

Abstract
Transient voltage-gated sodium currents are essential for the initiation and conduction of action potentials in neurons and cardiomyocytes. The amplitude and duration of sodium currents are tuned by intracellular fibroblast growth factor homologous factors (FHFs/iFGFs) that associate with the cytoplasmic tails of voltage-gated sodium channels (Navs), and genetic ablation of Fhf genes disturbs neurological and cardiac functions. Amongst reported phenotypes, Fhf2null mice undergo lethal hyperthermia-induced cardiac conduction block attributable to the combined effects of FHF2 deficiency and elevated temperature on the cardiac sodium channel (Nav1.5) inactivation rate. Fhf2null mice also display a lack of heat nociception, while retaining other somatosensory capabilities. Here we employ electrophysiological and computational methods to show that the heat nociception deficit can be explained by the combined effects of elevated temperature and FHF2 deficiency on the inactivation gating of sodium channels Nav1.7 and Nav1.8 expressed in dorsal root ganglion C-fibers. Hence, neurological and cardiac heat-associated deficits in Fhf2null mice derive from shared impacts of FHF deficiency and temperature towards Nav inactivation gating kinetics in distinct tissues.
1. Introduction
Cutaneous peripheral pain sensation is initiated within ganglionic nociceptive neurons bearing axons and receptive terminals that terminate in the epidermis of the skin. Noxious chemical, mechanical, or thermal stimulation opens transient receptor potential (TRP) cationic channels at afferent terminals 30, with sodium influx and depolarization triggering the activation of recessed voltage-gated sodium channels (Navs) that mediate initiation and orthodromic conduction of action potentials 15. Action potentials are generated through the essential actions of sodium channels Nav1.7 and Nav1.8. Loss-of-function mutations in genes encoding Nav1.7 and Nav1.8 are hypoalgesic, reducing or ablating peripheral nociceptor activation by noxious stimuli 1,3,6,12,20, while gain-of-function missense mutations in these channel genes can cause hyperalgesia 7,10,36.
The differential voltage-dependent gating properties of Nav1.7 and Nav1.8 enable these channels to act in concert to promote action potential initiation. Nav1.7 opens more rapidly and at more negative membrane potential than does Nav1.8, but Nav1.7 also inactivates at more negative voltages than Nav1.8 2,8. During a slow rise in voltage driven by stimulus-induced TRP channel currents at afferent terminals, a substantial fraction of Nav1.7 channels inactivate from the closed state, but the remaining available Nav1.7 fraction provides a rapid “primer” current that depolarizes the membrane sufficiently to allow Nav1.8 channels to open and further drive the upstroke of the initiating action potential 5. The orthodromic conducting action potential causes rapid passive depolarization of membrane that lies ahead of the advancing spike, allowing for a greater contribution of Nav1.7 sodium currents for spike conduction. Reduced availability of Nav1.7 near afferent terminals may explain why distal afferents harbor a greater density of Nav1.8 17.
Sodium channel surface density and gating are regulated by post-translational Nav modifications and by association with membrane-associated or cytoplasmic proteins. Fibroblast growth factor homologous factors (FHFs), also termed intracellular fibroblast growth factors (iFGFs), are small cytosolic proteins that bind to the carboxyl-terminal domain of Navs 13,18. Most FHF isoforms induce a depolarizing shift in voltage dependence of Nav inactivation without altering the voltage for activation 9,19,33. This voltage shift increases sodium current generated by Navs in three ways: 1) by reducing the fraction of channels rendered inactive at steady state resting potential, 2) by reducing the rate of closed-state Nav inactivation during membrane depolarization, and 3) by slowing rate of Nav inactivation from the open state 21,22.
Naturally occurring or engineered mutations in FHF genes result in disorders of excitable tissues, including ataxia 14,34, epilepsies 11,24,27,31, hypothalamus-induced obesity 29, hypoalgesia 35, and cardiac conduction block 21,22. Fhf2null mice are sensitive to ventricular cardiac conduction block when exposed to various stressors, including sodium or calcium channel blockers, uncoupling agents, or hyperthermia 21,22. FHF2 deficiency and elevated core body temperature collaborate to reduce availability and speed inactivation of cardiac Nav1.5 leading to failure of action potential conduction through myocardium 21.
Genetic ablation of mouse FHF2 in peripheral neurons also results in loss of cutaneous heat nociception 35. Corroborating these prior findings, we report here that Fhf2null mice are completely insensitive to cutaneous heat stimulation while retaining relatively normal mechanical nociception. We speculated that FHF2 in nociceptive neurons modulates Nav1.7 and Nav1.8 inactivation gating in a manner analogous to its effects on cardiac Nav1.5 inactivation gating. Such modulation would provide a transient sodium current reserve in heated afferent terminals. Indeed, we show here that FHF2 promotes a 15 mV depolarizing shift in the voltage dependence of steady state Nav1.7 inactivation and slows closed- and open-state Nav1.7 inactivation. Furthermore, there is an additive effect of FHF2 deficiency and elevated temperature towards speeding the rate of Nav1.7 inactivation. The voltage dependence of tetrodotoxin-resistant transient sodium current (Nav1.8) also displays a 6 mV hyperpolarizing shift in Fhf2null DRG neurons compared to wild-type neurons. As a consequence of accelerated Nav inactivation, Fhf2null saphenous nerves undergo rapid C-fiber action potential conduction block when heated above 41°C ex vivo, with rapid restoration of spike conduction upon cooling. Furthermore, through the use of wild-type and Fhf2null nociceptive neuron computational models that differ only with respect to Nav inactivation gating, we show that simulated heating of afferent terminals to 43°C fails to generate action potentials in the Fhf2null model. Overall, our findings show that the limiting of sodium currents by FHF2 deficiency and elevated temperature constitutes a common gating mechanism underlying heat nociception and cardiac conduction deficits in Fhf2null mice.
2. Materials and Methods
2.1 Mouse genetics and genotyping
The murine embryonic stem cell clone EPD0339-4-F09 (International Mouse Phenotype Consortium) carrying a knockout first with conditional potential cassette bears loxP sites flanking (floxing) Fhf2 exon 3 and Frt sites flanking the embedded G418-resistance and beta-galactosidase markers; these cells were used to derive mice carrying a Fhf2targeted allele by blastocyst injection and outbreeding 21. The Fhf2targeted allele was converted to an Fhf2null allele by injection of Cre recombinase-encoding plasmid into fertilized Fhf2targeted/+ eggs followed by reimplantation 21. Alternatively, the Fhf2targeted allele was converted to the conditional Fhf2flox allele by mating with mice that globally express a nuclear-targeted FLP recombinase ROSA26Sortm2(FLP*)Sor (Jackson Labs Stock #012930) 25 to excise the embedded G418-resistance and beta-galactosidase markers. The Fhf2null and Fhf2flox alleles were backcrossed for four generations onto the 129S2 strain background. By crossing heterozygous Fhf2flox/+ females with male transgenic Advillin-Cre driver mice (Jackson Labs Stock #032536), which express Cre recombinase exclusively in the peripheral sensory neurons 37,38, we were able to generate Fhf2flox/Y:Adv-Cre test mice and Fhf2+/Y:Adv-Cre negative control littermates. Fhf2flox mice have been made publicly accessible through Jackson Labs (Stock #036280).
