

Abstract
Studying proteins through the lens of evolution can help identify conserved features and lineage-specific variants, and potentially, their functions. MolEvolvR (http://jravilab.org/molevolvr) is a web-app that enables researchers to run a general-purpose computational workflow for characterizing the molecular evolution and phylogeny of their proteins of interest. The web-app accepts input in multiple formats: protein/domain sequences, homologous proteins, or domain scans. MolEvolvR returns detailed homolog data, along with dynamic graphical summaries, e.g., MSA, phylogenetic trees, domain architectures, domain proximity networks, phyletic spreads, and co-occurrence patterns across lineages. Thus, MolEvolvR provides a powerful, easy-to-use interface for computational protein characterization.
The rate at which molecular or biochemical functions are assigned to proteins greatly lags behind the rate at which new protein families are discovered1. This gap impedes identifying and characterizing the complete molecular systems involved in various critical cellular processes such as molecular pathogenesis, antibiotic resistance, or stress response. Several studies2–16 have demonstrated the power of molecular evolution and phylogenetic analysis in determining molecular functions of such proteins. There are many individual tools17–36 for protein sequence similarity searches or ortholog detection, delineating co-occurring domains (domain architectures), and building multiple sequence alignments and phylogenetic trees. However, there is a paucity of unified software or web frameworks that effectively integrate these approaches to comprehensively characterize proteins and help discern function by exhaustively identifying all members of relevant protein families (including remote homologs), mapping domains/domain-architectures, and tracing their phyletic spread across lineages.
Here, we present MolEvolvR, a web application that provides a streamlined, easy-to-use platform for comprehensively characterizing proteins (Fig. 1; accessible at http://jravilab.org/molevolvr). MolEvolvR performs homology searches across the tree of life and reconstructs domain architecture by characterizing the input proteins and each of their homologs and presents these results in the context of evolution across the tree of life. The computational evolutionary framework underlying MolEvolvR is written using custom R37–52 and shell scripts. The web application is built with an R/Shiny39,53,54 framework with additional UI customizations in HTML, Javascript, and CSS, and has been tested on Chrome, Brave, Firefox, and Safari browsers on Mac, Windows, and Linux operating systems.

A. MolEvolvR allows users to start with protein(s) of interest and perform the full analysis (1+3+4), only protein characterization (1+3), only homology searches (1+4), or start with external outputs from BLAST or Interproscan for further analysis, summarization, and visualization (2+3+4). MolEvolvR is interactive, queryable, and customizable. B. Multiple input options in MolEvolvR: FASTA, NCBI accession numbers, pre-computed MSA (in FASTA formats), analysis outputs from an external web or command-line BLAST or InterProScan runs. C. The different analysis options available in MolEvolvR are based on the chosen input formats in 1A, 1B.
To illustrate the full range of analysis made possible by MolEvolvR, we consider a researcher starting with a protein of unknown function. In Step 1, MolEvolvR resolves this query protein into its constituent domains and uses each domain for iterative homology searches18,19 across the tree of life (>7,000 complete genomes)55–58. This divide-and-conquer strategy will identify all proteins similar to each domain, including remote homologs that cannot be found by homology searches using the full-length protein sequences alone. In Step 2, to delineate molecular function, MolEvolvR reconstructs the domain architecture by characterizing the protein and each of its homologs by combining: 1) sequence alignment and clustering algorithms for domain detection17,22,23,59; 2) profile matching against protein domain and orthology databases30,31,60–63; 3) prediction algorithms for signal peptides31,64, transmembrane regions31,65–67, cellular localization31,66, and secondary/tertiary structures31,68–71. This analysis results in a detailed molecular characterization of each query protein and its homologs across the three kingdoms. MolEvolvR is versatile in many ways. First, the web-app accepts multiple types of queries. Researchers can start their searches using protein/domain sequences of single- or multi-protein operons (e.g., FASTA, NCBI protein accession numbers), homologous proteins (e.g., web or command-line BLAST output), or motif/domain scans (e.g., InterProScan output) [Fig. 1B]. Second, it enables tailored analyses to answer a variety of questions (e.g., determining protein features restricted to certain pathogenic groups to discover virulence factors/diagnostic targets) [Fig. 1C]. Finally, it provides multiple types of outputs (e.g., complete set of homologs/phylogenetic tree, domain architecture of query protein, most common partner domains) [previews in Fig. 1A]. Besides tables and visualizations for each result, MolEvolvR also provides graphical summaries that bring these results together in the context of evolution: i) structure-based multiple sequence alignments and phylogenetic trees; ii) domain proximity networks to consolidate results from all co-occurring domains (across homolog domain architectures; iii) phyletic spreads of homologs and their domain architectures; and iv) co-occurrence patterns and relative occurrences of domain architectures across lineages [Fig. 1A]. The web-app contains detailed documentation about all these options.
