data confirm previous anatomical predictions by showing that corticofugal transmission is on average stronger onto shell IC glutamate compared to GABA neurons. However, we also find that shell IC GABA neurons are densely contacted by potent, intra-collicular synapses from local glutamate neurons. As such, repetitive corticofugal activity drives strong polysynaptic excitation in a subset of shell IC GABA neurons and generates local inhibition. Our results identify a novel mechanism for the neo-cortex to drive recurrent computations in a sub-cortical circuit. More broadly, the data establish a biophysical context via which descending inhibitory control operates in apparent absence of a dense, monosynaptic convergence onto inhibitory networks.
Results
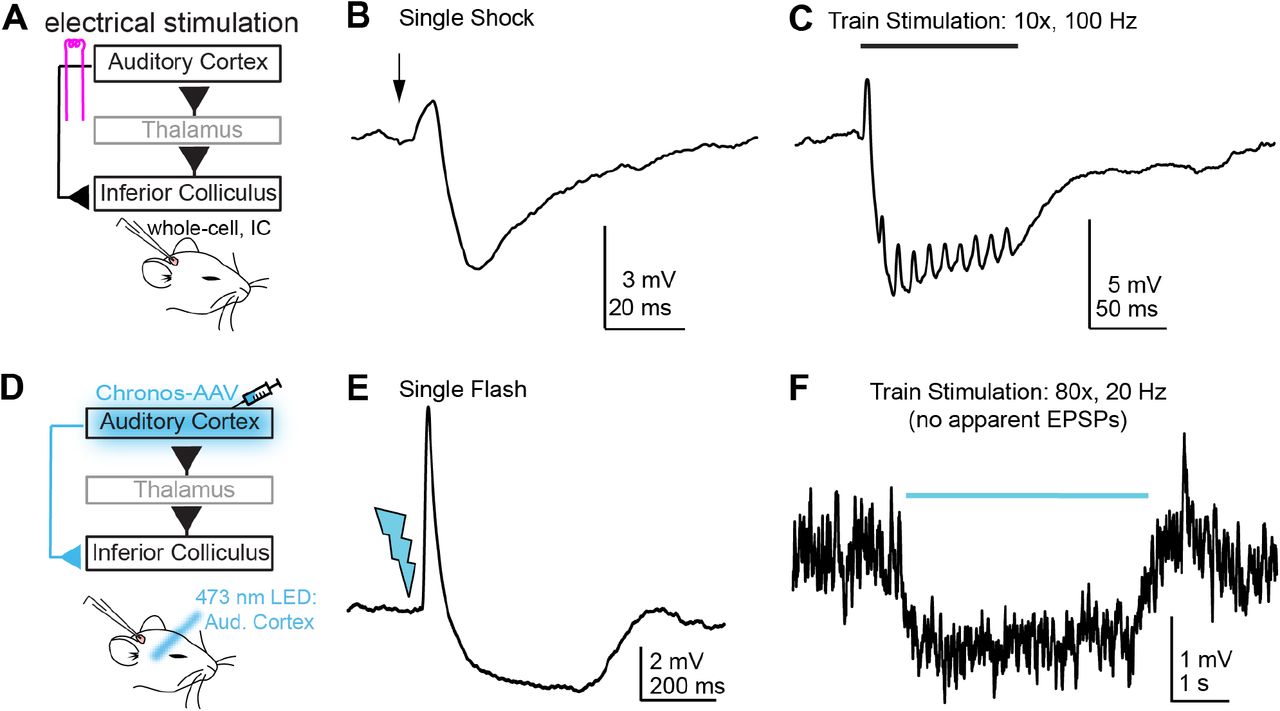
Cortical stimulation inhibits superficial IC neurons in vivo
Previous studies show that stimulating the auditory cortex profoundly reduces sound-evoked spiking in IC neurons. As a first test to determine if we could similarly observe descending inhibition of IC neurons in vivo, we electrically stimulated the auditory cortex of urethane anesthetized mice during whole-cell patch-clamp recordings from neurons in the cortico-recipient, superficial IC layers. Indeed, single shocks (Figure 1B) or train stimulation (Figure 1C; 10 shocks delivered between 10-200 Hz) drove brief excitatory postsynaptic potentials (EPSPs) followed by large inhibitory postsynaptic potentials (IPSPs) in n = 4/7 cells tested. Similar results were obtained with optogenetic stimulation of auditory cortex neurons expressing the light gated opsin Chronos: Single flashes of 473 nm light delivered to auditory cortex neurons drove EPSP/IPSP sequences in n = 6/21 cells tested (Figure 1E). Interestingly, inhibition was also seen in 5/17 IC neurons that did not receive any apparent excitation from auditory cortex. In n = 4 neurons, repetitive auditory cortical activity (80 stimuli at 20 Hz) caused a tonic or phasic hyperpolarization of the membrane potential (Figure 1F), whereas in 1 neuron tested, single light flashes generated a single, long-latency IPSP. Altogether these data indicate that in addition to the monosynaptic depolarizations mediated by glutamatergic corticofugal projections (Oberle et al., 2022), descending activity can also substantially hyperpolarize IC neurons, presumably via polysynaptic inhibition.

A) Cartoon of experiment. Whole cell recordings are obtained from superficial IC neurons in urethane anesthetized mice; A bipolar electrode is placed in auditory cortex to activate corticofugal fibers. B) Example average trace showing a brief EPSP followed by a large and long-lasting IPSP upon auditory cortex stimulation. C) Example average trace showing that synaptic inhibition summates upon repetitive stimulation of auditory cortex. Data in B and C are from the same neuron. Stimulation artifacts are blanked for clarity. D) Chronos is expressed in auditory cortex neurons via AAV injections and 2-4 weeks later, whole-cell patch-clamp recordings are obtained from superficial IC neurons. E) Example average traces showing an EPSP-IPSP sequence following single light flashes delivered to the auditory cortex. F) In a different experiment as that shown in E, repetitive stimulation of auditory cortex generates no apparent EPSPs, but rather causes a sustained hyperpolarization of the neuron’s membrane potential.
