

SUMMARY
Hemogenic endothelial cells (HECs) are specialized cells that undergo endothelial to hematopoietic transition (EHT) to give rise to hematopoietic progenitors. Though not defined as a hematopoietic organ, the lung houses many resident hematopoietic cells, aids in platelet biogenesis, and is a reservoir for hematopoietic stem and progenitor cells (HSPCs), but lung HECs have never been described. Using explant cultures of murine and human fetal lungs, we demonstrate that the fetal lung is a source of HECs that have the functional capacity to undergo EHT to produce de-novo HSPCs. Flow cytometric and functional assessment of fetal lung explants showed the production of HSPCs that expressed key EHT and pre-HSPC markers. scRNA-Seq and small molecule modulation demonstrated that fetal lung EHT is reliant on canonical EHT signaling pathways. These findings suggest that functional HECs are present in the fetal lung, thus establishing this location as a potential extramedullary site of de-novo hematopoiesis.
INTRODUCTION
Hematopoietic stem cells (HSCs) originate from a rare sub-population of arterial endothelial cells known as HECs. Making up just 1-3% of the total endothelial cell population in the AGM, HECs are commonly thought to be confined to a small window of gestation between embryonic days 8 to 11 (E8-11) in mice, and E27-40 in humans1,2. Within this time window, HECs can also be found in other hematopoietic organs and vessels including the yolk sac, placenta, and vitelline and umbilical arteries3–5,6(p1),7,8. Some studies suggest that HECs may not be restricted to these hematopoietic organs and developmental window. Work from others suggests that functional HECs may also be found in the embryonic head around E10-E11 as well as the perinatal chicken and murine bone marrow (BM)9,10. Collectively, these studies demonstrate that our spatiotemporal understanding of HECs remains limited and raises questions about the hematopoietic potential of other organs.
Recent studies have highlighted the presence and importance of various resident blood cell populations in the lung11. In particular, the lung was demonstrated to be a reservoir of HSPCs, but the origins of this population of progenitors remains unknown12–14. In-utero mechanical stimuli have been shown to be an important environmental cue for both HEC and pulmonary development. More specifically, cyclic stretch induced activation of Yes Activated Protein (YAP) signaling in the AGM promotes EHT15. Interestingly, rhythmic breathing movements begin occurring around E16 in fetal mice, and mechanical stimuli has a significant impact on fetal airway and alveolar epithelial development16–18. Beyond mechanical signaling, cell-cell extrinsic signaling within the AGM niche is critical for EHT. The ventral wall of the dorsal aorta is the most common site of EHT due to its proximity to the underlying mesenchyme which modulates EHT via Notch, BMP4, SHH, and Wnt pathways3,19,20. All of these pathways are similarly very important in pulmonary fetal development21.
Based on these parallels, we hypothesized that the fetal lung is another potential site of hemogenic endothelium with the functional capacity to produce hematopoietic progenitors. Employing fetal lung explant cultures, we demonstrate that the murine and human fetal lung are sources of putative HECs. Multipotent, fetal lung-derived HSPCs showed the canonical features of cells produced by EHT as determined by flow cytometry, single cell transcriptomics, functional assays and immunofluorescent histology. These findings highlight the fetal lung as another potential site of de-novo hematopoiesis, suggesting that the lung may have a greater role in instructing tissue specific hematopoiesis and/or overall hematopoietic development.
MATERIALS AND METHODS
Ethics Statement
All animal housing and experimental procedures were approved by the Boston University School of Medicine Institutional Animal Care and Use Committee (BUSM IACUC). Work involving human tissue samples was approved by Partners Human Research Committee (Protocol #2016P001106).
Tissue isolation and processing
Mouse
E17 timed-pregnant C57/BL6 mice were purchased from Jackson Laboratories. The fetal liver and lung were isolated from surrounding tissue by blunt dissection and set aside in a solution of 10% characterized fetal bovine serum in Hanks’ balanced salt solution (HBSS, Gibco). Using a 5mL syringe fitted with a 16-gauge needle, the fetal liver and fetal lung were drawn up and expelled several times to physically dissociate the tissue. Samples were subsequently placed in a digest buffer containing HBSS, 1mg/mL DNase I (Sigma), and 0.5mg/mL LiberaseTM (Sigma). This digest mixture was placed on a rocker at 37°C for 30-minutes to 1-hour. Post-digestion, lung samples were filtered and resuspended in RBC lysis for 5 min at 37°C, then washed, filtered, and resuspended in HSPC medium.
Human
All fetal samples were within the age range between 19-24 weeks post-conception. Lung lobes were dissected away from the main airways and minced with a scalpel before being placed in a digest buffer containing HBSS, 1mg/mL DNase I (Sigma), and 0.5mg/mL LiberaseTM (Sigma). This digest mixture was placed on a rocker at 37°C for 30-minutes to 1-hour. Post digestion, lung samples were filtered and resuspended in RBC lysis for 5 min at 37°C, then washed, filtered, and resuspended in HSPC medium.
Explant Cultures
Isolated cells suspended in HSPC medium were plated onto Matrigel coated plates. HSPC medium was made up of StemPro-34 Serum Free Medium, 50 µg/ml ascorbic acid, 400 nM monothioglycerol, 100µg/ml Primocin, 2 mM L-glutamine, and the following human or murine growth factors: 50 ng/ml vascular endothelial growth factor A (VEGFA), 100 ng/ml basic fibroblast growth factor (bFGF), 100 ng/ml stem cell factor (SCF), 100 ng/ml FMS-related tyrosine kinase ligand (FLT3L), 100 ng/ml thrombopoietin (TPO), 100 ng/ml Interleukin-6 (IL6). On the third day of all cultures, media was aspirated, rinsed once with PBS, and fresh media was applied.