A specific instance of the web-app, applied to study several Psp stress response proteins (present across the tree of life), can be found here: https://jravilab.shinyapps.io/psp-evolution2. MolEvolvR is a generalized web-server of this web-app. To demonstrate its broad applicability, we have applied the approach underlying MolEvolvR to study several systems, including proteins/operons in zoonotic pathogens, e.g., nutrient acquisition systems in Staphylococcus aureus4,5, novel phage defense system in Vibrio cholerae6, surface layer proteins in Bacillus anthracis7, helicase operators in bacteria8, and internalins in Listeria9.
Thus, MolEvolvR (http://jraviilab.org/molevolvr) is a flexible and powerful interactive web tool that researchers can use to bring molecular evolution and phylogeny to bear on their proteins of interest.
Supplementary Material
Supplementary Text
Here, we present a case study with the Lia operon (from Bacillus subtilis) involved in lantibiotic stress response, one of the better-studied phage shock protein (PSP) stress response systems2. The PSP systems are well-known for having a good mix of conserved components (e.g., PspA) and variant themes (e.g., centering on PspBC or Lia components). It is unclear upfront as to what the underlying building blocks (domains/domain architectures) might be that culminate in the stress response function and what their history is in the broader context of bacterial evolution (phylogeny, phyletic spreads). Therefore, we focus on a detailed characterization of the six Lia proteins, LiaIHGFSR, in this section; the detailed results (analyzed data and graphical summaries) are available here for interactive exploration: jravilab.org/molevolvr/?r=k0PsZM. We used MolEvolvR to conduct a comprehensive analysis across the three superkingdoms of life and delineate all occurrences of Lia proteins in terms of their underlying domain architectures and phyletic spreads.
Homologs
We first generated the homologs for each of these six proteins across all completed representative/reference genomes (from bacteria, archaea, and eukaryota) [Data tab, web-app]. The homolog data from these genomes are available in an interactive and queryable format in the MolEvolvR web-app with linked NCBI accession numbers, species, lineages, percentage similarities, and several protein- and similarity-search-related parameters [Fig. S1]. Best hits by species or lineage can be easily subsetted. The whole data table is available for download in a standard CSV format.
Domain architectures
Next, we determine the domain architectures (based on sequence-structure motifs/domains, disorder predictions, transmembrane regions, signal peptides, and cellular localizations) of the six Lia proteins and each of their homologs. In the ‘Domain Architecture’ tab, we first summarize our results for individual and across all query proteins [Fig. S2A, left]. In addition to the diverse domain architectures, we can also determine their phyletic spreads using a simple popup feature to determine widespread vs. lineage-specific domain architectures [Fig. S2A, right]. Using MolEvolvR, we can also visualize the diverse domain architectures and cellular localizations (e.g., Pfam, Gene3D, Phobius, MobiDB) of representative homologs of each Lia protein to discover both similar and dissimilar homologs, e.g., ones with novel partner domains, variants with altered localization [Fig. S2B]. We summarize our domain findings (by profile database/prediction algorithm) in the ‘Network’ section of the ‘Domain Architecture’ tab [Fig. S2C]. The proximity network consolidates the findings across proteins (or by protein) by connecting co-occurring domains within proteins by their frequency of occurrence across lineages (nodes, domains; edges, co-occurrence; node size and width of arrows, frequencies). This big picture view, along with the co-occurrence plots [Fig. S2D], helps discover new components and their relative frequencies across thousands of homologs. They also lead to serendipitous connections across homologs of the different Lia proteins, e.g., variants of the two-component LiaRS system that carry both the response regulator and histidine kinase domains, as well as predominantly lone domains, e.g., PspA_IM30 [Fig. S2D].
