

Abstract
Supergenes are sets of genes and genetic elements that are inherited like a single gene and control complex adaptive traits, but their functional roles and units are poorly understood. In Papilio polytes, female-limited Batesian mimicry is thought to be regulated by a ∼130kb inversion region (highly diversified region: HDR) containing three genes, UXT, U3X and doublesex (dsx) which switches non-mimetic and mimetic types. To determine the functional unit, we here performed electroporation-mediated RNAi analyses (and further Crispr/Cas9 for UXT) of genes within and flanking the HDR in pupal hindwings. We first clarified that non-mimetic dsx-h had a function to switch from male to non-mimetic female and only dsx-H isoform 3 had an important function in the formation of mimetic traits. Next, we found that UXT was involved in making mimetic type pale-yellow spots and adjacent gene sir2 removed excess red spots in hindwings, both of which refine more elaborate mimicry. Furthermore, downstream gene networks of dsx, U3X and UXT screened by RNA sequencing showed that U3X upregulated dsx expression and repressed UXT expression. These findings demonstrate that a set of multiple genes, not only inside but also flanking HDR, can function as supergene members, which extends the definition of supergene unit than we considered before. Also, our results indicate that dsx-H functions as the switching gene and some other genes such as UXT and sir2 within the supergene unit work as the modifier gene.
Article summary Supergenes are thought to control complex adaptive traits, but their detailed function are poorly understood. In Papilio polytes, female-limited Batesian mimicry is regulated by an ∼130kb inversion region (highly divergent region: HDR) containing three genes. Our functional analysis showed that doublesex switches the mimicry polymorphism, and that an inside gene UXT and an outside gene sir2 to the HDR work to refine more elaborate mimicry. We here succeed in defining the unit of mimicry supergene and some novel modifier genes.
Introduction
Batesian mimicry is a phenomenon in which a non-toxic species (mimic) escapes predation from predators such as birds by mimicking the appearance, colors, shape, and behavior of a toxic and unpalatable species (model) (1) and can be achieved only when multiple traits are properly combined. For example, the butterfly’s wing pattern is composed of various colors and complex patterns, and unless almost all wing pattern elements are similar to the model species, mimicry cannot be achieved successfully. In the Batesian mimicry butterflies, it is also known that not only wing patterns and shapes but also the flying behavior should resemble the model species (2, 3). In addition, some Papilio species show polymorphic Batesian mimicry, but few intermediate offspring between mimetic and non-mimetic types was observed (4–7). These facts indicate that the multiple sets of traits for the mimicry are inherited in tightly linked manners, which have led to the “supergene” hypothesis (8). It was originally considered that supergene is composed of multiple flanking genes which are linked in the same chromosomal loci and inherited tightly together (9–13). On the other hand, it has also been hypothesized that a single gene or a single regulatory element may regulate complex phenotypes such as mimicry by controlling multiple downstream genes (14, 15). Although many studies have reported that the supergene loci may be involved in the formation of complex phenotypes, no attempt has made to reveal the functions of multiple genes within the supergene locus.
In a swallowtail Papilio polytes, only females have the mimetic and non-mimetic phenotypes, and males are monomorphic and non-mimetic (Fig. 1A). The mimetic female of P. polytes has red spots on the outer edge of hindwings and pale-yellow spots in the center of hindwings, which mimics the unpalatable model butterfly, Pachliopta aristolochiae (6, 16, 17). The pale-yellow spots of the mimetic and non-mimetic forms differ not only in shape and arrangement, but also in the pigment composition (18, 19). Males and non-mimetic females fluoresce under the UV irradiation, whereas mimetic females and model species, Pachliopta aristolochiae, do not fluoresce under the UV irradiation (18, 19). It is also known that the mimetic female also resembles Pachliopta aristolochiae in the behavior of flight path (2). Previous studies have shown that mimicry is regulated by the H locus and that the mimetic female (H) is dominant to the non-mimetic female (h) according to the Mendelian inheritance (6). Recently, whole genome sequences and genome-wide association studies have shown that about 130 kb of chromosome 25 which includes doublesex (dsx) is responsible for the H locus (Fig. 1B) (20, 21). The direction of this region differs between the H allele and the h allele due to the inversions at both ends, suggesting that the H allele evolved from the h allele, given the conserved structure for the h allele type among lepidopteran insects (20, 21). It is thought that recombination between the two alleles is suppressed by the inversion, and the accumulation of mutations and indels over the years has resulted in a highly diversified region (HDR) with low sequence homology between H and h (21).