MethoCult Colony-Forming Unit (CFU) Assay
CFU assay was performed using the murine MethoCult GF M3434 (Stem Cell Techologies) or human MethoCult H4034 Optimum (Stem Cell Techologies) kit. Procedure was performed per manufacturer’s instructions.
Tissue Histology and Cell Imaging
A small portion of the human fetal lung was fixed in 4% paraformaldehyde for 2 hours, cryoprotected in 30% sucrose and embedded in Optimum Cutting Temperature embedding medium. 10-12µm tissue sections were made using a cryostat. Sections were rinsed with PBS prior to permeabilizing and blocking in a solution of 0.4% Triton X-100 and 10% NDS. A list of antibodies used for immunofluorescence can be found under Supplemental Table 1.
Imaging of cell cultures were performed using a Keyence BZ-X700 fluorescence microscope. Cytospins were stained using the Hema 3 Stat Pack (Fisher) and imaged using a Nikon Eclipse NiE.
Flow cytometry
All staining and washing steps were done using a solution of 1% BSA in PBS, 5mM EDTA. Single cell suspensions were stained with the appropriate master mix of antibodies on ice for 30 minutes. A table of antibodies used for analysis can be found under Supplemental Table 1. Fc-Receptor block treatment was performed using an anti-mouse CD16/32 antibody (BioLegend, Clone: 93) or anti-human Fc receptor block (biolegend, Human TruStain FcX) for 10 minutes prior to the addition of master mix. True-Stain Monocyte Blocker (BioLegend) and Brilliant Stain Buffer Plus (BD Biosciences) were included in master mix to minimize non-specific staining. Single stain controls were processed using cells and/or beads (Invitrogen, UltraComp eBeads Plus). Flow cytometry was performed using either a BD LSRII, a Stratedigm S1000EXI or a 5-laser Cytek Aurora spectral flow cytometer. Analysis was performed using FlowJo_v10.8 (FlowJo, LLC) software.
Single cell RNA sequencing
Adherent cells from days 4, 5 and 6 of explant cultures were released from Matrigel coated wells using Accutase. Collected cells were stained for VE-Cadherin-APC-Cy7 and resuspended in sort buffer (2% BSA in PBS) containing Calcein Blue AM (1:1000). Using a Beckman MoFlo Coulter Astrios, live-singlets and VECAD+ events were sorted into sort buffer (2% BSA in PBS with 5mM EDTA). Cells counts and viability after sorting were confirmed by hemocytometer using trypan blue. Suspension cells were left unsorted as VECAD expression is lost gradually as hematopoietic cells differentiate post-EHT emergence. VECAD+ sorted adherent cells and unsorted suspension cells were single cell captured using the 10X Genomics Chromium platform and prepared using the Single Cell 3’ v3 kit. Library preparation and sequencing was done at the Boston University Microarray and Sequencing Resource (BUMSR) Core using the Illumina NextSeq 2000 instrument.
Bioinformatic analysis
Reads were demultiplexed and aligned to the mouse genome assembly (GRCm38, Ensembl) with the STARsolo pipeline22. Further analyses were done using Seurat v. 3.1.423. After inspection of the quality control metrics, cells with more than 12% of mitochondrial content or less than 800 detected genes were excluded for downstream analyses. We normalized and scaled the UMI counts using the regularized negative binomial regression (SCTransform)24. Following the standard procedure in Seurat’s pipeline, we performed linear dimensionality reduction (PCA), and used the top 20 principal components to compute both the UMAP25 and the clusters (Louvain method26) which were computed at a range of resolutions from 1.5 to 0.05 (more to fewer clusters). Cell cycle scores and classifications were done using the Seurat method27. The cut-offs for independent filtering28 prior to differential expression testing required genes: a) being detected in at least 10% of the cells of either population and b) having a natural log fold change of at least 0.25 between populations. The tests were performed using Seurat’s wrapper for the MAST framework29. For a comparison on the performance of methods for single-cell differential expression see Soneson & Robinson et al.30.
Statistical analysis
Significance for Enrichr based pathway enrichment was determined by Fisher exact test31,32. Significance for pairwise scRNA-Seq gene expression comparisons was determined using the MAST framework29. Significance for flow cytometry assays performed in biological triplicate was determined by student paired t-test.
RESULTS
Murine fetal lung explant cultures produce HSPCs
To assess the functional capacity of potential lung HECs to produce blood progenitors, cells isolated from murine E17 lungs were plated onto Matrigel coated plates in an adapted hematopoietic differentiation media, termed HSPC medium33. This serum-free media was formulated to support the final stages of specification of HECs into HSPCs. The primary rationale for choosing E17 was that (1) this is during the window that fetal breathing movements are occurring16, and (2) E17 is a timepoint that is distant from the AGM EHT window (E8-11)1, thus helping to minimize contamination from AGM-EHT derived progenitors.