Corticofugal synapses are stronger onto glutamate compared to GABA shell IC neurons
The hyperpolarization of IC neurons observed during auditory cortex stimulation is intriguing, as anatomy data suggest that auditory cortico-collicular axons preferentially synapse onto excitatory glutamatergic IC neurons; far fewer descending synapses target GABA neurons in the IC (Nakamoto et al., 2013; Chen et al., 2018). We thus wondered if the functional strength of auditory corticofugal synapses differed between glutamatergic and GABAergic IC neurons in a manner consistent with anatomical, or rather, physiological data (e.g., Figure 1). To this end, we expressed the red fluorescent protein tdTomato specifically in GABAergic neurons by crossing crossed VGAT-ires-cre and Ai14 fl/fl mouse lines. We then virally transduced the optogenetic activator Chronos in auditory cortex neurons of these mice via stereotaxic injections, and 2-4 weeks later, prepared acute brain slices for whole-cell patch-clamp electrophysiology (Figure 2A,B). We targeted either tdTomato-positive or -negative somata in the dorso-medial shell IC to record from GABAergic or presumptive glutamatergic neurons, respectively (Supplemental Figure 1A,B). Single flashes of blue light delivered through the microscope objective activated Chronos-expressing auditory cortico-collicular axons and generated EPSPs that were >3-fold larger in glutamate compared to GABA neurons (Figure 2B,C. Median amplitude: 1.86 vs. 0.58 mV, n=29 glutamate neurons from N = 9 mice and n=37 GABA neurons from N=10 mice, respectively. p=0.0013, Kruskal-Wallis test). By contrast, median EPSP halfwidths were similar across the two neuron populations (37.5 vs. 41.0 ms, p=0.7223, Kruskal-Wallis test), indicating that amplitude differences were unlikely due to expression of postsynaptic glutamate receptors with differential glutamate affinity. Furthermore, these results were unlikely to reflect a preferential severing of GABA neuron dendrites during slice preparation, as instantaneous PSP rates were significantly higher in GABA compared to glutamate neurons (Figure 3A-C; median rate 15.9 vs. 7.4 Hz, n = 43 GABA neurons from N = 10 mice and n = 26 glutamate neurons from N = 9 mice, respectively. p < 0.001, Kruskal-Wallis test). Instead, our results imply a specific connectivity logic to the auditory cortico-collicular pathway whereby glutamate, rather than GABA neurons are the major recipients of monosynaptic corticofugal signals. By contrast, the differential background PSP activity suggests that GABA neurons may receive a substantial number of excitatory synapses from the intra-collicular circuit.

A) Tile scan of the IC in a VGAT-cre x Ai14 mouse. An area of interest is denoted by the dashed line and shown at higher magnification in panel B. Scale bar = 500 µm.
B) Magnification of dashed rectangle in panel A. The micrograph was contrast enhanced to highlight tdTomato-positive GABA neurons and tdTomato-negative, presumptive glutamate neurons visible as dark “shadows”. Scale bar = 100 µm.
C-E) Examples of distinct types of hyperpolarizing and depolarizing responses in GABA neurons encountered in the shell IC.
F-H) Same as A-C, but for glutamate neurons. Of note is the qualitative similarity in firing patterns and membrane properties of both neuron classes.

A) Diagram of experiment. The optogenetic activator Chronos is virally transduced into auditory cortex neurons of transgenic VGAT-cre x Ai14 mice. B) 2-4 weeks following surgery, corticofugal synaptic strength is assayed in presumptive GABA and glutamate neurons by recording optogenetically evoked EPSPs from visually targeted tdTomato-positive and negative neurons. Right, example average EPSPs evoked by single light flashes in a glutamate (black) and GABA (magenta) shell IC neuron. C,D) Summary data showing cumulative probability distributions for EPSP peak amplitudes (C) and half-widths (D) in glutamate and GABA neurons. Asterisks denote statistical significance.

A,B) Examples of spontaneous synaptic activity in single glutamate (A) or GABA (B) shell IC neurons. Scale bars apply to both panels. Of note is the higher rate and larger amplitude of spontaneous PSPs in the GABA neuron. C) Summary of instantaneous PSP rates for the two neuron classes.
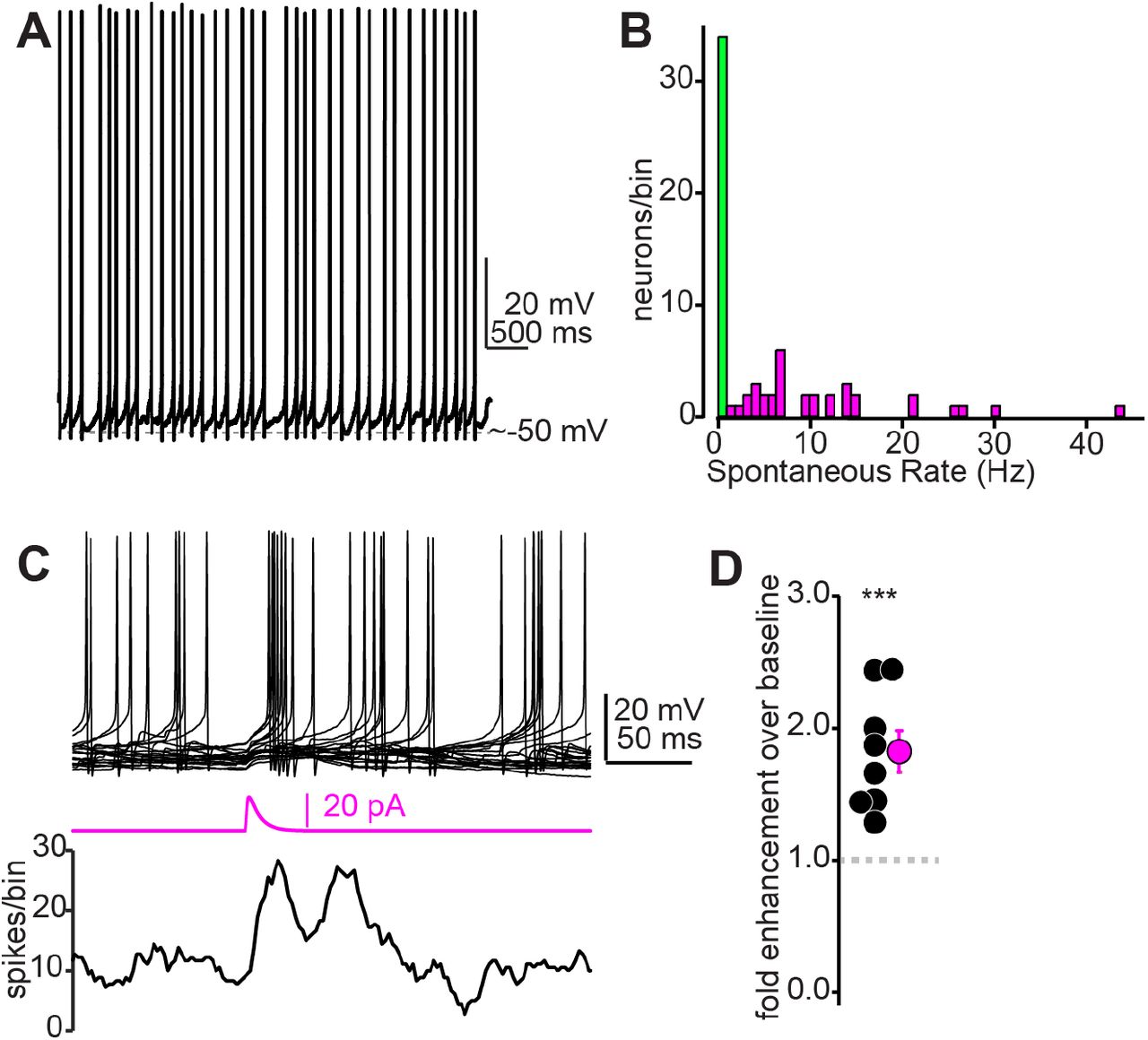
A subset of IC GABA neurons tonically fire action potentials
As expected from previous studies (Ono et al., 2005; Naumov et al., 2019), IC GABA and glutamate neurons displayed a range of overlapping cellular properties and could not be obviously distinguished based solely on membrane responses to current injections (Supplemental Figure 1C-H). However, we observed that GABA neurons were significantly more likely to tonically fire spikes at rest (Figure 4A,B. 34/68 GABA neurons fired tonically upon break-in compared to 1/25 glutamate neurons, χ2=17.14, p<0.001). Consequently, even sparse synaptic excitation might suffice to significantly increase the mean firing rate of IC GABA neurons. Indeed, injecting small excitatory postsynaptic current (EPSC)-like waveforms (10-20 pA peak amplitude) at the soma of tonically firing GABA neurons significantly increased spike rates 1.82 ± 0.16 fold relative to a pre-current injection baseline period Figure 4C,D; n = 8 cells from N = 2 mice, p=0.0012, one-sample t-test). Thus, a dense convergence of descending axons is not explicitly necessary to increase firing rates of shell IC GABA neurons.