(A) Wing patterns of adult male and non-mimetic and mimetic females of P. polytes, and toxic model, Pachliopta aristolochiae. Mimicry is regulated by H locus and mimetic allele (H) is dominant over the non-mimetic allele (h). (B) Detailed structure of mimicry HDR in P. polytes (21, 25). The direction of the HDR is reversed between h and H (i.e., inversion). Putative breakpoints of the HDRs are indicated by orange dotted lines. The breakpoint on the left side is located inside the 5’-untranslated region (UTR) of the ubiquitously expressed transcript (UXT) gene, and that on the right side is located just on the outer side of doublesex (dsx). (C) Expression levels of genes within and flanking the HDR in hindwings of mimetic (Hh) females at the wandering stage (W) of the late last instar larvae, 2 day after pupation (P2) and 5 day after pupation (P5). FPKM values by RNA sequencing are shown with the error bars of standard error. Different letters indicate significant differences (Tukey post hoc test, P<0.05). (D) Relative expression levels of genes within and flanking the HDR in hindwings of mimetic (Hh) and non-mimetic (hh) females and males (Hh) at P2 and P5 estimated by RT-qPCR. RpL3 was used as the internal control. Red, white, and black bars indicate mimetic and non-mimetic females and males, respectively. Error bars show standard error. Different letters indicate significant differences (Tukey post hoc test, P<0.05). Photo Credit: Shinya Komata, The University of Tokyo.
Nishikawa et al. (21) found in P. polytes, that knockdown of mimetic (H) type dsx (dsx-H) in the hindwings of mimetic females switched to a wing pattern similar to that of the non-mimetic females using the electroporation mediated RNAi method (22, 23). Knockdown of non-mimetic (h) type dsx (dsx-h) did not cause such a switch, suggesting that dsx-H is essential for the formation of mimetic patterns, but the functional role of dsx-h is unknown (21). The mimicry HDR contains not only dsx but also the 5’-untranslated region (UTR) portion of the Ubiquitously Expressed Transcript (UXT), a transcriptional regulator, and the long non-coding RNA Untranslated 3 Exons (U3X), present only in the HDR of the H allele (HDR-H) (Fig. 1B), but functions of UXT and U3X are still unclear (21). In P. memnon, which is closely related to P. polytes and exhibits female-limited Batesian mimicry, the locus responsible for mimicry (A) is a dsx-containing region of chromosome 25 and consists of two types of HDRs with low homology between A-allele and a-allele (24–26). The mimetic-type HDR (HDR-A) of P. memnon also contains the 5’-UTR portion of UXT in addition to dsx (25). Although an inversion is present in P. polytes and absent in P. memnon, the left-side breakpoint/boundary sites of the mimicry HDR is commonly located (25). Furthermore, also in the closely related species, P. rumanzovia, which possesses the female-limited polymorphism, the left-side boundary of the mimicry HDR is thought to be located at the same position, i.e., in the 5’UTR of UXT (26, 27). These suggest that the left-side breakpoint/boundary sites of the mimicry HDR, i.e., the 5’ UTR of UXT and its surrounding regions, may have an important role in the regulation of the polymorphism (27). Furthermore, in P. polytes, UXT and U3X are expressed in the hindwing, suggesting that these genes in the HDR may also be involved in the formation of mimetic patterns (21).