PCR genotyping was performed using PhireII Hot-Start DNA polymerase (Fisher Sci.). The Fhf2flox and Fhf2+ alleles were distinguishable upon PCR with primers IVS2-For (5’-GCCAGGAGTCTGCTCAACTCT) and IVS3-Rev (5’-GACTTTGGTGGGAGCATCCTGA), yielding products of 533 base pairs versus 388 base pairs, respectively.
2.2 Immunoblot detection of FHF2
Detergent-solubilized brain and DRG tissue lysates were electrophoresed through 4%–20% precast polyacrylamide gradient gel (Invitrogen), transferred to activated PVDF (Immobilion-P, Millipore), probed with rabbit anti-FHF2 polyclonal antibodies (Schoorlemmer 2002), and detected with peroxidase-conjugated secondary antibodies and enhanced chemiluminescence.
2.3 Footpad Immunofluorescence
Footpads tissue punch samples harvested from adult wild-type (Fhf2+/Y) and Fhf2null (Fhf2-/Y) mice were submersion fixed in 4% paraformaldehyde for 1 hour at 4°C, treated with cryoprotectant 30% sucrose in phosphate-buffer saline for 24 hours at 4°C, and embedded and frozen in OCT blocks for cryosections. Cryosections 30μm thick were permeabilized with 0.5% Triton X100, preabsorbed with 10% fetal calf serum, and incubated overnight at 4°C with chicken anti-PGP9 antibodies (EnCor Biotechnology) at 0.5 μg mL-1 each. After three wash cycles, the tissue was subsequently incubated with 1:200 dilution of flurophore-conjugated anti-chicken IgY at room temperature for 2 hours. Images were captured with a Leica TCS2 confocal microscope and images spanning 2-5 mm through the z-axis were merged.
2.4 Sensory assays
All mice were conditioned to test apparati, environments and modes of restraint for at least two consecutive days prior to data collection. Tail-flick: Heat nociception was evaluated using a tail-flick apparatus [Ugo Basile Tail Flick 7360]. Mice were held with tail resting unrestrained on disk heat source, which upon test initiation emitted infrared radiant heat 2 cm from the tip of the tail. The instrument measures the latency to tail withdrawal from the heat source. Paw pinch: Temperature independent nociception was assayed using a force-calibrated forceps pressure transducer [Ahlborn - Almemo 2450] to determine exactly how much pressure applied to the hind paw of a mouse elicited a pain induced withdrawal response. Mice were placed in a tubular restraint with their hind limbs exposed. Force was applied to each hind paw and the threshold response was recorded. Von Frey filament assay: Basal somatosensory response was measured using a set of sensory evaluator filaments [NC Medical - NC12775-99]. Mice were placed in an elevated mesh-floored cage and allowed to explore and adapt for 20-30 minutes. Stimulation was manually applied to the hind footpads of the mouse starting with the thinnest softest filament and proceeding through filaments of greater thickness, performing five trials for each filament. Paw withdrawal was scored as a positive response, while bending of the filament without paw withdrawal constituted a negative response.
2.5 Voltage clamp analysis of Nav1.7 sodium currents in transfected HEK293 cells
Human embryonic kidney HEK293 cells (QBiogene) were transiently co-transfected with a 2:1 mixture of Nav1.7-expressing plasmid 7 and a pIRES2-ZsGreen bicistronic plasmid (Clontech) expressing ZsGreen and mouse FHF2B proteins 9. The same pIRES2-ZsGreen plasmid without FHF2 coding sequence served as control. Cells were trypsinized 3 hours post-transfection, seeded onto gelatinized coverslips, and were used for recording after 48 hr. For sodium current recordings, coverslips were transferred to recording chamber containing carbogen-buffered bath solution (115 mM NaCl, 26 mM NaHCO3, 3 mM KCl, 10 mM glucose, 4 mM MgCl2, 2 mM CaCl2, 0.2 mM CdCl2, 3 mM myoinositol, 2 mM Na pyruvate, 7 mM NaOH-buffered HEPES pH 7.2) at 25 °C and green fluorescent cells were whole-cell patched with pipettes filled with 104 mM CsF, 50 mM tetraethylamine chloride, 10 mM HEPES pH 7.2, 5 mM glucose, 2 mM MgCl2, 10 mM EGTA, 2 mM ATP, 0.2 mM GTP and having 1-2 MΩ resistance. All voltage commands and current recordings were made using a MultiClamp 700 amplifier, digital/analog converter Digidata 1440, and pClamp10 software (Molecular Devices - Axon Instruments). For all recording protocols, voltage-gated current was isolated during data acquisition by P/N subtraction of leak and capacitive currents (N = −6). To ensure adequate voltage clamping during sodium channel activation was adequate cells were subjected to a 19-sweep series of voltage steps from a hold of −120 mV to between −80 mV and −10 mV in 5 mV increments. As criterion for adequate clamp, transient current peaks for all voltage commands were nested within the larger current trace of a following or preceding voltage step command.
Steady-state channel inactivation protocol
To determine the voltage dependence of steady state channel inactivation, a 19-sweep protocol used a −120 mV holding command, a 60 ms variable test voltage step (−120 + 5(n - 1) mV), followed by a 25 mV reporting pulse. Voltage ramp protocol: As a measure of closed state channel inactivation rate, a 10-sweep protocol used −120 mV hold command followed by depolarization to −30 mV either instantaneously (voltage step) or as a ramp ranging in time from 2ms (= 45 mV ms-1) to 18ms (= 5mV ms-1). After recording at 25 °C, temperature was ramped to 35 °C and later to 40 °C. At elevated temperatures, the cell was first tested as above to ensure maintenance of tight clamp, after which the voltage ramp protocol was conducted. For many cells, voltage ramp protocols could be successfully run at all three temperatures.