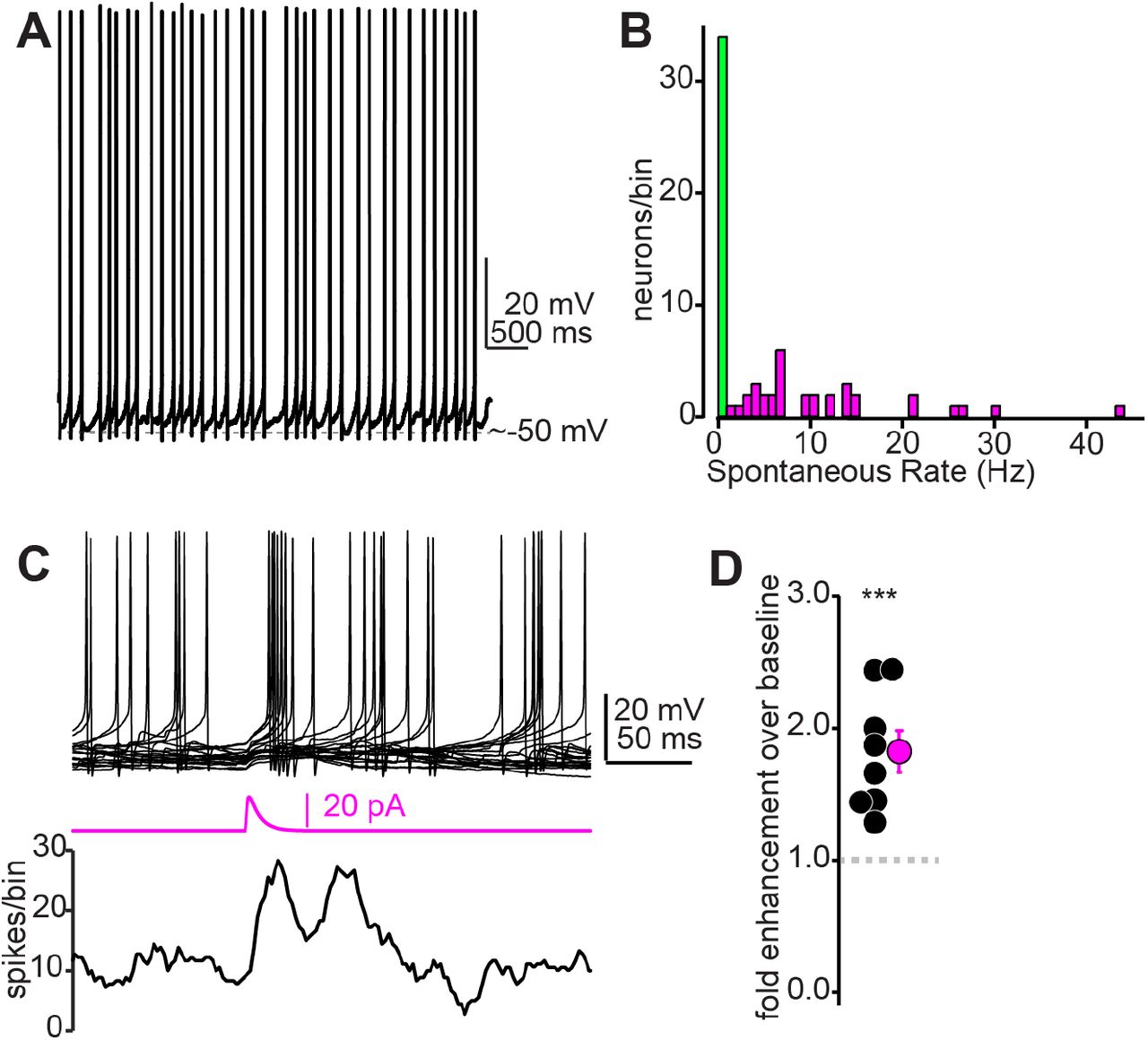
A) Example trace from a tonic firing IC GABA neuron recorded in a VGAT-cre x Ai14 mouse. B) Summary histogram of tonic firing rates in IC GABA neurons. Green bin denotes neurons that did not fire tonically. C) Top: Example overlaid traces from a tonically firing IC GABA neuron. Middle: An EPSC -like current waveform (20 pA peak amplitude) increases spike probability in the ∼50 ms following current injection, as exemplified in the spike (lower panel). D) Summary data from n=8 cells as in panel C, showing that small EPSC waveforms (10-20 pA) nearly double the spike probability of tonically firing GABA neurons.
Shell IC GABA neurons receive powerful intra-collicular excitation
The high rate of spontaneous PSPs in IC GABA neurons (Figure 3) might reflect a dense convergence of local, intra-collicular excitatory fibers. We tested this hypothesis by crossing G42 mice (Chattopadhyaya et al., 2004), which express GFP in a subset of IC GABA neurons (Supplemental Figure 2), with VGluT2-cre mice which express cre-recombinase in nearly all IC glutamate neurons (Chen et al., 2018). We then injected into the IC a cre-dependent virus packaging the red-shifted optogenetic activator ChrimsonR (Figure 5A). This approach enabled targeting our patch-clamp recordings to shell IC GABA neurons while selectively activating local glutamate neurons via an optic fiber coupled to a 625 nm LED (Figure 5B). We estimated the strength of unitary connections using a “minimal stimulation” paradigm designed to activate one or very few local inputs: The LED power level was titrated such that optogenetic stimulation fluctuated between synaptic successes and failures on a trial-by-trial basis (Figure 5C). Under these conditions, successful EPSPs had a mean amplitude of 2.8 ± 0.3 mV (Figure 5D, n = 11 cells from N = 6 mice). In addition, EPSP size grew monotonically with increasing LED power levels: EPSP amplitudes at maximal LED power had a median value 7.2 fold larger than EPSPs evoked with threshold stimulation (range: 2.2 to 37.7; Figure 5E,F. n=12 cells from N = 6 mice). These measurements likely under-estimate the extent of synaptic connectivity due to the severing of connections during the slicing procedure. Consequently, our values probably represent a lower bound for the extent of intra-collicular convergence onto shell IC GABA neurons.