Many supergenes show intraspecific polymorphism due to inversions, but in some cases, such as P. memenon, there is no inversion, but two types of HDR structures for mimetic and non-mimetic alleles are maintained (25). However, it has not been clear whether the functional unit of the supergene that regulates complex adaptive traits is limited in the area within the inversion or the region of low homology (i.e., HDR), or whether it extends to neighboring regions. Both in P. polytes and P. memnon, the external gene prospero, which is adjacent to the internal gene UXT in the HDR, has read-through transcripts only in mimetic females (21, 25). This suggests that some cis-regulatory element in the mimetic HDR may control the gene expression even in the external region, and that such a gene may be involved in the formation of the mimetic pattern.
In this study, we would like to elucidate the involvement of multiple genes other than dsx-H in the female-limited Batesian mimicry in P. polytes and the range of functional units in the supergene by examining the function of genes within and flanking the mimicry HDR. First, to search the allele- or phenotype-specific expression, the expression patterns of genes within and flanking the mimicry HDR were analyzed by RNA sequencing (RNA-seq) and reverse transcription quantitative PCR (RT-qPCR). Second, we explored the more detailed function of dsx: dsx-H is thought to switch mimetic and non-mimetic phenotypes, but the functional roles of dsx-h and three isoforms in dsx have been unclear. Third, to know the function of UXT and U3X other than dsx within the HDR-H, as well as prospero and sir2 in close proximity to the outside of the HDR, we performed RNAi by in vivo electroporation (22, 23) and Crispr/Cas9 knockout for UXT. In addition, RNA-seq was performed on the dsx-H, UXT, and U3X gene knockdown wings and the control wings to elucidate the regulatory relationship and downstream genes of the three genes.
Results
Comparison of the expression levels of genes inside and flanking the mimicry HDR-H
It is reported and that wing coloration occurs after day 9 of pupation (P9), and that the expression of dsx-H, which is thought to play a major role in the formation of mimetic patterns in P. polytes, peaked on days 1–3 of pupation (P1–P3), suggesting that the mimetic pattern formation is determined on days 1–3 of pupation (18, 21). Therefore, gene expression in the first half of the pupal stage was considered to be important for the mimetic wing pattern formation. In order to investigate whether each gene in and flanking the HDR-H is involved in these process, we examined the expression levels of the genes, prospero, UXT-H (UXT from H-allele), UXT-h (UXT from h-allele), U3X, dsx-H, dsx-h, Nach-like, and sir2, in the hindwing imaginal discs at the wandering stage (W) of the last instar larvae, P2 and P5, by RNA-seq (Table S1 shows the list of samples used). The results showed that Nach-like was not expressed at all in any developmental stage as reported in P. memnon (25). Other genes were expressed in mimetic females, non-mimetic females and males (Fig. 1C and fig. S1).
In the RNA-seq data of mimetic females, dsx-H and dsx-h showed contrasting expression patterns. dsx-H showed a peak expression in the P2 stage, while dsx-h was highly expressed in the P5 stage (Fig. 1C). The expression of U3X, which is only present in the H locus, tended to show the constant expression and relatively high in P2, while the data was not statistically significant (Fig. 1C). There was no significant difference in the expression pattern of UXT between UXT-H and UXT-h, and the expression level of UXT was higher in W and P2 and significantly lower in P5 (Fig. 1C). The expression pattern of prospero was significantly larger in P2 as in dsx-H, and that of sir2 was largest in W as in UXT, but not statistically significant (Fig. 1C), In the RNA-seq experiment, we used three or more samples at each stage (W, P2, P5) for mimetic females, but insufficient numbers of samples for some non-mimetic females and males (Fig. S1), and thus we further performed RT-qPCR using P2 and P5 samples for mimetic females (Hh), non-mimetic females (hh), and males (Hh) (Fig. 1D).