2.6 Voltage clamp analysis of TTX-resistant sodium currents in acutely dissociated DRG neurons
DRGs were plucked from spinal columns of 8-12 week-old mice and trimmed free of associated nerves. Neurons were dissociated by treatment with 1% trypsin and 150 μg/ml collagenase in Hank’s balanced salt solution at 37°C for 30 minutes followed by washing into Dulbecco’s modified Eagle’s medium (DMEM) + 10% fetal bovine serum (FBS) + 250 μg/ml DNaseI and trituration with fire-polished Pasteur pipettes. Dissociated cells were washed three times with DMEM + 10% FBS and seeded on to 12 mm diameter coverslips precoated with poly-D-lysine and laminin at a density of 3 x 104 cells/coverslip and cultured for 18-40 hours prior to recording. Recordings were conducted in extracellular solution consisting of 24 mM NaCl, 3mM KCl, 3 mM CsCl, 26 mM NaHCO3, 50 mM Choline Cl, 11 mM Glucose, 25 mM TEA-Cl, 2 mM Na Pyruvate, 3 mM Myoinositol, 10 mM HEPES, 5 mM 4-aminopyridine, 2 mM MgCl2, 1 mM BaCl2, 1 mM CaCl2 and 0.2 mM CdCl2 and 0.5 μM tetrodotoxin (TTX) at pH 7.2 using patch pipettes of 1-1.5 MΩ resistance when filled with 120 mM K gluconate, 4 mM NaCl, 5 mM KOH-buffered HEPES, 5 mM KOH-buffered EDTA, 15 mM glucose, 1 mM MgSO4, 3 mM Mg-ATP, 0.1 mM Na2-GTP at pH 7.2. The voltage dependence of TTX-resistant sodium channel steady state inactivation was assayed in a 19-sweep protocol used a −70 mV holding command, a 60 ms variable test voltage step (−70 + 5(n - 1) mV), followed by a 25 mV reporting pulse.
2.7 Temperature-induced excitation of acutely dissociated DRG nociceptive neurons
Voltage-sensitive dye recordings
DRG neurons grown on coverslips as described above were first treated with a blocking solution consisting of sterile HBSS with 10 mM HEPES + 1% horse serum for 5 minutes at room temperature followed by staining with HBSS/HEPES + 70 mM Di-8-ANEPPS (Biotium) for 15 minutes room temperature and recovery in HBSS/HEPES + 10% horse serum for 5 minutes. Coverslips of stained neurons were transferred to recording chamber containing carbogen-bubbled extracellular solution: 115 mM NaCl, 26 mM NaHCO3, 3 mM KCl, 1.2 mM KH2PO4, 3 mM glucose, 2 mM myoinositol, 2 mM Na pyruvate, 7 mM HEPES, 1.2 mM MgSO4, 2 mM CaCl2, 0.2 mM CdCl2 at pH 7.2. Using a 40X lens objective, SOLA-SE LED illumination (Lumencor) and a filter cube consisting of a 520±45 nm excitation interference filter, a dichroic mirror with 570 nm central wavelength, and 610nm barrier emission filter, fluorescence images were captured at 2 Hz with a NeuroCCD-SM camera (Redshirt Imaging) while heating the bath from 32°C to 44°C at a rate of approximately 4 degrees C per minute. Action potentials were detected as transient fluorescence changes of 2-3% resting fluorescence. Excitable cells were then imaged by high-resolution bright field illumination in order to assess the neuronal cell diameter.
Current-clamp recordings
Recordings were performed using the same extracellular solution as above and patch pipettes of 2-3 MΩ resistance when filled with 120 mM K gluconate, 4 mM NaCl, 5 mM KOH-buffered HEPES, 5 mM KOH-buffered EDTA, 15 mM glucose, 1 mM MgSO4, 3 mM Mg-ATP, 0.1 mM Na2-GTP at pH 7.2. Cells approximately 12 microns in diameter were selected for patching based upon prior optical imaging that had determined the size of heat responsive cells. Excitability was first assessed at 32°C by applying 200 millisecond current pulses ranging from 0-400 pA in 25 pA intervals. Excitable cells were then held with 0 pA current injection while heating the chamber to 45°C at approximately 4 degrees C per minute. Voltages were recorded at 10 kHz acquisition rate.
2.8 Temperature-induced modulation of C-fiber compound action potential conduction
Saphenous nerve segments approximately 10 mm in length were used for recording C-fiber compound action potentials (CCAPs) as previously described 17. Each end of a nerve was drawn into a glass pipette and electrically isolated from the central nerve with petroleum jelly to establish high resistance electrical seals. The central nerve was bathed in continuously replenished carbogen-bubble synthetic interstitial fluid. One nerve end was stimulated with a 100 microsecond current pulse every nine seconds, while a recording electrode placed at the other end of the nerve monitored evoked potentials. Recordings were initiated at 27°C with the stimulus strength adjusted sufficient to yield a maximal evoked response. Thereafter, a heating element in the buffer flow path was used to heat the bath up to 45°C at a rate of approximately 0.2 degrees Celsius per second, and then cooled back to 27°C at the same rate. Only CCAPs from the declining temperature ramp were used for analysis.
2.9 Computational modeling of wild-type and Fhf2null nociceptor excitation
A computational model of a nociceptive neuron with realistic afferent terminal branching and TTX-sensitive and -resistant transient voltage-gated sodium conductances constructed on the NEURON simulation platform 4,16 was adapted for our construction of temperature-responsive wild-type and Fhf2null nociceptor models. For all gated ion conductance models within the neuron, we applied a rate scaling factor Q10 of 3^log10[localtemp(°C) - 37], where localtemp is the temperature in a particular segment of the neuron, thereby allowing for localized temperature modulation. The Fhf2null nociceptor model was identical to the wild-type model, except for altering the parameters within the TTX-sensitive and -resistant sodium conductances to induce left shifts in V1/2 inactivation and accelerated inactivation to an extent consistent with experimental findings. These wild-type and Fhf2null nociceptor models will be deposited at the publicly accessible repository ModelDB (https://senselab.med.yale.edu/modeldb/).
To assess axonal conduction of action potentials, a segment of the main axon was stimulated with a 100 microsecond 300 pA current pulse while setting the entire neuron to either 37°C or 43°C. TRP channel activation was simulated as inward current ramps delivered into all termini of the afferent tree. For simulation of mechanosensation, the entire neuron was maintained at 37°C, while for simulation of heat nociception, all segments of the afferent tree within 250 microns of termini were raised to 45°C before injection of current ramps. In order to simulate heat stimulation of an acutely dissociated model neuron, the axon was disconnected from the soma, and a current ramp was injected into the soma set to 45°C.