A) Confocal micrograph from the IC of a G42-GFP mouse. Scale bar = 500 µm. Dashed rectangle denotes area of interest in the dorso-medial shell IC.
B) Area of interest from panel A shown at higher magnification. Scale bar = 100 µm.
C) Example current-clamp recording from a GFP+ neuron in the dorso-medial IC of a G42 mouse. The majority of GFP+ neurons recorded in G42 mice fired spontaneously when recorded in current-clamp with 0 pA bias current (8/11 neurons tested).
D) Example trace from the same neuron in panel C, but hyperpolarized with -30 pA bias current. A +40 pA current injection step reveals an adapting firing pattern. 18/21 neurons tested had similar adapting spike patterns when injected with current steps following membrane hyperpolarization to prevent spontaneous spiking. The remaining GFP+ neurons either fired a transient burst of spikes atop a T-type like hump (n=2 cells; See also Figure 3E from Oberle et al., 2022), whereas n = 1 cell showed minimal spike rate adaptation.

A,B) Cartoon of experiment. A flex-ChrimsonR virus is injected in the IC of G42 x VGluT2-cre mice. 2-4 weeks later, patch-clamp recordings are targeted to GFP+ neurons in the shell IC and glutamate neurons are optically stimulated with 625 nm light via an optic fiber positioned above the slice (not shown). C) Example recording from a shell IC GABA neuron showing transmission successes (black) and failures (grey) following optical activation of ChrimsonR-expressing glutamate neurons. D) Summary data plotting the average peak amplitude of putative unitary EPSPs during threshold stimulation. Black is individual experiments, magenta is mean ± SEM. E,F) Increasing LED power beyond threshold generates larger EPSPs, indicating that multiple presynaptic glutamate neurons converge onto a single postsynaptic GABA neuron. Data are from a different experiment as the one in C.
Repetitive corticofugal activity drives polysynaptic excitation onto IC GABA neurons
The dense convergence of intra-collicular excitation suggested by Figures 3 + 5 implies that corticofugal activity might drive spiking in IC glutamate neurons, subsequently causing polysynaptic EPSPs in IC GABA neurons. In this context, local recurrent excitation would amplify auditory corticofugal signals onto IC GABA neurons independently of strong monosynaptic innervation. We tested this hypothesis by repetitively stimulating Chronos-expressing corticofugal axons (25 flashes at 50 Hz) while recording from GABA or glutamate neurons in VGAT-cre x Ai14 mice; these stimulation parameters are within the physiological range of sound-evoked firing rates in corticofugal neurons (Williamson and Polley, 2019).
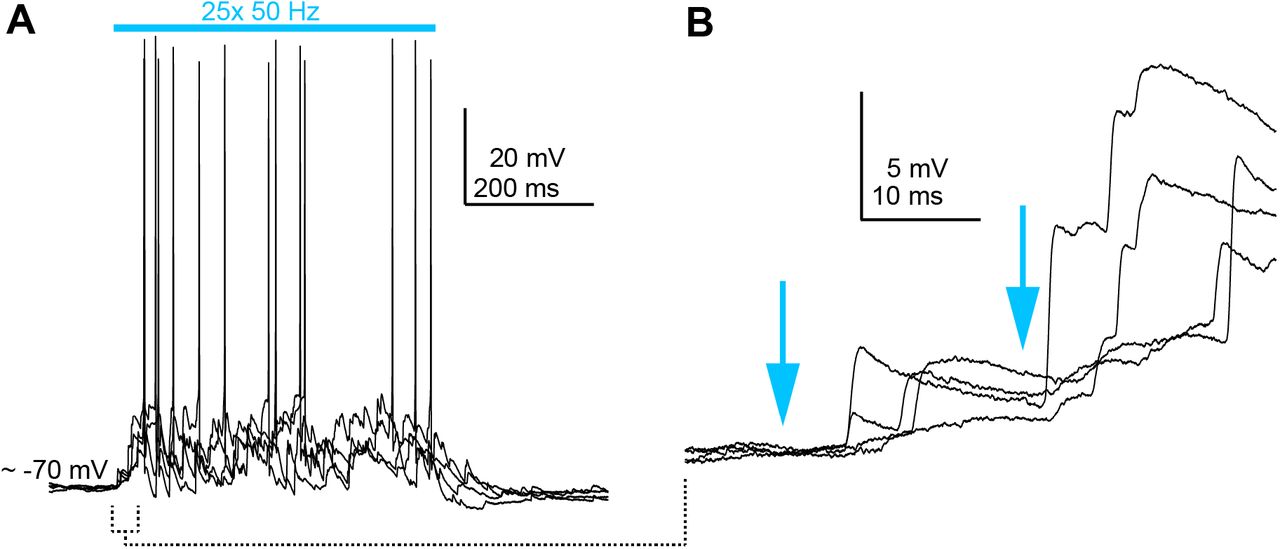
In a subset of GABA neurons (n = 21 neurons from N = 16 mice), train stimuli triggered asynchronous barrages of large amplitude EPSPs that could be evoked even when single light flashes resulted in either weak or inapparent EPSPs (Figure 6A,B). Strikingly, these EPSP barrages were sometimes powerful enough to drive spikes even when the membrane potential was hyperpolarized with bias current to prevent tonic firing (Supplemental Figure 3). Close inspection of the raw traces indicated that these asynchronous EPSPs are generated by polysynaptic activity rather than monosynaptic inputs from auditory cortex: EPSPs occurred with a variable delay and displayed considerable temporal jitter in their onset following individual light flashes (Figure 6C). Additional examples of polysynaptic EPSPs in IC GABA neurons are provided in Supplemental Figure 4. Interestingly, EPSP barrages with long and variable latencies were less common in glutamate neurons (n = 2 neurons from N = 2 mice). Instead, single and train stimuli almost exclusively generated short latency EPSPs that were temporally locked to individual light pulses (Figure 6D-F; n = 14 neurons from N = 8 mice), as expected from dense monosynaptic connectivity of corticofugal axons onto IC glutamate neurons.

A) Example recording where repetitive corticofugal stimulation triggered spiking in an IC GABA neuron. Records are multiple overlaid single trials.