In contrast to fetal liver explants where immediate expansion of floating cells is observed due to an expanding hematopoietic progenitor population, fetal lung explants initially developed a robust adherent layer. Discrete clusters of suspension cells are observed by day 3, which expand to robust colonies between days 4-6 (Figure 1A). To visually determine cell identity based on their morphology, cytospins of day 6 suspension cells were performed, which revealed cells with a progenitor-like morphology as well as various differentiated hematopoietic cells including macrophages/monocytes, neutrophils and megakaryocytes (Figure 1B). This diversity of hematopoietic cells suggests that hematopoietic progenitors are arising from these cultures. To examine this, suspension cells were functionally assessed for progenitor potential by MethoCult assay. Day 4-6 suspension cells showed similar potential to form all types of colony forming units (CFUs), but this potential was no longer detected at Day 8 (Figure 1C, D). Progenitor phenotyping was further assessed by flow cytometric assessment of the broad murine HSPC markers Lin-/Sca+/Kit+ (LSK) and the SLAM markers CD48 and CD150. A greater fraction of CD45+ hematopoietic cells isolated from the adherent cell layer were LSK-HSPCs and CD48-/CD150+ SLAM marker defined HSCs (Figure 1E). Collectively, these data suggest that the adherent layer of cells from fetal lung explants gave rise to HSPCs.

(A) Images of a single position between days 3 to 6 of a murine fetal lung explant culture. (B) Cytospin of fetal lung derived suspension cells. Images are annotated for cell type: MK = Megakaryocyte, N = Neutrophil, M = Monocyte/Macrophage, P = Progenitor. (C) Images of representative colonies from a CFU assay. (D) Colony counts from CFU assays performed with day 4, 5, 6 and 8 suspension cells. (E) Representative plots of flow cytometric assessment of LSK and SLAM marker defined HSPC populations. Suspension represents cells that were floating in media, and Adherent represents cells that were collected post-treatment with Accutase. Error bars represent standard deviation.
Murine fetal lung explants exhibit the dynamics of EHT
Time-lapse capture of live explant cultures from days 5-6 showed adherent cells transitioning to suspension cells (Supplemental_Video). These observations closely mimic AGM explant cultures and in-vitro based hematopoietic differentiations showing the transition of HECs into hematopoietic cells34–37. The resultant pre-HSPCs that are birthed from AGM derived HECs retain some endothelial markers and are marked by co-expression of the endothelial marker vascular endothelial cadherin (VECAD) and the broad hematopoietic marker CD4538. Fetal Lung explants gave rise to VECAD+/CD45+ cells, and a gradual reduction in VECAD expression coincided with the expression of the more differentiated marker CD45 (Figure 2A). The fraction of CD45+ cells that were VECAD+ also decreased with successive days of culture (Figure 2B). Time matched fetal livers, which house HSPCs but not HECs39, were cultured under the same conditions and did not result in the production of a population of VECAD+/CD45+ cells (Supplemental Figure 1A). This suggests that fetal lung derived VECAD+/CD45+ pre-HSPCs are not resultant from the expansion of pre-existing progenitors. SLAM marker defined HSCs were predominantly present amongst the VECAD+ suspension cells (Figure 2C). With time, a gradual reduction in the fraction of VECAD+ HSCs was observed, which coincided with the expansion of the more lineage restricted hematopoietic progenitor cell 1 (HPC-1) and HPC-2 populations.

(A) Flow cytometric assessment of VECAD and CD45 expression on day 5 fetal lung explants. (B) CD45 and VECAD expression across days 4 to 6. VECAD expression is gradually lost as endothelial cells complete their transition to CD45+ hematopoietic cells, and this process diminishes across days 4-6 of culture. (C) Assessment of SLAM marker defined HSPC populations stratified by VECAD expression across multiple days in culture. (D) Assessment of pre-HSC and EHT markers on VECAD+/CD45+ defined progenitors and separated by adherent versus suspension cell populations.
Beyond VECAD, successive staging from a pro-HSC to a pre-HSC phenotype is marked by the expression of CD41 and CD43, respectively40. EPCR and CD44 have also been previously demonstrated as markers of pre-HSPCs40–42. Fetal lung derived VECAD+/CD45+ cells showed expression of CD41, CD43, CD44, and EPCR (Figure 2D). There was a notable reduction in CD41, CD43 and EPCR expression as these cells transitioned into a suspension state. Intensity of EPCR expression also decreased as VECAD+/CD45- cells matured away from an endothelial signature (Supplemental Figure 1B). In contrast, CD41 expression transiently peaked at the VECAD+/CD45+ pre-HSPC stage. Under these experimental conditions, however, no notable changes in CD44 expression were observed regardless of physical and maturational state. We also observed significantly less expression of pre-HSC markers on fetal liver VECAD+/CD45+ cells suggesting that in contrast to those from the fetal lung, these cells are likely not pre-HSPCs (Supplemental Figure 1A).
Human fetal lung explants undergo EHT to produce HSPCs
In an effort to understand if this phenomenon is conserved in humans, cells collected from human fetal lungs isolated from embryos aged between 19-24 weeks post-conception were cultured under the same conditions described above. Similar to murine fetal lung explants, human fetal lung explants initially developed an adherent layer and gave rise to suspension cells between days 7-10 (Figure 3A). Cytospins and flow cytometry of suspension cells showed progenitor-like cells and various differentiated cell types, including megakaryocytes, red blood cells, monocytes/macrophages and neutrophils (Figure 3B, C). Suspension cells assessed by Methocult assay also showed CFU capacity (Figure 3D). These data collectively suggest that human fetal lung explants are giving rise to HSPCs capable of multilineage differentiation.