3. Results
FHF2 Deficiency in Sensory Neurons Causes Loss of Heat Nociception with Lesser Effects on Other Somatosensory Modalities
Fhf2null mice (Fhf2-/Y male and Fhf2-/- female) maintained on the 129S2 background strain are viable and fertile, but are sensitive to cardiac conduction block when core body temperature rises above 41°C 21. When Fhf2-/Y and Fhf2-/- mice were tested for heat nociception by tail flick assay, they were uniformly nonresponsive over 12 second exposure of the tail to the infrared heating beam, whereas wild-type Fhf2+/Y and Fhf2+/+ mice sensed heat and flicked their tails in 3.2 +/- 0.3 seconds (Fig. 1A). In order to determine whether heat nociception requires FHF2 expression in nociceptive neurons, we mated Fhf2flox/+ mice heterozygous for a loxP-flanked (flox) Fhf2 conditional allele (see Methods) with Advillin-Cre transgenic mice that express Cre recombinase exclusively in peripheral neurons 37,38. Male Fhf2flox/Y:AdvCre progeny showed complete absence of FHF2 protein in DRG neurons while retaining normal FHF2 levels in the brain (Fig. 1B). When tested in the tail flick assay, Fhf2flox/Y:AdvCre mice were uniformly nonresponsive, while control animals retaining peripheral FHF2 expression (Fhf2flox/Y and Fhf2+/Y:AdvCre) responded to heat stimulus in 3.0 +/- 0.2 seconds (Fig. 1C).

Somatosensation in mice harboring global or peripheral neuron-specific Fhf2 gene ablation. (A) Tail flick assay of heat nociception in wild-type and Fhf2null mice. Cumulative percentage plot for tail flick as function of tail heating time for wild-type female (Fhf2+/+), wild-type male (Fhf2+/Y), Fhf2null female (Fhf2-/-) and Fhf2null male (Fhf2-/Y) mice. Male and female Fhf2null mice were not responsive to heating. n, number of mice. (B) FHF-2 protein expression in wild-type and mutant mice. Post-nuclear protein extracts from whole brain, cerebral cortex, and DRGs were subjected to SDS polyacrylamide gel electrophoresis and immunoblotting with antibodies directed against the C-terminus of FHF2 shared by all FHF2 isoforms. Lower molecular weight FHF2 (FHF2B) and higher molecular weight FHF2 isoforms were present in wild-type, but not Fhf2null, cerebral cortex (left). Male mice carrying a floxed Fhf2 allele (Fhf2fl/Y) and the advillin-Cre transgene (Adv-Cre) showed complete loss of FHF2B expression in DRG, while retaining FHF2 expression in brain. (C) Tail flick assay in mice with DRG-specific ablation of FHF2 expression.
Cumulative percentage tail flick plot shows that mice with DRG-specific ablation of FHF-2 expression (Fhf2fl/Y:Adv-Cre) were insensitive to heating, while control mice (Fhf2+/Y:Adv-Cre and Fhf2fl/Y) all rapidly responded to tail heating. n, number of mice. (D) Paw pinch assay of mechanical nociception in wild-type and Fhf2null mice. For each animal, the footpad was squeezed with a force-calibrated forceps until a withdrawal response was elicited. n.s., not significant impairment of mechanical nociception. (E) Von Frey filament assay of touch sensation in wild-type and Fhf2null mice. Five male wild-type (Fhf2+/Y) and mutant (Fhf2-/Y) mice were placed without restraint on a mesh platform, a hind paw footpad was touched with nylon filaments of graded thickness/force, and sensation was scored by paw withdrawal. Five trials were conducted for each filament per mouse (25 trials per genotype cohort). Graph plots number of trials (out of 25) that a response was elicited. *, P < 0.004; **, P < 0.0003. (F) Footpad sensory fiber innervation in wild-type and Fhf2null mice. Sensory fibers in footpad cryosections were visualized by immunofluorescence using antibodies to PGP-9. Dashed line indicates outer surface.
Fhf2null and wild-type mice were tested for mechanical nociception by the paw pinch assay, which entails slowly increasing the compression force on the footpad until a response is elicited. Fhf2+/Y mice responded to compression at a force of 199 +/- 25 gm (n = 7), while Fhf2-/Y mice responded at 252 +/- 30 gm (n = 9; P < 0.21)(Fig. 1D). Therefore, Fhf2null mice retain relatively near-normal mechanosensitivity. Fhf2null and wild-type mice were also tested for paw sensitivity to touch using graded diameter Von Frey nylon filaments. Fhf2-/Y mice were moderately but significantly less sensitive to touch compared to Fhf2+/Y littermates (Fig. 1E). In order to qualitatively assess sensory fiber distal innervation, cryosections of Fhf2+/Y and Fhf2-/Y footpads were stained with antibodies for the neural marker PGP9.5. Figure 1F shows similar projection of afferents through the pad and close to the surface in wild-type and mutant animals.
In summary, Fhf2null mice have multiple somatosensory deficits, with heat nociception completely absent. FHF2 is functionally required in heat sensitive, nociceptive neurons, with mutant afferents showing apparently normal projections. Our findings are consistent with those reported previously for nociception when peripheral neurons were rendered Fhf2null on a different background strain 35.
FHF2 Modulation of Inactivation Gating For Sodium Channels Expressed in Nociceptive Neurons
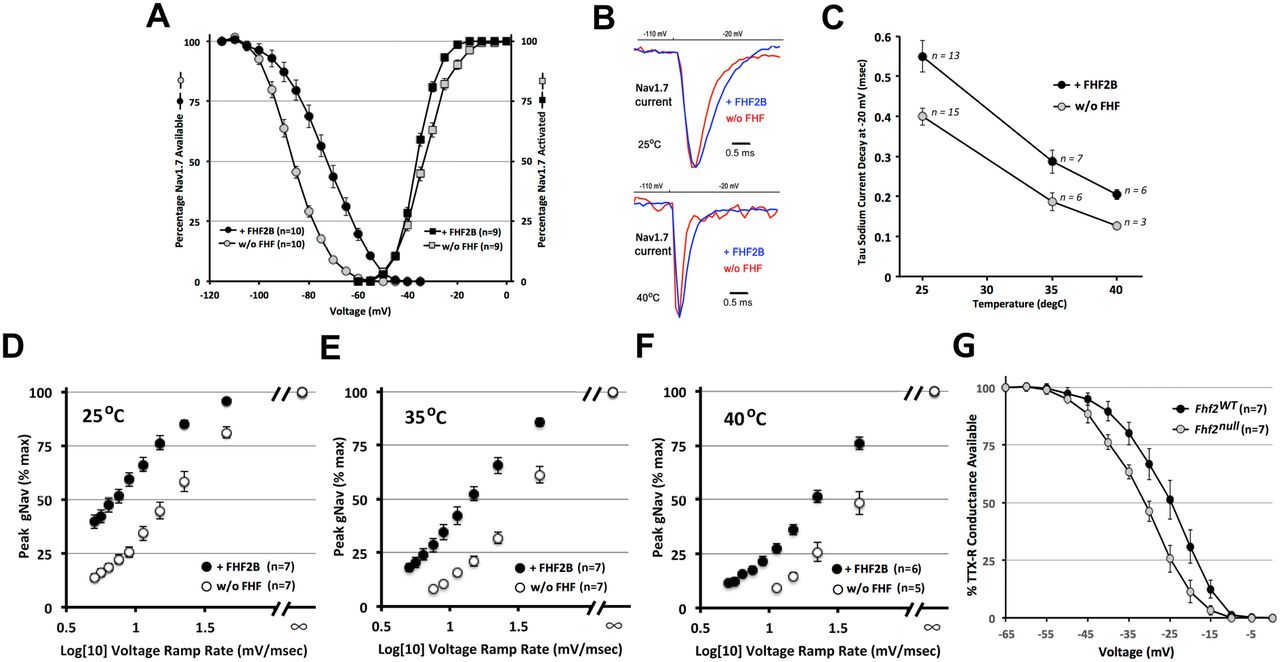
Nav1.7 is the most abundantly expressed sodium channel in nociceptive neurons 5, and the Nav1.7 cytoplasmic tail has been shown to bind FHF2 35. Indeed, the crystal structure of FHF2 in complex with the tail of cardiac Nav1.5 revealed that the channel residues that interface with FHF2 are conserved within all nine mammalian sodium channel alpha subunits 32. In order to assess potential effects of FHF2 on Nav1.7 inactivation gating, HEK293 cells (from QBiogene) were cotransfected with Nav1.7 expression plasmid 7 together with either an FHF2B/GFP bicistronic expression plasmid or GFP-only plasmid 9. Fluorescent cells were selected for patching and voltage clamp recording of sodium currents (see Methods). The short B isoform of FHF2 protein was chosen for our analyses based upon immunoblotting data showing that FHF2B predominates in peripheral neurons, whereas higher molecular weight FHF2 species are more abundant in the brain (Fig. 1B) and heart 21.