RT-qPCR showed that the dsx-H expression was significantly high in P2 of mimetic females but low in P5 and males in every stage (Fig. 1D). The dsx-h expression was significantly high in P5 of non-mimetic females compared to other stages, mimetic females and males (Fig. 1D). It is noteworthy that the expression of dsx was low in males at all stages (Fig. 1D). The highly expression of dsx-H in P2 of mimetic females is consistent with RNA-seq results (Fig. 1C) and previous studies, which may be related to the mimetic color pattern formation (21, 28). U3X was not detected in non-mimetic females (hh) because it is present only in the H locus, and was expressed in mimetic females and males at P2 and P5 stages (Fig. 1D). For UXT-H, there was no significant difference in expression levels among mimetic females and males in any stages (Fig. 1D). The expression of UXT-h was significantly greater in non-mimetic females (hh), probably because they are h homozygous, but it was particularly high in non-mimetic females in P5, about five times higher than the expression of UXT-h in mimetic females (Fig. 1D). The expression of sir2 and prospero in mimetic females was similar to that of RNA-seq results, and their expression was also observed in non-mimetic females and males (Fig. 1D). The expression of sir2 was significantly higher in males in P5 than in mimetic or non-mimetic females (Fig. 1D).
To summarize the results of the expression analyses by RNA-seq and RT-qPCR, dsx-H and dsx-h appear to be regulated separately, as dsx-H and dsx-h showed contrasting expression patterns. The trend of high expression at P2 as well as dsx-H was observed in prospero and U3X, and the trend of gradually decreasing expression at W, P2, and P5 was observed in UXT-H, UXT-h, and sir2. Only dsx-h showed a tendency to increase expression at P5.
Expression and functional roles of dsx-h and three isoforms of dsx
It is important to clarify the functional roles of dsx-H and dsx-h in the evolution of mimicry supergene. Although the involvement of dsx-H in the formation of mimetic traits has been shown, the function of dsx-h has been unclear (19, 21, 29). The hindwing patterns of non-mimetic females and males are almost the same in the bright field, but there are minute differences when observed under the UV irradiation. In non-mimetic females, the innermost and second innermost pale-yellow spots do not fluoresce, whereas in males the innermost one fluoresces slightly and the second one fluoresces completely (see Nontreated in Fig. 2, A and B). We injected siRNA of the target gene (i.e., dsx in this case) into the hindwing immediately after pupation (Day 0 of pupation: P0) and performed electroporation to cover most of the hindwing, which induces RNAi only in the target area (22, 23). When dsx was knocked down in non-mimetic females (hh), the second pale-yellow spot fluoresced, showing a similar pattern to males (Figs. 2A and S2). In males, however, there was no clear change after knockdown (Figs. 2B and S2), indicating that dsx-h maintains its original function of sexual differentiation in the hindwing as well as the mimetic pattern formation.

(A, B) Knockdown of dsx-h in the left hindwings of non-mimetic (hh) female (A) and male (B). The siRNA was injected into the left pupal hindwing immediately after pupation and electroporated into the dorsal side. Pale-yellow spots are numbered starting from the inside. The numbered spots in red indicate those whose UV response was altered by knockdown. In non-mimetic females (A), knockdown of dsx changed the second spot to produce UV fluorescence like males (hh) (B). Scale bars, 1cm. Supplementary Figure S2 show other replicates. (C) Gene expression levels of each dsx isoforms in mimetic (Hh) females. FPKM values were calculated by RNA-seq at the wandering stage (W) of the late last instar larvae, at 2 day after pupation (P2) and at 5 day after pupation (P5). Orange bars indicate the expression levels of dsx isoforms from mimetic (H) allele and gray bars indicate from non-mimetic (h) allele. F1, F2 and F3 means female isoform 1, 2 and 3, respectively. There was no statistically significant difference among isoforms. (D–F) Knockdown experiments of dsx F1, F2 and F3 in the hindwings of mimetic (Hh) females. siRNA was injected into the left pupal hindwing immediately after pupation and electroporated into the ventral side. No phenotypic changes were observed by knockdowns of dsx F1 and F2 (D and E), but knockdown of dsx F3 changed the mimetic pattern to the similar of the non-mimetic pattern (F). Red and white arrowheads represent the changed red and pale-yellow regions, respectively. Scale bars, 1 cm. Supplementary Figure S4 show other replicates. (G–I) Gene expression levels of each isoform in the knockdown wings of mimetic (Hh) females at 2 days after pupation. When F1 was knocked down, only F1 was down-regulated (G), when F2 was knocked down, only F2 was down-regulated (H), and when F3 was knocked down, only F3 was down-regulated (I). Gray and red bars shown the expression in nontreated and treated hindwings, respectively. We estimated the gene expression levels by RT-qPCR using RpL3 as the internal control. Error bars show standard error of three biological replicates. *P < 0.025 for one-tailed paired t-test. Photo Credit: Souta Shinozaki, The University of Tokyo.