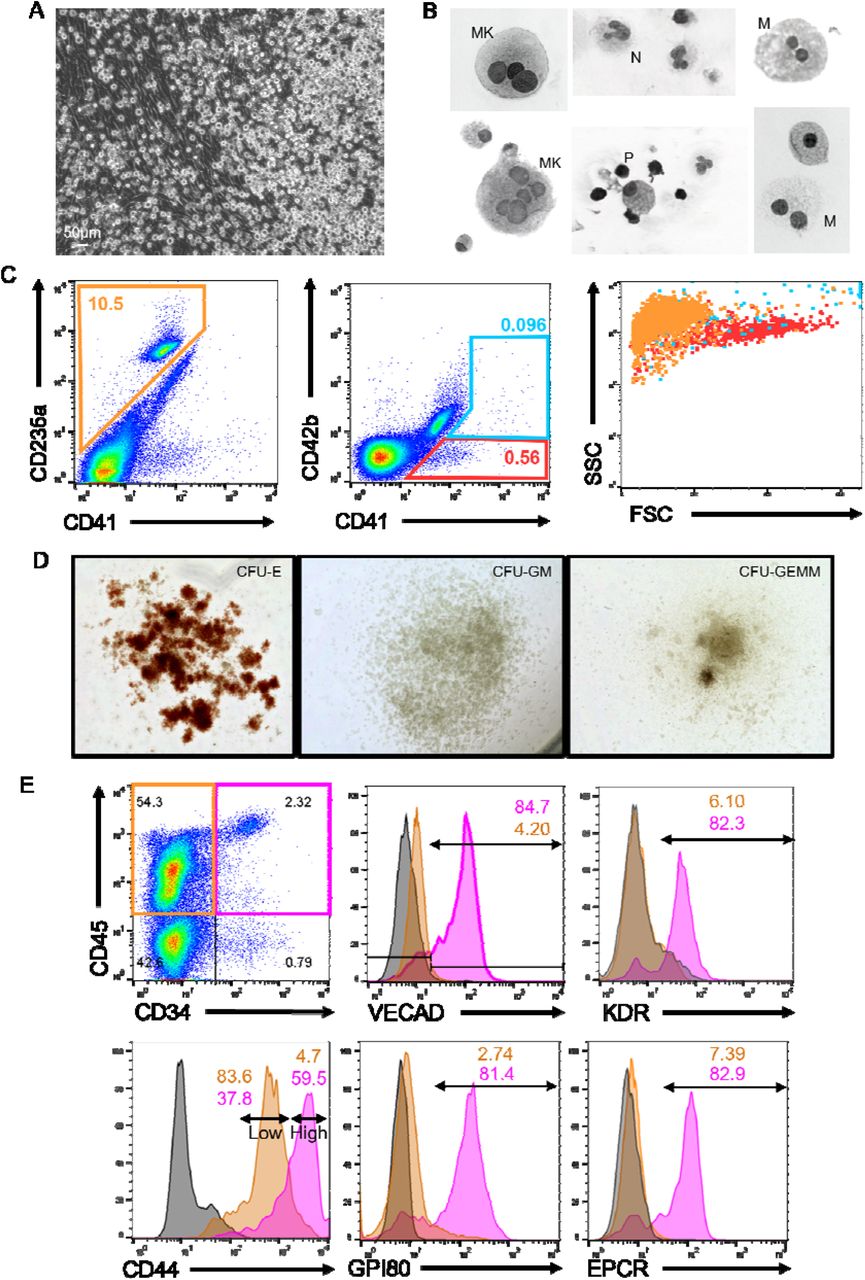
(A) Image of a human fetal lung explant culture showing a robust population of floating hematopoietic cells against a background of adherent cells. (B) Cytospin of human fetal lung suspension cells. (C) Flow cytometric assessment of differentiated erythrocyte and megakaryocyte populations. (D) Images of representative colonies from a CFU assay. (E) Assessment of pre-HSC and EHT markers on CD34+/CD45+ defined progenitors versus CD34-/CD45+ differentiated hematopoietic cells.
To assess whether these suspension cells were derived via EHT, flow cytometric assessment of endothelial and HSPC markers was performed on human fetal lung explants (Figure 3E). Human HSPCs were broadly defined by co-expression of the progenitor marker CD34 and the hematopoietic marker CD45. The majority of fetal lung derived CD34+/CD45+ progenitors co-expressed VECAD, which was downregulated as these cells differentiated and lost expression of CD34. Additionally, fetal lung derived HSPCs expressed the HSPC markers KDR, GPI80, CD44 and EPCR. Reduction in the expression of these pre-HSPC/HSPC markers coincided with the loss of CD34 expression. Notably, human fetal lung derived HSPCs were enriched with a distinct CD44-high population, which others have reported is a marker of type II pre-HSPCs found within the intra-aortic hematopoietic clusters of the AGM41.