The voltage dependence of steady-state Nav1.7 inactivation was V1/2 = −86.8 +/- 1.0 mV in the absence of FHF2B (n=10) and was right-shifted to V1/2 = −72.0 +/- 1.9 mV when FHF2B was present (n=10, P<0.00001) (Fig. 2A). By contrast, FHF2B did not significantly alter the voltage dependence of Nav1.7 activation (Fig. 2A). We then examined Nav1.7 inactivation rate from the open state at 25°C, 35°C and 40°C in the presence vs. absence of FHF2B upon depolarization to −20 mV. At 25°C, Nav1.7 inactivated more slowly in the presence of FHF2B (τ = 0.55 +/- 0.04 msec) vs. its absence (τ = 0.40 +/- 0.02 msec, P<0.006, Fig. 2B,C). Nav1.7 inactivated progressively faster at 35°C and 40°C, but at each temperature, inactivation was significantly slowed by the presence of FHF2B (Fig. 2B,C). Hence, the shortest duration of Nav1.7 current we measured was at 40°C in the absence of FHF2B (τ = 0.13 +/- 0.05 msec, Fig. 2B,C) due to the independent effects of elevated temperature and FHF vacancy.

FHF-2 modulation of sensory neuronal sodium channel inactivation. (A) Voltage dependence of Nav1.7 steady-state inactivation and activation with or without FHF2B at 25°C. The presence of FHF2B induced a 15 mV depolarizing shift in V1/2 inactivation without significantly impacting activation. n, number cells analyzed. (B) Nav1.7 current traces with or without FHF2B at 25°C and 40°C.. (C) Nav1.7 current decay time constants at −20 mV +/- FHF2B at 25°C, 35°C, and 40°C. FHF2B slows channel inactivation at all temperatures.. (D-F) Nav1.7 peak conductance upon ramped vs. step depolarization +/- FHF2 at (D) 25°C, (E) 35°C, and (F) 40°C. Slower voltage ramp rates reduce sodium channel opening via closed-state inactivation, with greater reduction in the absence of FHF2B. (G) Steady state inactivation of TTX-resistant sodium channels in wild-type and Fhf2null DRG neurons. V1/2 inactivation of TTX-resistant currents undergoes a hyperpolarizing shift in the absence of FHF2.
The rate of closed-state Nav1.7 inactivation was assayed at different temperatures in the presence vs. absence of FHF2B by a voltage ramp protocol, as previously described 21. The voltage clamp was raised from −110 mV to −20 mV either instantaneously (step) or linearly over times ranging from 2 to 18 msec (ramps), and the Nav1.7 peak sodium conductance for each voltage ramp was expressed as percentage of the peak conductance that had been achieved by direct voltage step. Under each transfection and temperature condition, Nav1.7 peak conductance diminished as voltage ramp speed was reduced (Fig. 2D-F), reflecting increased closed-state inactivation. At any voltage ramp speed, inactivation was more pronounced by both elevated temperature and the absence of FHF2B (P<0.003)(Fig. 2D-F). More than 50% of channels underwent closed-state inactivation during a rapid 45 mV/msec ramp at 40°C in the absence of FHF2B (Fig. 2F).
In small-diameter nociceptive neurons, Nav1.7 is the principal TTX-sensitive sodium channel, while Nav1.8 is responsible for the transient TTX-resistent sodium current. Transient TTX-resistant sodium currents in acutely dissociated wild-type and Fhf2null DRG neurons (inferred to be Nav1.8 currents) were analyzed by voltage clamp. Steady-state inactivation occurred with V1/2 = −31 mV (n = 7) in wild-type neurons vs. V1/2 = −24.5 mV (n = 7, P<0.025) in Fhf2null neurons (Fig. 2G), demonstrating that FHF2 modulates inactivation gating of the two principal sodium channel isoforms in nociceptive neurons.
FHF2 Is Not Required For Heat-Induced Excitation of Acutely Dissociated DRG Neurons
FHF2 is required in peripheral neurons for heat nociception and FHF2 deficiency facilitates inactivation of the Navs expressed in DRGs. Therefore, we first sought to analyze heat-induced excitation of acutely dissociated Fhf2WT and Fhf2null DRG neurons.