Phylogeny
Finally, we used MolEvolvR to study the evolution of the Lia proteins [Phylogeny tab, MolEvolvR]. We recorded the presence of homologs, by lineage, through interactive sunburst plots [Fig. S3A; Phylogeny tab, web-app] and heatmap for all query proteins [Fig. S3B; Data tab, web-app]. We generated multiple sequence alignments and phylogenetic trees based on key representatives from diverse species, lineages, and domain architectures [Fig. S3C-D, Phylogeny tab, web-app].
Through these comprehensive analyses, we first characterized the proteins encoded by the Lia operon in terms of their domain architectures, with LiaH, a PspA stress response effector protein, two transmembrane proteins LiaI and LiaG, two globular domains of unknown function with Toastrack-like domains in LiaF and LiaG, and a two-component system LiaRS with response regulator/receiver domain and histidine kinase. We discovered several homologs in lineages outside Firmicutes for each of the Lia proteins and domains, including the widespread two-component system, while other domains (PspA, DUF2154, DUF4097) were predominantly present in only Firmicute genomes. We also identified new connections within the more extensive Lia proximity network based on homologs of the LiaRS proteins, with proteins that carried domains from both response regulator and histidine kinase domains (e.g., in proteobacteria, planctomycetes), and rare DUF4097-containing homologs within Firmicutes that carry an additional N-terminal DUF1700 or a second DUF4097 domain.