B) EPSPs evoked by the first two light flashes in (A) are shown at a faster time base to highlight the onset jitter and polysynaptic nature of the EPSPs.

A) Example overlaid traces (10 consecutive trials) from an IC GABA neuron. Blue bar denotes stimulation of corticofugal fibers.
B) Traces in A expanded to highlight EPSPs evoked during the first 10 light pulses. Blue ticks indicate onset of individual light flashes.
C) Latency histogram for EPSPs of the neuron of A+B, showing two distinct peaks.
D-F) Same as panels A-C but for a different GABA neuron.
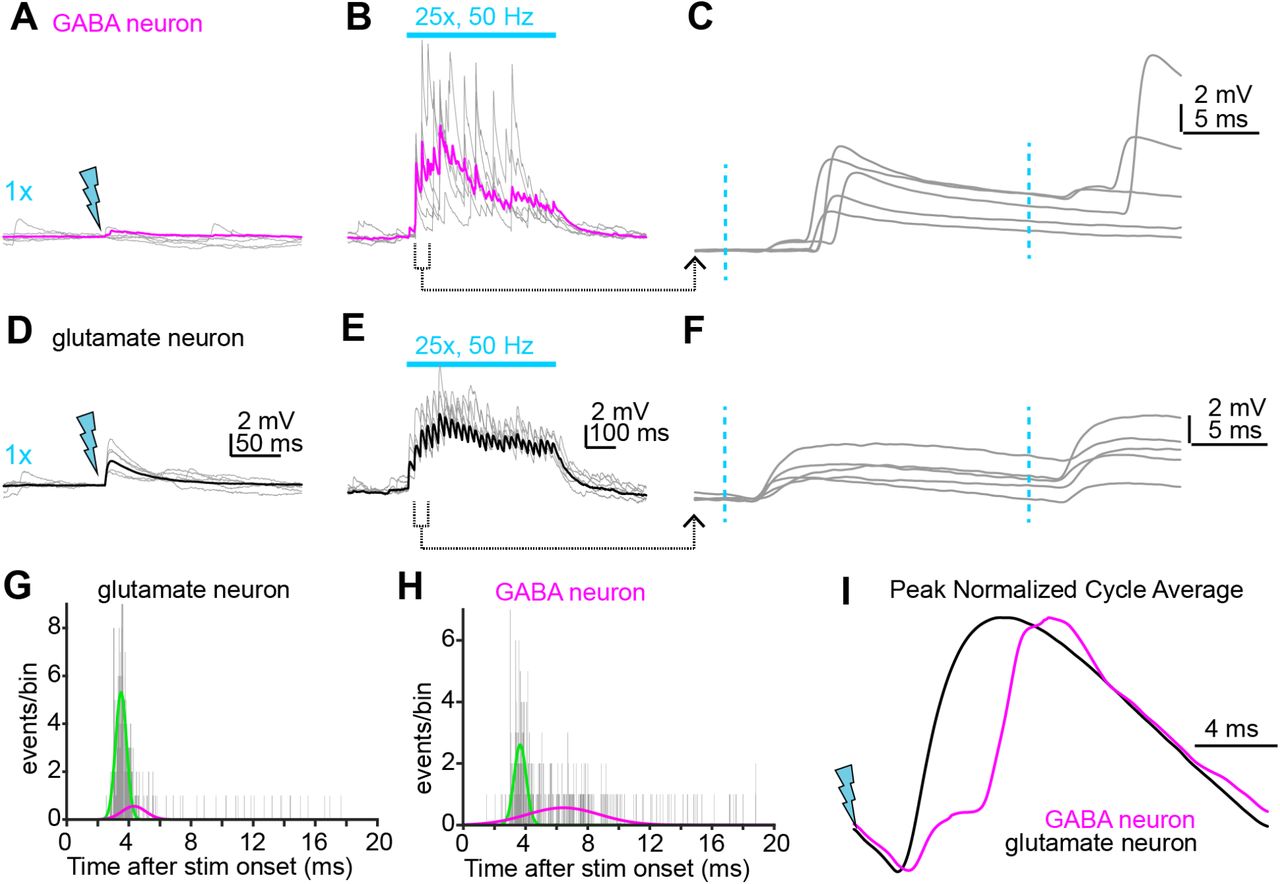
A,B) Example EPSPs in a GABA neuron during single (A) or train (B) stimuli delivered to corticofugal axons. While single light flashes generate negligible synaptic depolarization in this example, train stimulation drives powerful EPSP barrages. Gray traces are individual trials, magenta is average of multiple trials. C) The second and third stimuli from panel B are shown at faster timebase. Blue dotted lines denote light flashes. Of note is the long latency and jittered onset of the large EPSPs, a hallmark of polysynaptic origin. D,E) Same experimental approach as in A-B, but for a glutamate neuron. Gray traces are individual trials, black is average. Scale bars in D and E also apply to panels A and B, respectively. F) The second and third stimuli in the train from panel E are shown at a faster time base. Of note is the short latency and low onset jitter. G,H) Histograms of the EPSP onset latency after each of the 25 stimuli in the trains for the GABA and glutamate neurons shown in A-C and D-F, respectively.I) The membrane voltage following each stimuli in the 50 Hz trains is averaged and peak normalized. Black and magenta traces are data from the example glutamate and GABA neuron, respectively. Of note is the dual component EPSP in the GABA neuron, suggesting a monosynaptic corticofugal input followed by a much larger recurrent EPSP from the local circuit.
The difference in EPSP timing between GABA and glutamate neurons is further apparent when the EPSP latencies after each pulse in the stimulus train are plotted as latency histograms for the two example cells in panels A-F: Whereas EPSPs in the glutamate neuron reliably occur ∼3-4 ms following each pulse (Figure 6G), EPSP onsets in the GABA neuron are temporally dispersed over 10 ms following light stimuli (Figure 6H). Consequently, double gaussian fits to the data reveal two clear peaks at ∼3 and ∼7 ms in the histogram from the GABA neuron, but not in the data from the example glutamate neuron (see also Supplemental Figure 4C,F). A qualitatively similar result can be observed when averaging the membrane potential waveform of the example glutamate and GABA neurons following each light pulse in the 50 Hz train (Figure 6I): The cycle averages from the glutamate and GABA neurons show one and two peaks, respectively. We interpret the first short-latency peak as reflecting the monosynaptic input from corticofugal synapses, while the later, more temporally-jittered EPSPs reflect recurrent excitation from local IC glutamate neurons driven to spike by monosynaptic corticofugal inputs. Altogether, these data show that repetitive corticofugal activity triggers powerful recurrent excitation in shell IC GABA neurons.