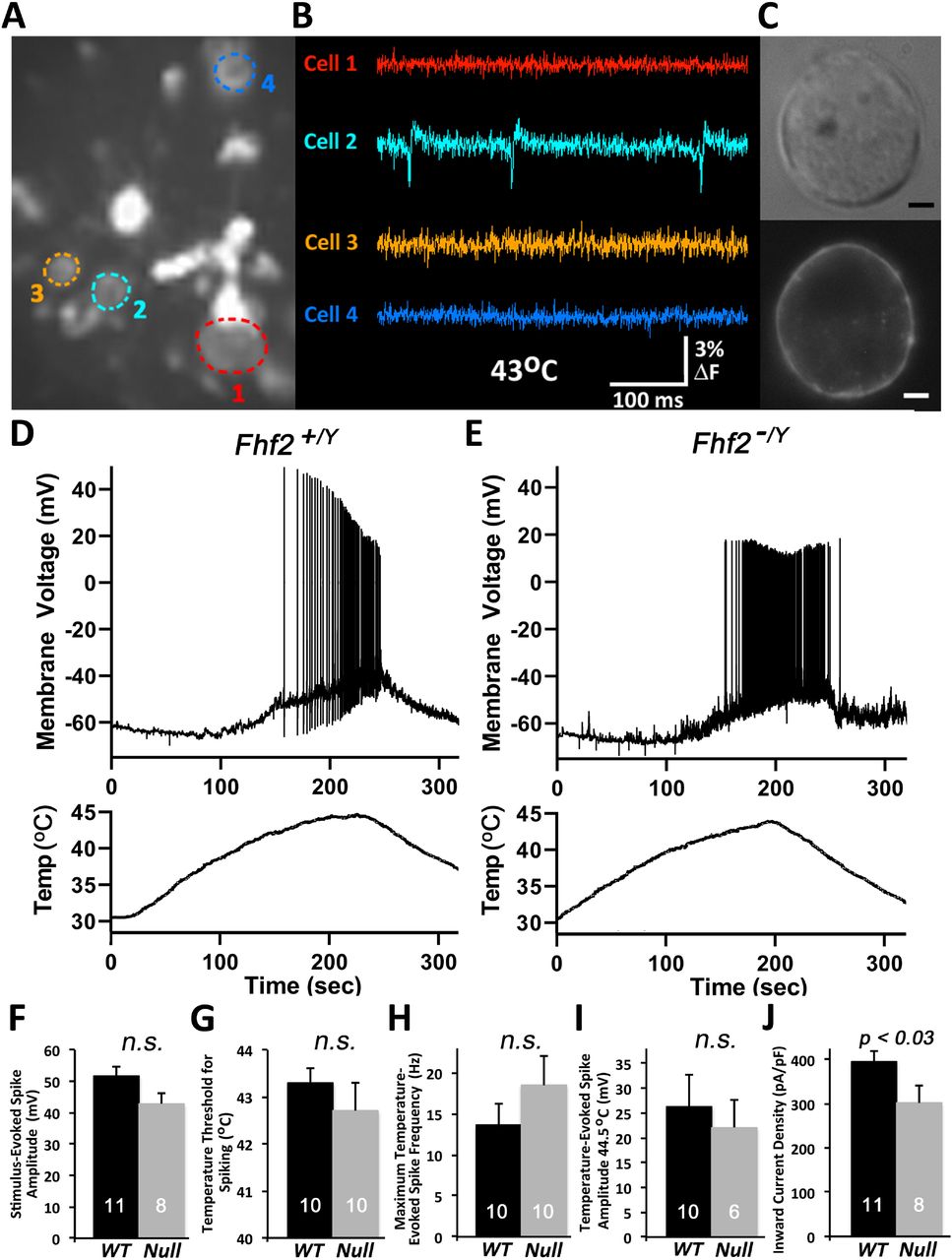
To determine which cell sizes in heterogeneous DRG population to focus on for patch clamp analysis, we first surveyed fields of adult acutely dissociated Fhf2WT neurons for heat-induced fluorescence spikes following extracellular staining with voltage-sensitive dye Di8-ANEPPS. In one visual field containing several stained cells (Fig.3A), one of four cells commenced firing action potentials upon heating to 43°C (Fig. 3B). This cell (Fig. 3C) and other optically identified heat excitable neurons had a diameter of approximately 12 microns.

Excitation by heat of acutely dissociated DRG neurons. (A-C) Optical detection of heat sensitive nociceptor excitation. Acutely dissociated wild-type DRG neurons were cultured overnight and stained with di-8-ANEPPS. A field of cells was imaged with high speed CCD camera while heating to 43°C. (A) A single image shows fluorescence of four neurons (color circled) along with debris including myelin. (B) Fluorescence intensity monitored at 2 kHz shows action potentials generated in cell 2. (C) Bright field and fluorescent high magnification imaging of cell 2. Scale bar, 2 microns. (D) Voltage recording of a patched 12 micron diameter wild-type neuron during heating and cooling. (E) Voltage recording of a patched 12 micron diameter Fhf2null neuron. (F-J) Excitation properties of wild-type and Fhf2null heat sensitive cells. (F) Stimulus-evoked spike amplitude, (G) temperature threshold for spike initiation, (H) maximum temperature-evoked spike frequency, (I) spike amplitude at 44.5°C, and (J) inward current density upon voltage clamp step from −70 mV to 0 mV. Number of recorded cells and standard errors are indicated for each recording parameter. n.s., not significant.
For electrical recordings, Fhf2WT DRG neurons were dissociated, cultured overnight, and ~12 micron diameter cells selected for patching. Patched cells were first analyzed in voltage clamp for inward sodium and outward potassium currents in response to command depolarization, followed by excitability in response to inward current injection. Current-excitable cells were then subjected to temperature ramping from 32°C to 45°C followed by cooling while recording voltage. Figure 3D shows an example of a heat-excitable Fhf2WT neuron which commenced firing at 42°C. Figure 3E shows heat-induced excitation of a Fhf2null neuron. The fraction of Fhf2WT neurons that were excitable by heating (39 out of 96 cells) was statistically indistinghishable from the fraction of heat-excitable Fhf2null neurons (13 out of 31 cells). By a range of criteria, including current stimulus-evoked spike amplitude (Fig 3F), temperature threshold for spike induction (Fig 3G), maximum temperature-evoked spike frequency (Fig 3H), and temperature-evoked spike amplitude (Fig 3I), Fhf2null neurons were as excitable as their wild-type counterparts. The only parameter significantly affected by Fhf2 deletion was a small reduction in the amplitude of peak inward current upon voltage-clamp depolarization from −70 mV to 0 mV (Fig 3J). This likely reflects a hyperpolarizing shift in V1/2 steady state inactivation of Nav1.7, rendering some of the channels inactivated at −70 mV prior to depolarization.
We suspect that acutely dissociated Fhf2null DRG neurons retain heat-induced excitability due to the transient reorganization and expression of nociceptor- and voltage-gated channels after the severing of axonal processes (see Discussion).
Rapid and Reversible Heat-Induced Conduction Failure in Fhf2null C-Fibers
If altered voltage dependence and kinetics of sodium channel inactivation in Fhf2null nociceptors account for loss of heat nociception, it should be possible to detect a rapidly reversible impairment in Fhf2null sensory action potential generation or conduction upon heating. Towards this end, we next tested the ability of unmyelinated sensory C-type fibers to conduct action potentials at different temperatures. Electrical stimulation of Fhf2WT and Fhf2null saphenous nerves ex vivo 17 generated C-fiber compound action potentials (CCAPs) (Fig. 4A,B). Each nerve was then stimulated every nine seconds while being warmed to 45°C and cooled back to 27°C, and CCAPs from the cooling phase of the temperature ramp were analyzed. Elevated temperature increased CCAP conduction velocity and decreased amplitude (Fig. 4C-F), reflecting faster rates of voltage-gated channel activation and inactivation. In Fhf2WT nerves, CCAP amplitudes declined linearly from 27°C to 44°C (Fig.4C,E,F). CCAP amplitudes in Fhf2null nerves showed a similar decrement up to 40°C, but declined precipitously above 40°C (Fig. 4D-F), reflecting conduction block or severe attenuation in at least half of all mutant fibers. Figure panels 4D,E illustrate that Fhf2null CCAP impairment was reversed within 27 seconds (3 stimuli) as nerves were cooled from 44°C to 40°C.