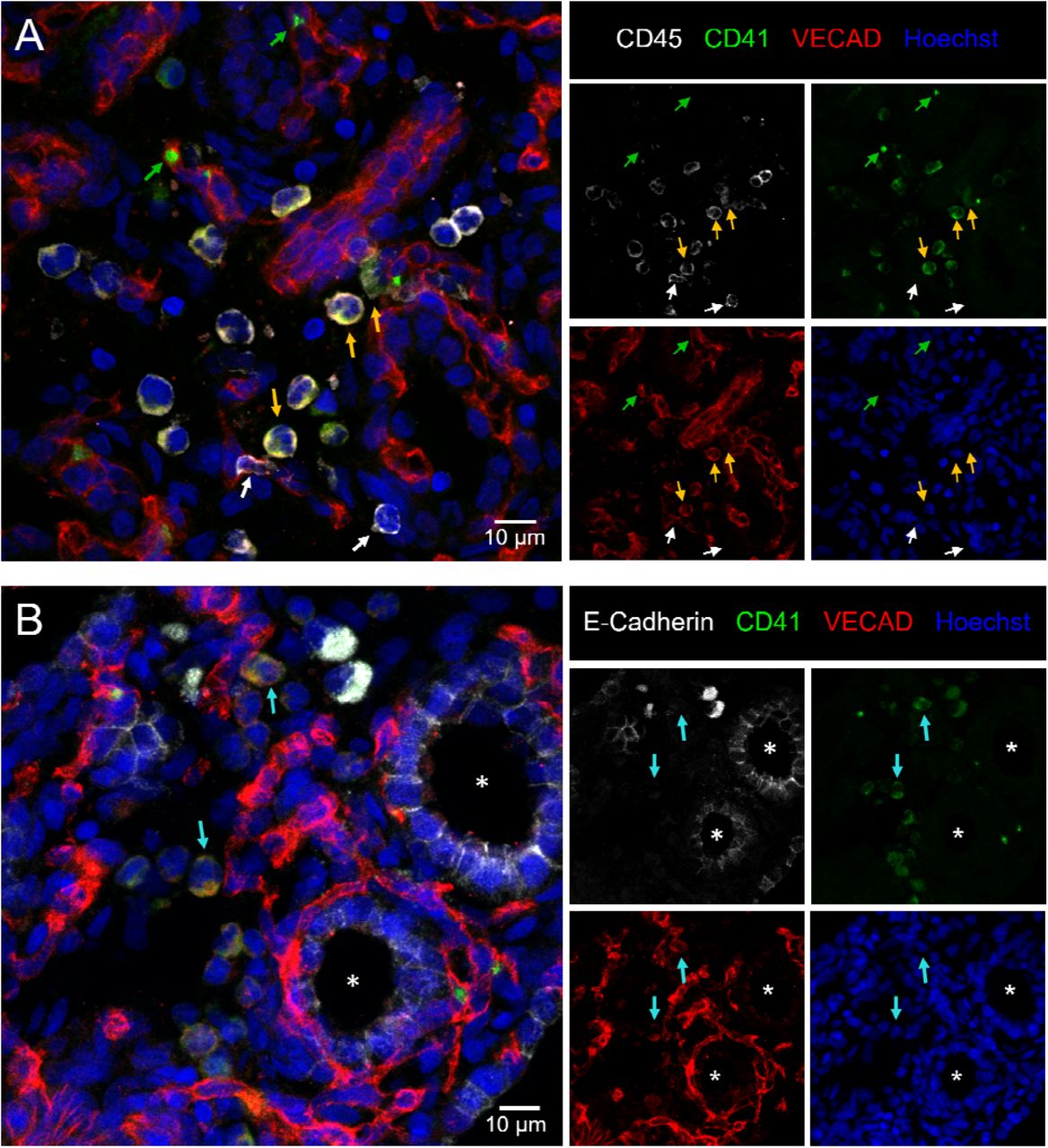
Hematopoietic clusters are a common histological hallmark of EHT occurring within the AGM and can be found in other organs that exhibit EHT including the placenta, and the umbilical and vitelline arteries8. To investigate whether fetal lung EHT can occur in-vivo we performed immunofluorescent staining of fixed-frozen human fetal lung sections. Immunofluorescent analysis revealed distinct regions of colocalized cells co-expressing the broad hematopoietic marker CD45, the endothelial marker VE-Cadherin, and the pre-HSC marker CD41a (Figure 4A). Additional staining for E-Cadherin suggests that these regions of EHT are not localized within the developing lung epithelium (Figure 4B).

12µm sections of a fixed frozen post-conception week 20 human fetal lung stained with the broad hematopoietic marker CD45, the EHT marker CD41, the endothelial marker VECAD, the epithelial cell marker E-Cadherin, and the nuclear stain Hoechst. (A) Cells co-expressing VECAD, CD41 and CD45 are highlighted with yellow arrows. Anucleate platelets single positive for CD41 are highlighted with green arrows. Differentiated hematopoietic cells only positive for CD45 are highlighted with white arrows. (B) Cells co-expressing VECAD and CD41 are highlighted with teal arrows, demonstrating they are not in developing E-Cadherin+ epithelial spaces, which are highlighted by a white asterisk.
Single cell transcriptomic mapping of murine fetal lung EHT
To map the developmental trajectory of cells produced in the murine fetal lung explant model system and to illustrate the repertoire of cells involved in the process, Single cell RNA sequencing (scRNA-Seq) was performed. The optimal time window for EHT in our model was determined to be from days 4-6 with the peak of CD45+/VECADlo cells on day 5 (Supplemental Figure 2A). Since cells that have recently undergone EHT transiently retain expression of VECAD, adherent cells were collected into a single cell suspension and sorted for VECAD+ to enrich for both endothelial cells and cells undergoing transition (Supplemental Figure 2B). To ensure the capture of progenitor cells as well as differentiated cells that have downregulated VECAD post-EHT, suspension cells were collected for sequencing but left unsorted. The single cell capture and processing was performed using the Chromium 10x Genomics platform, and subsequently sequenced using the Illumina NextSeq 2000 platform. A schematic of the sequencing methodology as well as cell capture numbers and read depth can be found in Supplemental Figure 2C, D.
SPRING analysis revealed a trajectory of cells transitioning from an adherent to a suspension state through 3 distinct clusters: endothelial, transitional, and hematopoietic (Figure 5A, B). Supervised gene expression analyses of canonical endothelial markers showed robust expression and subsequent downregulation in the endothelial and transitional populations, respectively (Figure 5C). Analysis of genes previously described to be important during EHT, many of which were recently reviewed3, highlighted the expression of these key markers at a discrete stage of EHT. For example, Notch1, Jag1, Dll4, and Sox17 were largely isolated to the endothelial cell stage. In contrast, Yap1 and Tead1 demonstrated sustained expression through the transitional cell stage. These results are in agreement with other reports demonstrating that Notch1 and Sox17 are critical in the early patterning of HECs, but their continued expression throughout EHT can be restrictive43,44. In contrast, Yap was previously shown to be critical in the maintenance of EHT, but not its initiation15. Lastly, expression of Lyve1 was predominantly expressed during the endothelial stage, which suggests the production of a definitive wave of hematopoiesis45(p1).