Corticofugal activity preferentially triggers recurrent excitation in GABA neurons
Our data thus far suggest that corticofugal transmission drives monosynaptic excitation of IC glutamate neurons, which in turn generate polysynaptic EPSPs in IC GABA neurons. This hypothesis predicts that the primarily polysynaptic EPSPs in IC GABA neurons should be uniquely sensitive to experimental manipulations that dampen the feedforward spiking of IC glutamate neurons during corticofugal stimulation. By contrast, the mostly monosynaptic corticofugal EPSPs onto IC glutamate neurons should be comparatively less reduced by the same manipulation. We tested this prediction by quantifying how a sub-saturating concentration of the AMPA receptor antagonist NBQX (50 nM) differentially impacts repetitive corticofugal activity (25 stimuli at 50 Hz) in IC glutamate and GABA neurons. This concentration of NBQX blocks 20-50% of the AMPA receptor charge transfer (Randle et al., 1992; Diamond and Jahr, 1997) and should decrease the probability that glutamate neurons fire spikes during corticofugal stimulation, thus non-linearly reducing polysynaptic EPSPs in GABA neurons. Accordingly, 50 nM NBQX reduced the total voltage integral during stimulus trains by 22±7% in glutamate neurons (Figure 7A,C; n=10 cells from N = 8 mice). By contrast, 50 nM NBQX caused a significantly greater reduction in GABA neurons showing long-latency and asynchronous EPSPs (61±3% reduction, Figure 7B,C; n = 11 cells from N = 8 mice; p<0.0001 compared to glutamate neurons, paired t-test). Altogether these data argue that while auditory cortical activity drives spikes in GABA neurons (Supplemental Figure 3), much of the underlying depolarization is driven by local excitatory inputs rather than monosynaptic connections from corticofugal axons.

A,B) Example average train EPSPs from a shell IC glutamate (A) or GABA (B) neuron before and after bath application of 50 nM NBQX (black and magenta traces, respectively). Lower traces are the cumulative integral of the EPSP waveform. C) Summary data showing the fraction remaining in 50 nM NBQX for the for the cumulative integral of the EPSP waveform. Black and magenta symbols are individual experiments and mean ± SEM, respectively. Asterisks denote statistical significance.
Corticofugal inputs generates polysynaptic inhibition
Our data show that auditory cortical activity strongly depolarizes IC GABA neurons via a combination of direct and polysynaptic excitation. If IC GABA neurons project their axons locally, stimulating auditory cortico-collicular axons should thus suffice to generate feed-forward inhibitory postsynaptic currents (IPSCs) in the shell IC. We tested this hypothesis in Chronos-injected wild-type mice by voltage-clamping the somata of shell IC neurons near the reversal potential for excitation (∼ +5 to +10 mV) and stimulating corticofugal axons with trains of blue light flashes (25x, 50 Hz). Recordings under these conditions were characterized by a flurry of spontaneous, outward inhibitory postsynaptic currents (IPSCs) that were preferentially sensitive to the GABAA receptor antagonist SR95531 (5-10 µM; Supplemental Figure 5. Median IPSC rate: 13.5 vs. 0.0557 Hz in control and SR95531, p=0.0156, sign-rank test, n = 7 cells from N = 3 mice). Strikingly, stimulating auditory cortico-collicular axons caused a ∼2-3 fold increase in IPSC rate relative to baseline (Figure 8A,B), as revealed by a main effect of time in a two-way, repeated measures ANOVA (F(14,126)=11.7, p<0.0001, n = 10 cells from N = 5 mice). Importantly, the IPSC rate increase was abolished by bath application of glutamate receptor antagonists NBQX (10 µM) and/or R-CPP (5 µM), indicating that it was mediated by polysynaptic activation of IC GABA neurons (Timepoint x drug interaction: F(14,126)=13.38, p<0.0001).

A) Example voltage-clamp recording from a shell IC neuron held at +8 mV. Of note are the spontaneous IPSCs often occurring in bursts, suggesting they are mediated by action potentials in local GABA neurons. Asterisk denotes one such IPSC burst shown at a faster time base in the inset.
B) In the same cell as shown in panel A, bath application of the GABAA receptor antagonist SR95531 (10 µM) blocks the majority of spontaneous IPSCs. Although quite rare, a few small amplitude events nevertheless remained in the presence of SR95531 (asterisk and inset; median IPSC rate in SR95531: one event per 17.95 seconds). These SR95531 resistant outward currents may reflect long-range glycinergic inputs from the ventral nucleus of the lateral lemniscus (Moore and Trussell, 2017).
C) Summary data for n = 7 cells showing that SR95531 profoundly reduces spontaneous IPSC rates, indicating that inhibitory transmission in shell IC neurons is mostly GABAergic.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Example recording from a shell IC neuron voltage clamped near the reversal potential for synaptic excitation, before and after bath application of glutamate receptor blockers. Gray traces are overlays of individual trials, black is average. Of note is that the powerful increase in IPSCs during corticofugal stimulation (blue bar) in control conditions is abolished by blocking glutamate receptors. B) The normalized IPSC rate is binned every 100 ms for statistical comparisons. Asterisks denote p<0.001 following Sidak’s post-hoc tests comparing appropriate control and drug timepoints. Of note is that significance is limited to timepoints during optical stimulation of corticofugal axons. C) Average traces showing temporal integration of corticofugal EPSPs in current-clamp before (black) and after (magenta) bath application of the GABAA receptor antagonist SR95531. Shading is ± SEM. D) Summary data for the effect of SR95531 on corticofugal transmission current-clamp. Values are the ratio of voltage integrals in drug and control conditions. Asterisk denotes statistical significance.
Functionally, this feedforward inhibition controlled the temporal integration of corticofugal excitation: Blocking GABAA receptors significantly increased corticofugal EPSP summation during repetitive stimulation in current clamp, measured as a significant increase in the voltage integral following bath application of 5-10 µM SR55931 (Figure 8C,D. Fractional change relative to control: 1.18 ± 0.06, n = 12 cells from N = 11 mice. p=0.013, one sample t-test). Interestingly, we also noted a minor, but statistically significant increase in membrane resistance following SR95531 (386 ± 61 vs. 397 ± 62 MΩ in control and SR95531, respectively. Fractional change: 1.04 ± 0.014, p = 0.0204, one sample t-test). Although this observation argues that the high rates of spontaneous IPSCs from the local circuit contribute a certain level of effectively tonic inhibition (e.g., Supplemental Figure 5. See also Brickley et al., 1996, 2001), this small change in membrane resistance in SR95531 is unlikely to account for our observed effect on EPSP summation: The relative increase in EPSP integral was more than 4-fold larger than the change in membrane resistance, and there was no significant pairwise correlation between the magnitude of EPSP enhancement and change in membrane resistance after SR95531 application (Pearson’s coefficient ± standard deviation from 100 bootstrapped iterations: -0.25 ± 0.36, p = 0.43). Thus, auditory cortico-collicular transmission generates intra-collicular inhibition that regulates how IC neurons integrate descending signals over time.