In addition, dsx has three female isoforms (F1, F2, F3) both in dsx-H and dsx-h, and one isoform in dsx-H and dsx-h in males (20, 21). To investigate the function of the three isoforms in females, we performed expression analysis by RNA-seq and knockdown experiment by RNAi. There was no significant difference in the expression levels of F1, F2, and F3, but F3 showed relatively higher expression levels (Figs. 2C and S3). RNAi by in vivo electroporation showed that only the F3-specific knockdown of dsx-H changed the mimetic pattern to the similar of the non-mimetic pattern (Figs. 2, D–F and S4): red spots became smaller, and the non-mimetic specific pale-yellow spots appeared (Figs. 2F and S4). In the RNAi experiment, we confirmed that only the target isoform was down-regulated by RT-qPCR (Fig. 2, G–I). These results indicate that only isoform 3 of dsx-H has an important function in the formation of mimetic traits.
Functional analysis of UXT and U3X in the HDR
In the UXT knockdown by siRNA injection in the hindwing, the pale-yellow spots were reduced, and the shape of pale-yellow region was flattened like non-mimetic phenotype, and the red spots were reduced or disappeared (Figs. 3A and S5). Knockout of UXT by Crispr/Cas9 was also performed. guide RNA was designed to target the functional domain of UXT, the prefoldin domain (Fig. S6A), and was injected into eggs immediately after egg laying together with Cas9 protein (CP-01, PNA Bio). A total of 294 eggs were injected and 21 adults were obtained (Mimetic female: 8; nonmimetic female: 5; male: 8, Fig. S6B). Eight of the mimetic females were subjected to PCR, cloning, Sanger sequencing to confirm the introduction of mutations (Fig. S7), and phenotypic observation. The hatching rate (7.1%) of the injected individuals was very low, suggesting that UXT may have an important function in survival. Genotyping using DNA extracted from the abdomen, head and wings of emerged individuals yielded five types of sequences in which mutations were introduced (Fig. S7). We observed individuals with the mosaic knockout, in which the pale-yellow spots were flattened as in the non-mimetic form, and the red spots were reduced or disappeared (Fig. 3B). In only one individual, the pale-yellow spots were changed, but the red spots were reduced in four individuals (Fig. S8). These results indicate that UXT is involved in mimetic pattern formation in both pale-yellow and red spots. In pale-yellow spots, both dsx-H and UXT are involved in the mimetic pattern formation, but dsx-H acts to suppress the two pale-yellow spots characteristic of non-mimetic female in outer side of hindwings (Fig. 2F), whereas UXT is thought to be involved in the overall shape of the pale-yellow spots (Fig. 3A).

(A) Knockdown of UXT in the hindwings of mimetic (Hh) females. siRNA was injected into the left pupal hindwing immediately after pupation and electroporated into the ventral side. Red and white arrowheads represent the changed red and pale-yellow regions, respectively. Supplementary Figures S5 show other replicates. (B) Mosaic knockout of UXT by Crispr/Cas9. Dorsal and ventral views of one representative of the eight individuals observed are shown. Red and white arrowheads represent the changed red and pale-yellow regions, respectively. In this individual, phenotypic changes were observed mainly on the left hindwing in dorsal view. Scale bars, 1 cm. Supplementary Figure S8 shows other replicates. (C) Knockdown of U3X in the hindwings of mimetic (Hh) females. The red arrow indicates the area where the red spot has expanded, and the area circled by red line indicates the original area of pale-yellow spots. On the treated side, the area of pale-yellow spots has slightly extended. Supplementary Figures S9 show other replicates. Photo Credit: Shinichi Yoda and Yûsuke KonDo, The University of Tokyo.