(A, B) SPRING plot trajectory of EHT clusters. (C) Heatmap of supervised gene expression analysis of endothelial, EHT and hematopoietic commitment markers. (D) Heatmap of unsupervised analysis of the top 50 differentially expressed genes. Representative gene ontology analysis is highlighted here along with the associated genes enriched in these biological processes. Full DGE analysis can be found under supplemental data.
As cells transitioned to hematopoietic commitment, they began expressing key pre-HSPCs genes including Cdca7, Myb, Gfi1 and Spn (CD43) (Figure 5C). Expression of Cdca7 was previously demonstrated to be isolated to pre-HSC populations found within AGM intra-aortic hematopoietic clusters46. Myb is critical in HSC maintenance and proliferation47, Gfi1 is critical in mediating the loss of an endothelial identity48(p1), and Spn (CD43) is a known marker of early pre-HSCs40,49. Notably, expression of these genes was primarily isolated to early progenitor clusters and decreased along the trajectory of maturation (Supplemental Figure 3A), which is in agreement with other reports showing that their downregulation is necessary to allow for competent differentiation.
Complementary to these supervised analyses, unsupervised analysis of the top 50 differentially expressed genes showed that transitional cells retain the expression of genes involved in the downregulation of angiogenesis as they differentiate away from an endothelial fate (Figure 5D, Supplemental Figure 3B, Supplemental_Data). This coincided with the upregulation of genes critical in extracellular matrix (ECM) organization as these cells undergo a physical transition from an adherent to a suspension state. In agreement with these findings, others have demonstrated that the emergence of hematopoietic cells from HECs relies on the Runx1 mediated upregulation of genes involved in ECM organization, cell adhesion and cell migration50(p1). Many of the same genes, including Runx1, were upregulated predominantly within the transitional cluster (Figure 5C, D).
Fetal Lung EHT is functionally reliant on canonical developmental pathways
To functionally assess the dependence of fetal lung EHT on Notch signaling, the Notch inhibitor, Compound E, was applied to explant cultures. Application of Compound E led to a significant reduction in VECAD+/CD45+ pre-HSPCs (Figure 6B, C). This is in line with other reports demonstrating that in-vitro Notch inhibition and in-vivo transgenic knockout of Notch1 specifically blocks EHT, but the proliferation, differentiation, and maintenance of HSPCs is conserved43,51.

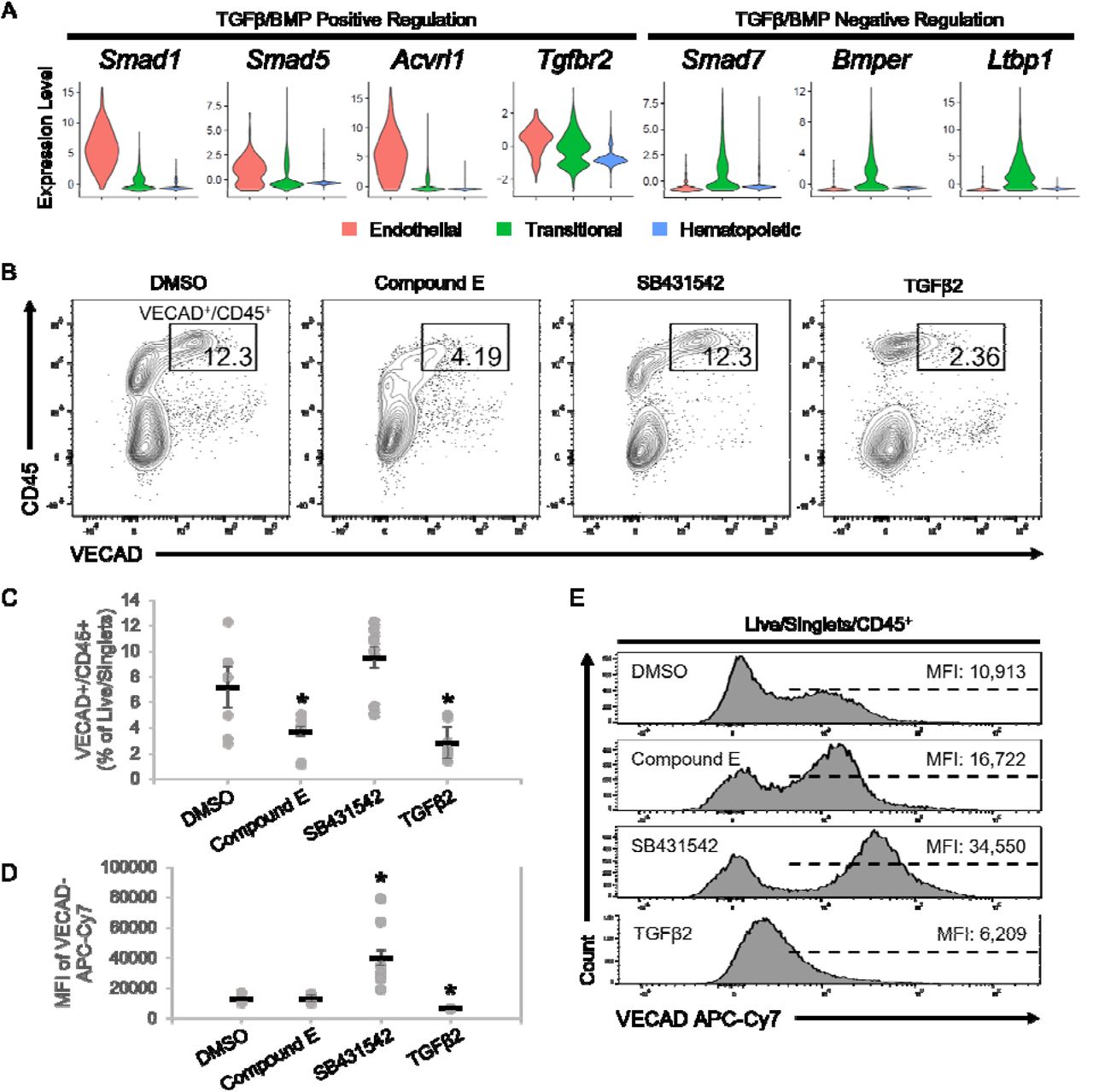
(A) Violin plots of expression of TGFβ/BMP pathways genes. (B) Representative flow cytometry plots demonstrating the change in CD45+/VECAD+ progenitors post-treatment with the Notch inhibitor Compound E, the TGFβ inhibitor SB431542, and recombinant TGFβ2. (C) Counts of CD45+/VECAD+ progenitors post-treatment. (D) Average MFI of VECAD expression post-treatment. (E) Representative histograms of VECAD expression post-treatment. Error bars represent standard deviation. *P < 0.01 compared to DMSO control.