Discussion
Although decades of in vivo physiology show that auditory cortical activity inhibits IC sound responses, the underlying synaptic mechanisms have been difficult to pin down. Indeed, anatomy data suggest that auditory cortical axons preferentially target glutamate, rather than GABA neurons in the IC (Nakamoto et al., 2013; Chen et al., 2018). This anatomical motif is in striking contrast to the often studied pyramidal neuron-based microcircuits of sensory cortices and hippocampus, where long-range excitation powerfully drives feed-forward inhibition to sharpen temporal fidelity of afferent signals: Glutamatergic synapses onto GABAergic interneurons in these circuits are typically more numerous, have higher synaptic conductance, and/or higher release probability than onto pyramidal projection neurons (Acsády et al., 1998; Lawrence et al., 2004; Gabernet et al., 2005; Cruikshank et al., 2007). At first glance, the anatomical constraints of the auditory cortico-collicular pathway suggest that descending inhibition of the IC is unlikely to operate via a classical feedforward recruitment of locally projecting IC GABA neurons. Instead, auditory cortex may effectively recruit long-range inhibitory afferents to the IC (e.g., dorsal nucleus of the lateral lemniscus; Budinger et al., 2000), or alternatively may provide monosynaptic inhibition via a newly discovered, long-range projection from VIP GABAergic neurons (Bertero et al., 2021).
However, our experiments reveal cellular and circuit mechanisms that enable the auditory cortex to trigger intra-collicular inhibition in absence of a dense innervation onto IC GABA neurons. Consistent with the anatomical data (Nakamoto et al., 2013; Chen et al., 2018), corticofugal synapses tended to generate smaller EPSPs in IC GABA compared to glutamatergic neurons. Nevertheless, two cellular mechanisms enabled corticofugal activity to effectively increase firing rates of IC GABA neurons. First, approximately half of shell IC GABA neurons encountered in our study fired tonically, similar to findings in the NPY-positive GABA neurons of the central IC (Silveira et al., 2020). Thus, even apparently weak descending EPSPs sufficed to increase the basal firing rates of GABA neurons. Second, repetitive firing of corticofugal axons drives robust polysynaptic excitation in IC GABA neurons due to local excitatory circuitry, thereby orchestrating a multisynaptic cascade leading to intra-collicular inhibition.
We did not observe direct monosynaptic GABAergic inputs from auditory cortex VIP neurons to the shell IC (Bertero et al., 2021). In vitro, the IPSC barrages recruited during auditory cortex stimulation were entirely blocked by glutamate receptor antagonists, indicating the necessity for intermediary, excitatory synapses onto IC GABA neurons with locally projecting axons. This result is perhaps surprising, as our stimulation paradigm was quite strong (25 light flashes at 50 Hz) and likely sufficed to activate descending synapses with low release probability. Furthermore, Bertero et al. showed that VIP-GABA axons terminate in the dorsomedial IC shell where our recordings were obtained. However, an absence of evidence is not conclusive evidence of absence: This newly discovered pathway may instead operate via non-canonical mechanisms such as extrasynaptic diffusion of GABA, or via release of the neuropeptide VIP. These synaptic properties would be difficult to quantify with our whole-cell patch-clamp recordings. In addition, selective viral tropism may have hindered the expression of our optogenetic construct in auditory cortical VIP neurons; extensive follow-up experiments are necessary to clarify these issues. Rather, our results show that intra-collicular mechanisms suffice to explain at least some of the auditory cortex’ inhibitory actions on IC sound responses.
Functionally, the polysynaptic activity driven by descending transmission could enable a myriad of “high-level” computations in the shell IC. For example, previous work in piriform cortex shows that recurrent excitatory synapses from principal cells onto interneurons trigger feedback inhibition and truncates the duration of principal cell responses to incoming activity from the olfactory bulb. Although odorant signals from the bulb’s lateral olfactory tract are graded with respect to odorant concentration, the output of piriform cortex principal cells is consequently rendered concentration invariant by this feedback inhibition which powerfully limits spiking to odor onset (Bolding and Franks, 2018). Our findings reveal a conceptually similar microcircuit architecture in the shell IC. Corticofugal projections, which potentially transmit behaviorally relevant information, could thus enhance IC glutamate neurons’ ability to trigger feedback inhibition and promote a level-invariant representation of behaviorally relevant sounds in the IC. Such a computation may be particularly useful in creating invariant neural responses necessary for sound localization in complex, reverberant environments (Hartmann, 1983; Rakerd and Hartmann, 1986). Indeed, the onset spiking response of IC neurons represents directional signals that are less corrupted by reverberant reflections and inter-aural decorrelation than later, sustained responses (Devore et al., 2009). It is not unreasonable to assume that onset spiking in glutamate neurons could recruit feedback inhibition that minimizes sustained activity during the reverberant tail of sounds, and that this mechanism is enhanced by “top-down” cortical signals during periods of increased vigilance and attention. However, extensive future studies are required to elucidate the behaviorally relevant conditions that trigger descending inhibitory control, and to understand how corticofugal projections might flexibly switch between amplifying and inhibiting tectal sound responses.
Methods
The experiments were approved by the University of Michigan’s IACUC and performed according to NIH’s Guide for the care and use of laboratory animals. In vitro experiments were performed on 5-8 week old male or female offspring of VGAT-ires-cre x Ai14 fl/fl or VGluT2-cre x G42 breeder pairs from our colony (Figures 2-7) or 5-8 week old C57/Bl6J mice ordered from Jackson labs (Figure 8). For in vivo experiments with wild-type mice, we used 4-7 week old offspring of CBA x C57Bl6/J matings (Figure 1B,C) or 7-10 week old C57/BL6J mice ordered from Jackson labs (Figure 1E,F). The raw data from some of these in vivo recordings were also included in a previous set of distinct analyses (Oberle et al., 2022).
Surgery for intracranial virus injections
Our virus injection protocol is described in detail in Oberle et al., 2022. Briefly, mice were anesthetized with 4-5% isoflurane in O2, mounted in a stereotaxic frame, and isoflurane was lowered to 1-2% while body temperature was maintained near 37-38° C using a heating blanket. Mice were administered 5 mg/kg carprofen as a pre-surgical analgesic, a small incision was made in the scalp, and 2% lidocaine was then applied to the wound margins. A 200-400 µm craniotomy was carefully drilled over the left auditory cortex (−2.75 mm from Bregma, centered on the lateral ridge) or left IC (0.9 mm caudal and 1 mm lateral from lambda suture) to inject 100-200 nL of pAAV-Syn-Chronos-GFP (Addgene #59170-AAV1) or pAAV-syn-flex-rc[ChrimsonR-tdTomato] (Addgene #62723-AAV5) virus. At the end of the surgery, the craniotomy was filled with bone wax and the skin was sutured. Immediately following surgery, mice received an analgesic injection of buprenorphine (0.03 mg/kg, s.c.) and recovered on a heating pad before returning to their home cage. A post-operative dose of carprofen was administered ∼24 hours later.