A more comprehensive analysis of the phage shock protein (PSP) system and its partner domains across the tree of life using a MolEvolvR-like approach leading to multiple novel discoveries has been described in a recent article2. The PSP system is a great use case to characterize using molecular evolution and phylogeny due to the variety of domain architectures, cellular localizations, and phyletic spreads of each of these protein families and operons. The PSP work, including this Lia operon use case, as well as other recent diverse biological applications2–11 indicate that the MolEvolvR approach is invaluable in characterizing new protein families of interest in the context of evolution.
Supplementary Figures
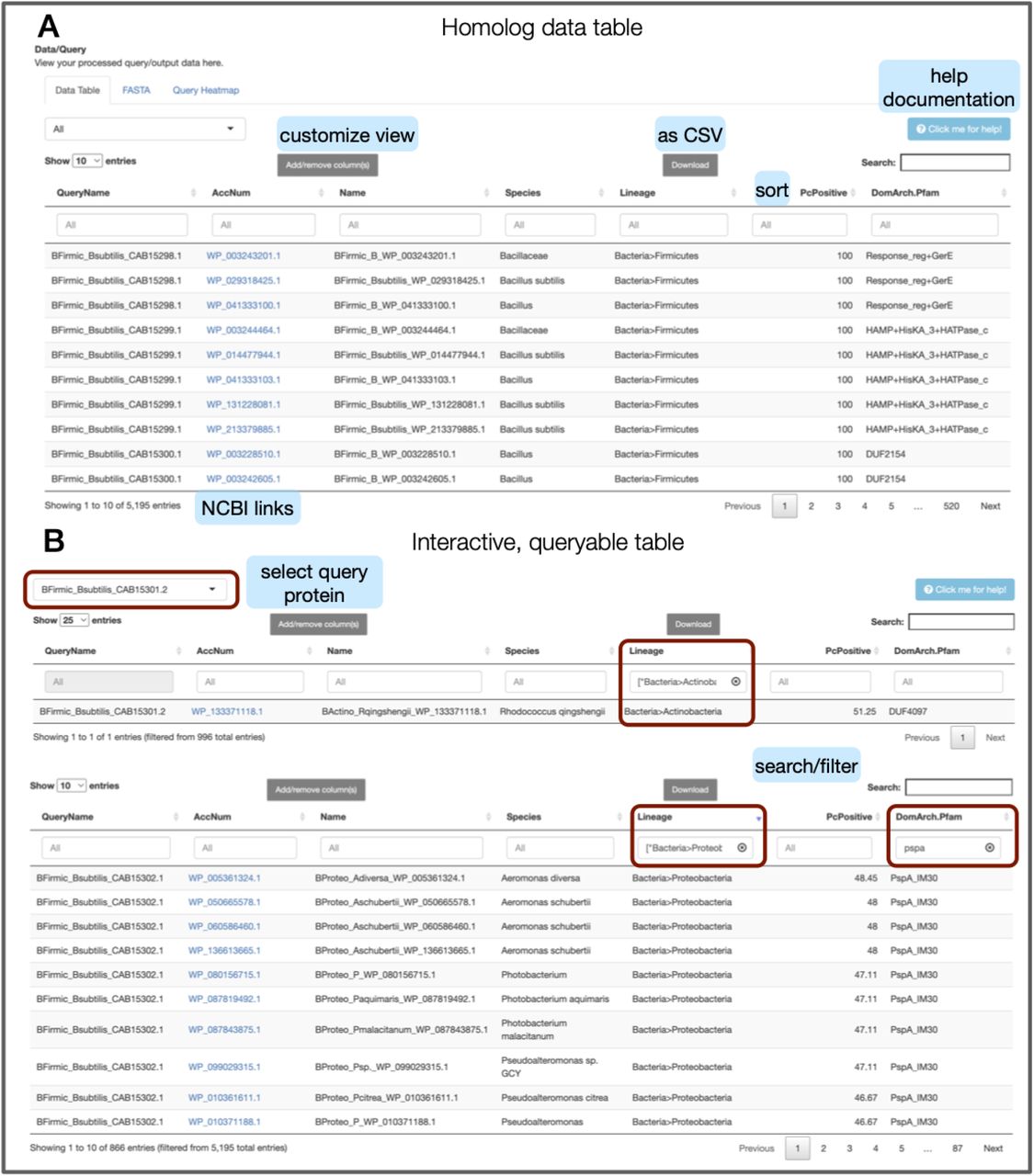
Homolog data table with the best hits from all superkingdoms of life (queried across all RefSeq genomes). For each homolog, we tabulate details across all query proteins, including genome, species, lineage, sequence homology (BLAST parameters), and domain architecture information in a sortable, queryable, and interactive manner. The ‘Add/remove column(s)’ button allows users to access the full list of columns and add/remove column(s) as needed. ‘Download’ lets the user download the filtered subset of the table or the entire table for further local analysis (as CSV). Results across and within individual protein searches can be viewed (using the dropdown menu on the top right). The accession number for each homolog is hyperlinked to its corresponding NCBI protein page. A shows a snapshot of the best hits across all query proteins with the default columns. B shows examples of the Interactive, queryable table with homologs being filtered based on specific lineages (e.g., actinobacteria) AND top: query protein (BFirmic_Bsubtilis_CAB15301.2) or bottom: domains (e.g., PspA). Light blue boxes with text are not part of the screenshot; they have been added to highlight key features.