Temperature modulation of C-fiber compound action action potential conduction. (A) Stimulus response of wild-type saphenous nerve. The nerve maintained at 30-31°C was stimulated at one end at time = 0 msec. The recording electrode detected an instantaneous stimulus artifact, fast arriving voltage deflections (1-3 msec) reflecting activation and conduction of myelinated axons, and slow C-fiber compound action potential (CCAP) from 10-20 msec post-stimulus. (B) Stimulus response of Fhf2null saphenous nerve. A similar CCAP was detected at 30-31°C. (C) Superimposed CCAP traces of a wild-type nerve stimulated every 9 sec during cooling from 44.3°C down to ~30°C. CCAPs displayed a near-linear increase in amplitude and decrease in velocity across the temperature range. (D) Superimposed CCAP traces of an Fhf2null nerve stimulated every 9 sec during cooling from 44.3°C down to ~30°C. The amplitude of the CCAP negative deflection fell precipitously when nerve temperature exceeded 41°C. (E) Plot of CCAP amplitudes versus temperature. For each of four wild-type and four Fhf2null nerves, the positive-to-negative peak-to-peak CCAP amplitudes at each temperature were plotted as a percentage of the amplitude recorded at 27°C. Wild-type nerves all showed near-linear amplitude decrement from minimal to maximal temperature, while Fhf2null nerves displayed enhanced decrements above 41°C. (F) Histogram representation of CCAP amplitude decrements with increasing temperature. For each of seven wild-type and six Fhf2null nerve recordings, CCAP amplitude ratios for 32°C vs. 28°C, 36°C vs. 32°C, 40°C vs. 36°C, and 44°C vs. 40°C. The decrement in CCAP amplitude is significantly greater for Fhf2null nerves versus wild-type nerves for 44°C vs. 40°C.
The rapid reversibility of conduction deficit upon nerve cooling is strong evidence that sodium channels in Fhf2null axons at high temperature are not denatured or internalized, but fail to generate sufficient inward current for impulse propagation due to accelerated inactivation. These findings parallel prior demonstration that hyperthermia-induced cardiac conduction block in Fhf2null mice is ameliorated upon cooling 21.
Heat Excitation Deficit in a Computational Model of Fhf2null Nociceptor
In order to assess the requirement of FHF2 in mature heat sensitive nociceptors, we employed computational modeling and simulations. For these studies, the wild-type (Fhf2WT) model was taken from the nociceptor model cell described by Barkai et al 4. Their model has a highly branched terminal axon with uniformly distributed leak and voltage-gated channels except for the distal afferent terminals, which lack voltage-gated sodium channels but can generate inward currents mimicking the opening of TRP receptor channels 4. The compartmental segregation of excitatory currents from voltage-gated channels was based upon recordings showing that action potentials initiate at recessed sites on nociceptive afferents 15. We simulated FHF2 loss-of-function by modifying the gating of the embedded TTX-sensitive (Nav1.7) and TTX-resistant (Nav1.8) conductances to reflect shifts in inactivation rates and voltage dependence commensurate with our recording data (Figure 2) without altering conductance densities. All channel gating transitions in both the Fhf2WT and Fhf2null nociceptor models, including Nav1.7 and Nav1.8, were accelerated with increasing temperature by employing Q10 rate scaling factors.
We first tested whether the Fhf2null model axon suffers conduction block at elevated temperature, analogous to ex vivo conduction block (Figure 4). Wild-type and Fhf2null model axons were point-stimulated by current pulse and action potential conduction was monitored over a distance of 5 millimeters. The wild-type model axon conducted an action potential at either 37°C (Fig. 5A) or 43°C (Fig. 5B). In contrast, the Fhf2null model axon conducted at 37°C (Fig. 5C), but suffered conduction block at 43°C (Fig. 5D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Computational modeling of wild-type and Fhf2null nociceptors. Wild-type and Fhf2null nociceptor models differ only in the inactivation gating parameters of the TTX-sensitive and TTX-resistant voltage-gated sodium conductances. All gated conductances have temperature-dependent state transition rates. (A-D) Simulation of axonal conduction. The long axon of each model was stimulated at one point with a 100 μsec current pulse, and membrane voltage was monitored at 0, 0.2, 0.4, 1.1, and 5.0 mm from the site of stimulation. (A) Wild-type model at 37°C. (B) Wild-type model at 43°C. (C) Fhf2null model at 37°C. (D) Fhf2null model at 43°C; note conduction failure beyond 0.4 mm from point of stimulation. (E-G) Simulation of mechanical nociception. (E) Branching morphology of the afferent terminal. The terminal architecture, as published elsewhere 4, was incorporated into wild-type and Fhf2null models. Temperature throughout nociceptor was set to 37°C. Arrows at all terminal branch tips indicate sites of up-ramping current injection delivered from 0.5 to 2.5 seconds of simulation followed by down-ramping from 2.5 to 3.5 seconds. This protocol simulated opening of TRP channels at branch tips due to mechanical stimulation. (F) Central axonal voltage recording of wild-type model neuron during simulation in (E). The current ramp evoked a train of action potentials that initiated peripherally and propagated antidromically. (G) Voltage recording of Fhf2null model neuron during simulation in (E). A train of action potentials was generated, though fewer in number than in the wild-type model. (H-J) Simulation of heat excitability. (H) Terminal branching architecture. Branch segments within 200 microns of tips were set to 43°C (red), while remainder of neuron was maintained at 37°C. Current ramp injection into branch tips simulated heat stimulation. (I) Central axonal voltage recording of wild-type neuron during simulation in (H). Stimulation evoked a train of antidromic action potentials. (J) Voltage recording of Fhf2null neuron during simulation in (H). The mutant neuron was not excited by stimulation, simulating the heat nociception deficit observed in vivo. (K-M) Simulation of heat-induced excitation in “acutely dissociated” model soma. (K) Model soma were disconnected from axons, somatic temperature was set to 45°C (red), and up-ramped somatic inward current simulated heat-induced excitation of the isolated soma. (L) Wild-type and (M) Fhf2null isolated soma fired trains of action potentials.