In vivo electrophysiology
Mice were deeply anesthetized with isoflurane, mounted in the stereotaxic frame, and craniotomies were carefully opened over the left IC and auditory cortex as described above and in Oberle et al. 2022. For optogenetic stimulation, we left the dura intact and implanted a cranial window over the auditory cortex. For electrical stimulation we made a small slit in the dura and the craniotomy was sealed with silicone elastomer. The IC craniotomy was plugged with silicone elastomer, a titanium headbar was implanted with dental cement, and the mouse was removed from the stereotax before being re-anesthetized with urethane (1.5 g/kg, i.p.) and head-fixed in a custom-made sound attenuation chamber. Body temperature during the recording session was maintained at 37-38° C with a custom designed, feedback-controlled heating blanket. Optogenetic stimulation was performed with a 0.5 NA, 400 µm core optic fiber (Thorlabs M45L02) coupled to a 470 nm LED (Thorlabs M470F3) positioned <1 mm away from the auditory cortex cranial window. For electrical stimulation experiments, a bipolar platinum-iridium electrode (FHC 30210) was carefully inserted ∼800 µm into auditory cortex at an angle roughly perpendicular to the cortical layers and shocks were delivered via a custom stimulus isolator. Whole-cell current-clamp recordings were obtained ∼100-400 µm from the dura with pipettes containing (in mM): 115 K-Gluconate, 4 KCl, 0.1 EGTA, 10 HEPES, 14 Tris-Phosphocreatine, 4 Mg-ATP, 0.5 Tris-GTP, 4 NaCl, pH 7.2-7.3, 290 mOsm (open tip resistance: 5-10 MΩ).
In vitro electrophysiology
2-4 weeks following viral injections, mice were deeply anesthetized with isoflurane and 200-300 µm brain slices containing both IC hemispheres were prepared in warm (∼34° C) oxygenated ACSF containing (in mM): 119 NaCl, 25 NaHCO3, 3 KCl, 1.25 NaH2PO4, 15 glucose, 1 MgCl2, 1.3 CaCl2, 1 ascorbate, 3 pyruvate. A small cut was typically made in the lateral portion of the right cerebellum or right IC to visually identify the un-injected hemisphere. Slices were then incubated at 34° C in a holding chamber filled ACSF for 25-30 min and then stored at room temperature until use. During experiments, slices were mounted in a submersion chamber continuously perfused with oxygenated ACSF heated to 32-34° C (2-4 mL/min; chamber volume: ∼ 1 mL). Neurons in the dorso-medial shell IC were visualized via DIC or Dodt contrast optics using a 40x or 63x objective (Zeiss Axioskop 2 FS Plus or Olympus BXW51 microscope). GABA neurons were identified from tdTomato or GFP fluorescence. Presumptive glutamate neurons were identified based on lack of fluorescence in VGAT-cre x Ai14 mice. Whole-cell current-clamp recordings were obtained using pipettes filled with the same K+ rich internal solution as employed for in vivo recordings (open tip resistance: 3-6 MΩ). In some experiments, 30 µM Alexa 488 or 0.1% biocytin were added to the internal solution to visualize neuronal morphology via online fluorescence or post-hoc histological reconstruction. Voltage clamp recordings were obtained with a Cs+ based solution containing (in mM): 110 Cesium Methanesulfonate, 10 QX-314-Bromide, 0.1 EGTA, 10 HEPES, 0.5 Tris-GTP, 4.5 MgATP, 5 TEA-Cl, 10 Tris-phosphocreatine. Experiments were generally conducted within 3-4 hours following slice preparation. Chronos-expressing axons were stimulated via 2-5 ms wide-field flashes of blue light delivered through the microscope objective. ChrimsonR-expressing IC neurons were stimulated via 0.5-1 ms flashes from a 400 µm diameter optic fiber (0.5 NA) coupled to a 625 nm LED <1 mm above the slice.
Data Acquisition
Data were acquired using a Molecular Devices Multiclamp 700B or AM Systems model 2400 patch-clamp amplifier, online filtered at 2-10 kHz, and digitized at 50 kHz with a National Instruments PCI-6343 card + BNC2090A interface controlled by Matlab based acquisition software (Wavesurfer). As shown in Figure 4, many GABA neurons fired tonically in vitro. Consequently, negative bias current (−5 to -200 pA) was often injected to hyperpolarize neurons and prevent spontaneous or optogenetically triggered spiking when recording EPSPs in GABA neurons.
Data analysis
All experiments were analyzed using custom scripts written in Matlab. Analyses in Figures 1, 2, 5, 7, and 8 were run on averages of multiple trials (typically >10 trials per condition) after baseline subtraction and lowpass filtering at 1 kHz. EPSP peak amplitudes were calculated by averaging data points ± 0.1 ms around the local maximum following optogenetic stimulation. Halfwidths were defined as the full width at half-maximum of the peak. Voltage integrals were calculated using the Matlab functions trapz() or cumtrapz(). Event detection analyses in Figures 3, 4, 6, and 8 were conducted by differentiating the membrane voltage or current traces, smoothing the data (1-5 ms sliding window), and using threshold crossing or peak detection algorithms to identify synaptic events. Synaptic current waveforms (Figure 4C,D) were generated in Wavemetrics Igor Pro using the Neuromatic package (Rothman and Silver, 2018).
Statistics
In the text, n and N refer to the number of neurons and mice, respectively. Sample sizes were not explicitly pre-determined prior to data collection. However, the number of cells and mice in these experiments are in accordance with commonly accepted standards in the field. Data were tested for normality using a Lilliefors test prior to statistical comparisons. Parametric tests were used for normally distributed data, while non-parametric tests were used if one or more of the datasets deviated from normal. Alpha is corrected for multiple comparisons in post-hoc significance tests following ANOVA. Statistics were run in Matlab or Graphpad Prism 9. Pearson’s coefficient and associated bootstraps were calculated in Matlab with the functions corr() and bootstrp(), respectively.
Author Contributions
HMO + PFA conducted and analyzed experiments of Figures 1 + 8. ANF performed stereotaxic injections with contributions from HMO. HMO + ANF conducted histology and confocal imaging. PFA performed and analyzed experiments of Figures 2-7, interpreted the results, and wrote the paper.
Acknowledgments
Funding was provided by the Whitehall Foundation, Hearing Health Foundation, and NIH/NIDCD R01DC019090 to PFA, as well as T32DC005356 and T32DC000011 to HMO. We thank Dr. Michael Roberts for critical comments on the manuscript, Dr. Kevin Jones for providing the G42 mouse used for histology in Supplemental Figure 2, Dr. Meike Rogalla for the illustrations in Figure 1, and Jordyn Czarny for technical assistance with histology.
References