A. Domain architecture summary table with phyletic spreads. The table shows the top (most predominant) domain architecture by query protein (or across all queries, as in this case) with the frequency of occurrence and lineages in which they occur. The slider allows users to pick the top hits, representing 98% of all homologs in this case. The second snapshot shows the popup that appears when each domain architecture row is clicked; it elaborates on the ‘LineageCount’ by showing the frequencies of occurrence by individual lineage for the selected domain architecture. B. Representative domain architectures. The figure shows cartoon depictions of representative domain architectures (Pfam) and cellular localizations (Phobius) of the 6 query proteins. The Pfam and Phobius annotations for each domain prediction (by colour) are shown in the legend. C. Domain proximity network. The network captures co-occurring domains within the top 98% of the homologs of all the ‘query’ Psp members and their key partner domains (after sorting by decreasing frequency of occurrence). The size of the nodes (domains) and width of edges (co-occurrence of domains within a protein) are proportional to the frequency of their occurrence across homologs. The query domains (original proteins/domains of interest) and other commonly co-occurring domains are indicated in red and grey. The complete network, as well as the domain-centric ones, are available on the web-app. D. Phyletic spread of the predominant domain architectures across query proteins. The heatmap shows the presence/absence of homologs of all query proteins across key lineages (columns) for each predominant domain architecture (rows). The colour gradient indicates the highest number of homologs in a particular lineage. The heatmap gives the full picture. Rows: Top domain architectures across all homologs. Columns: The major archaeal, bacterial, eukaryotic, and viral lineages with representative sequenced genomes. Blue boxes with text are not part of the screenshot; they have been added to highlight key features.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Lineages of homologs. Sunburst plot shows the phyletic spread of all the homologs. The legend shows the lineages (inner ring, kingdoms; outer ring, phyla) that carry homologs. Note: The sunburst plots only display lineages of >0.1 % fraction of total proteins. B. Phyletic spread of the homologs by query protein. The heatmap shows the presence/absence of homologs across key lineages (columns) for each query (rows). The colour gradient indicates the highest number of homologs in a particular lineage. The heatmap gives the whole picture. Rows: Query proteins are queried against all sequenced and completed genomes across the three major kingdoms of life. Columns: The major archaeal, bacterial, eukaryotic, and viral lineages with representative sequenced genomes. C. The multiple sequence alignment is overlaid on the phylogenetic tree. In the tree generated for LiaS (BFirmic_Bsubtilis_CAB15299.1), each leaf (protein) is named with its kingdom (e.g., ‘B’ for bacteria), phylum (first six letters, e.g., ‘actino’ for actinobacteria), Genus, species (represented as ‘Gspecies,’ e.g., ‘Bsubtilis’ for Bacillus subtilis), and NCBI protein accession number (e.g., CAB15298.1), resulting in a uniquely identifiable name for the protein, ‘KPhylum_Gspecies_AccNum,’ e.g., BFirmic_Bsubtilis_CAB15299.1. Key: the colours in the multiple sequence alignment depiction correspond to different amino acids. D. Snapshot of the multiple sequence alignment of representative homologs of LiaS (BFirmic_Bsubtilis_CAB15299.1) by lineage. The generated MSA, with representative homologs from each lineage, species, or domain architectures, is available to users as a downloadable searchable PDF. Light blue boxes with text are not part of the screenshot; they have been added to highlight key features.
Funding
We would like to thank our funding sources: Endowed Research Funds from the College of Veterinary Medicine, Michigan State University, NSF-funded BEACON funding support, and Michigan State University start-up funds awarded to JR; NSF-funded REU-ACRES summer scholarship to SZC.
Author Contributions
JR conceived the study; JR designed the study; JTB, SZC, LMS, and JR acquired the data, performed all the analyses, and made the figures and tables, specifically JTB, SZC, LMS, JR wrote the backend code, JTB, SZC, JR built the R/Shiny web-app, JBJ set up the server backend for the web-app; LMS and JR wrote the first draft of the manuscript; JTB, LMS, and JR revised the manuscript.
Data Availability and Reuse
All the data, analyses, and visualizations are available in our interactive and queryable web application: http://jravilab.org/molevolvr. Text, figures, and the webapp are licensed under Creative Commons Attribution CC BY 4.0.
Acknowledgments
We would like to thank members of the JRaviLab (Elliot Majlessi, Ethan Wolfe, Karn Jongnarangsin, Kewalin Samart) for testing the web-app at various stages and providing the authors with several iterations of constructive feedback. We are also extremely grateful to Krishnan Raghunathan, Premal Shah, Arjun Krishnan, and members of the Krishnan lab (Kayla Johnson, Nathaniel Hawkins) for early feedback on MolEvolvR and the manuscript. We have benefited from several diverse use cases and challenges brought to us by our collaborators that helped fine-tune the functionality of the web-app, thanks to collaborations with L Aravind, Neal Hammer, Christopher Waters, Antonella Fioravanti, Jonathan Hardy, Kayla Conner, Christina Stallings, Helen Blaine, and Stephanie Shames. We appreciate timely help from Kellen Reason, our current system administrator, for maintaining the backend of our web-server.
Footnotes
References