Simulations were next run by current injection into afferent terminals of nociceptor models at 37°C, which recapitulates the effects of noxious chemical or mechanical stimulation (Fig. 5E). The Fhf2WT model fired a train of action potentials in response to such stimulation (Fig. 5F). The Fhf2null nociceptor model also generated action potentials, albeit fewer in number, during current injection (Fig. 5G), consistent with preservation of mechanosensation (Fig. 1C) and chemosensation 35 in Fhf2null mice. Heat nociception was simulated by injecting current into afferent terminals while the temperature of distal axon segments were elevated to 43°C while leaving the rest of the model neuron at 37°C (Fig. 5H). Under these conditions, the Fhf2WT nociceptor model still generated a stream of action potentials (Fig. 5I), while the Fhf2null model failed to be excited (Fig. 5J).
In an attempt to explain why acutely dissociated Fhf2null nociceptor neurons display relatively normal heat-induced excitation, we simulated acute dissociation of nociceptive neurons by severing the entire axons in the nociceptor models and performing simulated somatic current injection at 45°C (Fig. 5K). Under these conditions, both wild-type and Fhf2null models generated a train of action potentials during current injection (Fig. 5L,M). These findings emphasize the importance of FHF2 to preserve sodium channel availability at elevated temperature during passive propagation of depolarization from nociceptor axon terminal to trigger zone and during conduction down the axon.
4. Discussion
We have shown here that FHF2 is a powerful inactivation gating modulator for the principle TTX-sensitive and TTX-resistant sodium channel isoforms expressed in nociceptive neurons. FHF2 induces depolarizing shifts in V1/2 inactivation for Nav1.7 and Nav1.8, and slows the rate of Nav1.7 inactivation from closed and open states. Elevating temperature acts as an independent accelerant of inactivation, speeding closed and open state Nav1.7 inactivation regardless of FHF2 association. Hence, the transient sodium flux upon membrane depolarization is most compromised at elevated temperature in the absence of FHF2. These gating principles offer a plausible explanation for the specificity of heat nociception deficit in Fhf2null mice, despite the broad expression of FHF2 in all classes of sensory neurons. These principles also provide a rationale for preservation of mechanosensation in Fhf2null mice, which is mediated, at least partially, by the same nociceptors that respond to noxious temperatures 30.
Loss of heat nociception was observed following either global or peripheral neuron-restricted genetic ablation of the Fhf2 gene, demonstrating the requirement of FHF2 function in heat sensitive nociceptors. We were initially surprised to find that heat-induced excitability of acutely dissociated Fhf2null DRG neurons was unimpaired. This paradox is likely explained by the fact that acutely dissociated neurons lack axonal processes and are electrotonically compact; with heat-inducible TRP channels and voltage-gated channels comingled on the somatic plasma membrane, inward TRP channel currents directly depolarize the somatic membrane bearing voltage-gated sodium channels. By contrast, depolarization of heated nociceptor afferent terminals produces retrograde axial current that must passively depolarize a peri-terminal trigger zone 15 subject to cable property delay, reducing Nav1.7 availability in the absence of FHF. Furthermore, reduced sodium current duration in the Fhf2null afferent may provide insufficient charge to ensure spike conduction, consistent with our observed failure of unmyelinated Fhf2null sensory axons to sustain C-fiber compound action potentials upon heating. Our computational modeling further supports this hypothesis. While current influx at afferent terminals of the Fhf2null model neuron failed to evoke action potentials at 45°C, simulated injection of the same current density into the heated axon-severed soma was able of to trigger excitation. It is also possible that axotomy accompanying preparation of acutely dissociated DRG neurons triggers upregulation of channel expression to provide sufficient sodium current in mutant cells at elevated temperature.
While peripheral sensory axons express FHF2, myelinated and unmyelinated axons in the central nervous system show little or no FHF expression 9. This natural deficiency may explain why spike conduction in brain axons is blocked under hyperthermal conditions 23.
We employed plasmid-transfected HEK293 cells to assay the effects of FHF2B expression on the inactivation of Nav1.7. The voltage dependence of Nav1.7 steady-state inactivation was markedly right-shifted by 13 mV from V1/2 of −86 mV in the absence of FHF2B to −73 mV in its presence. As discussed above, strong modulation of Nav inactivation by FHF2 is central to the heat-associated nociception and cardiac deficits in Fhf2null mice. Our findings are in sharp contrast to Yang et al. (2019), who reported only a minor shift in Nav1.7 V1/2 inactivation from −76 mV in HEK cells without FHF cotransfection to −73 mV in presence of FHF2B. We suspect these disparate findings reflect the expression of endogenous FHF2 protein in some sublineages of HEK293 cells that result in a significant depolarizing shift in Nav1.7 V1/2 inactivation in the absence of FHF transfection. Indeed, we have identified a different sublineage of HEK293 cells with verified endogenous FHF2 expression that yields Nav1.7 inactivation data comparable to those by Yang et al. (A. Lesage, M. Sarlandie, C. Marra, C. Marrionneau, M. Goldfarb, unpublished data). It is possible that the failure by Yang and colleagues to detect a strong inactivation modulatory effect of FHF2 led them to emphasize other mechanistic bases for the Fhf2null heat nociception deficit 35.
In the central nervous system, FHF2 is expressed in both excitatory and inhibitory neurons 24. Fhf2+/- female mice show enhanced susceptibility to epileptic seizures, with most juvenile mice undergoing seizures when core body temperature was elevated above 42°C 24. As FHF2 expression is subject to X-linked chromosome inactivation, 50% of all Fhf2+/- neurons are functionally Fhf2null. We suggest that core hyperthermia combined with FHF2 deficiency triggers seizures by impairing firing of some inhibitory neurons by the same Nav inactivation gating principles that dictate heat and FHF2 modulation of cardiac and sensory sodium channels.
5. Conclusions
FHF2 expression in sensory neurons is absolutely essential for heat nociception. FHF2 modulation of Nav1.7 and Nav1.8 inactivation gating allows for sufficient inward sodium currents in response to temperature elevation and opening of TRP channels to enable axonal initiation and propagation of antidromic action potentials. This conclusion is supported by voltage clamp analysis of sodium currents, by C-fiber action potential conduction, and by initiation and conduction of action potentials in a nociceptive neuron computational model as a function of temperature and FHF2 expression status. The heat nociception deficit is mechanistically equivalent to cardiac conduction deficit in Fhf2null mice exposed to core hyperthermia.
Conflicts of Interest
The authors have no conflicts of interest regarding the research described in this paper.
Acknowledgments
We wish to thank Sergio Solinas (University of Sassari, Sardinia) for expert assistance in implementing modifications to the nociceptor neuron computational model of Barkai et al 4. We thank Glenn I. Fishman (NYU Langone Medical Center) for his encouragement and support. This work was supported by R01HL142498 to G.I.F., a CUNY research grant to M.G., and the Blaustein Pain Research Foundation and the Neurosurgery Pain Research Institute at Johns Hopkins (to M.R.).
References