In addition to the Notch family genes described previously, gene expression analysis demonstrated that fetal lung HECs exhibit the dynamic regulation of TGFβ/BMP pathways required for EHT. Similar to Notch, TGFβ/BMP pathways are important in the early patterning of HECs, but their subsequent inhibition is required for EHT44,52–54. Expression of key mediators of TGFβ/BMP signaling, including Smad1, Smad5, Acvrl1 and Tgfbr2, peaked at the endothelial stage and was progressively downregulated throughout EHT (Figure 6A). Driving this downregulation was the concurrent upregulation of the TGFβ/BMP inhibitors Smad7, Bmper and Ltbp1.
To assess the dependence of TGFβ signaling in fetal lung EHT, the TGFβ receptor inhibitor, SB431542, and recombinant TGFβ2 were applied to explant cultures. Application of recombinant TGFβ2 led to a significant reduction in VECAD+/CD45+ pre-HSPCs (Figure 6B, C). In contrast, SB431542 did not significantly affect the production of VECAD+/CD45+ pre-HSPCs. However, SB431542 induced a significant increase in the intensity of VECAD expression, while TGFβ2 caused a reduction (Figure 6D, E). While others have shown that SB431542 treatment can enhance the production of HSPCs44,55, these findings suggest that there is already sufficient inhibition from the upregulation of TGFβ/BMP inhibitors discussed previously.
DISCUSSION
Although not defined as a hematopoietic organ, the lung houses many resident blood cells that carry out both broad and tissue specific hematopoietic functions. Immune surveillance in the lung is carried out via unique resident immune cells, including dendritic cells, T-cells, B-cells, alveolar macrophages, interstitial macrophages, innate lymphoid cells and natural killer cells11,56. Supporting these classical immune cells are lung-resident megakaryocytes, which have a unique immune phenotype in addition to carrying out platelet biogenesis57,58. Lastly, the murine lung has been demonstrated to house HSPCs12–14. Complementary to these prior findings, we have demonstrated here that the murine and human fetal lungs are a source of functional HECs capable of giving rise to HSPCs. This suggests that the developing lung may have a more direct role in hematopoiesis than previously thought.
Although we have demonstrated the presence of fetal pulmonary HECs, the physiological significance of de-novo hematopoiesis occurring outside of the AGM and in the lung remains unclear. The potential need for the fetal lung to have an alternative source of blood may stem from the fact that most blood is shunted away from the fetal lung because oxygenation is provided via the mother. As a result, the developing murine lung only receives about 16% of total circulating blood59. The de-novo generation of blood in the fetal lung may act as an additional in-situ source of essential blood products for organogenesis. This is an important consideration when accounting for the role of macrophages and platelets in both organogenesis and angiogenesis60–63. Notably, the origins of many lung resident blood cells are not fully understood, but to date, the origins of resident macrophages are the most well studied.
Lung macrophages are derived from three distinct developmental waves originating from the yolk sac, fetal liver and BM64. Each wave persists into adulthood and occupies unique niches within the lung65. Recent work also suggests that such developmentally and phenotypically distinct subsets of macrophages may also extend to the heart, liver, kidney, and brain66. These findings demonstrate the complex diversity seen throughout hematopoietic development and how each phase individually makes significant physiological contributions. As such, the differentiation capacity of lung HECs requires further investigation to determine whether lung HECs contribute to the in-situ development of lung hematopoietic populations.
While often discussed as the precursor to HSCs, HECs also give rise to other blood progenitors. A subset of placental HECs lineage traced by Hoxa13 preferentially form placental macrophages, termed Hofbauer cells, that remain solely in the placenta67. This finding also suggests that differentiation capacity and bias may already be predetermined at the HEC stage, which others have also proposed. THBS1 marks a subset of human embryonic stem cell derived endothelial cells that are biased towards megakaryopoiesis68. Ly6a versus Tek expression marks HECs with HSC versus EMP potential, respectively69. CXCR4 expression differentiates AGM derived HECs with HSC versus MPP potential70.
Others have described the presence of functional HSCs in E16 murine fetal lungs. However, LSK progenitors only make up on average about 0.02% of the total fetal lung cell isolate, compared to 0.75% in the adult BM14. This low number of HSPCs suggests that the role of fetal lung HECs may not contribute significantly to the progenitor cell pool and instead are skewed towards the immediate production of differentiated cell populations.
While more in-vivo work is required to determine the developmental trajectory and potency of lung HECs, the contributions of hematopoiesis that are not reliant on long-term HSCs are important to consider. Indeed, the majority of embryonic hematopoiesis is maintained by a pool of yolk sac derived erythro-myeloid progenitors (EMPs) that reside in the fetal liver71,72. EMPs give rise to the majority of tissue resident macrophages that persist into adulthood73, and embryonic erythrocyte production is sustained primarily from EMPs74. In addition to EMPs, transient HSCs that do not persist into adulthood have also been demonstrated to be involved in the establishment of the developing hematopoietic system. Tie2-Cre lineage tracing studies suggest that fetal HSCs that contribute to establishment of the developing hematopoietic system differentiate rapidly, which is in stark contrast to adult long term HSCs that are relatively quiescent75. Studies utilizing the Flk2-Cre tracer model (FlkSwitch) also demonstrate the existence of a transient population of HSCs with a lymphoid bias76,77. Although fulfilling the classic definition of an HSC based on their capacity for long-term multi-lineage reconstitution, these transient HSCs are only present from E10.5 until P14.
Notably, the fetal lung HECs described here were isolated from E17 mice, which is outside of the window of EHT within the AGM (E9.5-11.5). Work from Yvernogeau et al. also suggests that EHT can occur beyond this AGM window within the perinatal bone marrow10. They noted that perinatal sources of hematopoiesis occur at a time when HSC expansion in the fetal liver has stopped (∼E17) and the BM niche is still maturing in order to support the long-term residency of adult HSCs. Later stage EHT in the lung may thus aid in minimizing the need for long term HSC-dependent hematopoiesis during early development.
Niche interactions are another critical component of driving EHT4,78. Induction of key developmental pathways via secreted ligands is spatially organized within the AGM and influence the potency of resultant progenitors20. Replicating these conditions is thus critical in simulating EHT in an in-vitro setting. Most other employed methods of ex-vivo HEC cultures are reliant on the utilization of a supportive feeder cell layer, such as OP9 cells or immortalized AKT endothelial cells, to instruct EHT51,79. Notably, fetal lung explants undergo EHT without such feeders, which suggests that the lung independently has the cellular makeup to support EHT. However, further investigation is required to determine the cell-specific interactions of the in-vivo microenvironment where EHT would occur and how the lung niche instructs local HEC development.
Altogether, the findings described here demonstrate the existence of functionally validated HECs in the fetal lung, a population which is also conserved in humans. Given the diversity in HEC development and the significance of each distinct wave of hematopoiesis, the physiological influence of lung HEC development in-vivo needs further investigation. Expanding our overall understanding of HEC location and timing will also aid towards the goal of fully understanding the biological cues required for the development of functional HECs. Such findings can eventually be harnessed for the common goal of producing putative HSCs and functionally mature hematopoietic cells from pluripotent stem cells.
LIMITATIONS OF STUDY
In this study, explant cultures were the primary methodology employed to investigate lung HECs. This ex-vivo methodology removes these cells from their microenvironment, thus limiting our understanding of these cells in their native in-vivo context. Of note, we do not believe we have induced the direct conversion of endothelial cells as direct conversion involves the forced overexpression of select transcription factors to competently induce transdifferentiation3. However, this still leaves many questions about whether these cells under steady state conditions in-vivo will naturally undergo EHT. Many tracing methods were considered to investigate these questions but also have significant limitations. While many lineage tracing models have been developed to trace either endothelial cell or HSPC populations individually, all models exhibit a significant amount of recombination events across both populations, especially during embryonic development80. As such, application of these methods would still not sufficiently answer such questions when considering the rarity of HECs and the existence of circulating HSPCs81, especially during the embryonic migration of HSPCs exiting the fetal liver. An inducible barcoding model was also considered82. However, others have previously noted that EHT is not the result of asymmetric cell division37. As such, a parent endothelial cell to trace the resultant hematopoietic progeny back to would not exist and negate the utility of such a method. Development of novel technologies to trace organ specific endothelial cell populations will greatly support these endeavors.
AUTHORSHIP CONTRIBUTIONS
A.K.Y. performed experimental design, data collection, data analysis, interpretation of data and manuscript preparation. G.J.M. performed and supervised experimental design, interpretation of data and manuscript preparation. C.V.M. and J.L.V. performed the bioinformatic/computational analysis and supported the experimental design and manuscript preparation. A.C.B, K.V., T.W.D., A.B.Y., V.V., G.M. and A.B.B assisted with data collection and manuscript preparation.
DISCLOSURES
The authors of this manuscript have no conflict of interest to disclose.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (NIH) Training Grant for Hematology (5T32HL7501-36) and the Predoctoral NRSA for MD/PhD Fellowships (1F30HL154552-01). The authors would like to thank the following for technical support: Brian R. Tilton of the Boston University Flow Cytometry Core Facility; Dr. Yuriy Alekseyev and Ashley LeClerc from the Boston University Microarray and Sequencing Resource (BUMSR) Core; Dr. Michael T. Kirber from the Boston University Cellular Imaging Core.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.
- 36.
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.
- 46.↵
- 47.↵
- 48.
- 49.↵
- 50.
- 51.↵
- 52.↵
- 53.
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}