In the U3X knockdown, the pale-yellow spots were extended downward (Figs. 3C and S9), and the red spots below the innermost pale-yellow spots, which were only slightly visible in the control, were enlarged (indicated by red arrow in Fig. 3C). Phenotypic changes were observed by U3X knockdown, but not simply a change from mimetic to non-mimetic phenotype. Since U3X is a non-coding RNA, the phenotypic changes observed upon knockdown of U3X may be due to changes in the expression of other genes that are regulated by U3X. The expansion of red and pale-yellow spots upon knockdown of U3X suggests the existence of genes that play a role in suppressing the excessive appearance of these spots.
The regulatory relationship and downstream genes of the three genes inside the HDR-H
Since all three genes in the HDR-H, dsx-H, U3X, and UXT, were found to be involved in the formation of mimetic patterns, we decided to examine their regulatory relationships and downstream genes. On the second day after injection (P2), siRNA un-injected hindwings (control) and injected hindwings (knockdown) were sampled for RNA extraction. The extracted RNA was subjected to RT-qPCR to confirm the decreased expression of the knocked-down gene (Fig. 4A), and RNA-seq was performed to compare the expression levels in the control and knockdown sides (the list of samples used for RNA-seq is shown in Table S2). First, we confirmed siRNA injections of dsx-H, UXT and U3X reduced the expression levels of the target genes (Fig. 4A). According to the comparative analyses, about 500 to 1500 differentially expressed genes (DEGs) were extracted as genes whose expression was decreased or increased when each gene was knocked down (Fig. 4B).

(A) Measurement of knockdown effect using RT-qPCR. We compared the expression levels of dsx-H, UXT and U3X between nontreated (grey bar) and treated hindwings (red bar) by RT-qPCR using RpL3 as an internal control. Values and error bars denote the mean and standard deviation of three biological replicates. *P < 0.025 for one-tailed paired t-test. (B) The number of differentially expressed genes (DEGs) identified by dsx-H, UXT and U3X knockdowns. (C) Venn diagram depicting the abundance of DEGs (P < 0.05) for each comparison between three genes by untreated and siRNA-treated samples and shows only the number of transcription factors and signal factors. (D) Knockdown of rn in the hindwings of mimetic (Hh) females of Papilio polytes. siRNA was injected into the left pupal hindwing immediately after pupation and electroporated into the ventral side. Knockdown of rn changed the pale-yellow spots to produce UV fluorescence. UV fluorescence is not originally seen in mimetic females. White arrowheads represent the changed pale-yellow regions by knockdown. Scale bars, 1cm. Supplementary Fig. S12 show other replicates. (E) Fold Change of FPKM values (knockdown/control sides) of genes around H locus during knockdowns of dsx-H, UXT and U3X. FPKM fold changes of dsx-H, dsx-h, UXT, U3X, prospero, sir2, and rad51 are shown. rad51 is a gene adjacent to sir2 (Fig. 1B), but its involvement in mimetic pattern formation has not been investigated to date. The value is 1 when the FPKM values of the siRNA treated and untreated sides are equal. Wald-test, paired; ***P<0.001, *P<0.05. Photo Credit: Souta Shinozaki, The University of Tokyo.
We focused on the transcription factors and signaling factors whose expression is promoted by dsx-H, UXT, and U3X (Figs. S10 and S11), and found that wnt1, wnt6, and rotund (rn) were commonly down-regulated by knockdown of dsx-H, UXT, and U3X (Fig. 4C). wnt1 and wnt6 have been reported to be involved in the mimetic pattern formation (29). When we knocked down rn, there was no characteristic change in the mimetic pattern, but there was an overall change in the color of the black, red, and pale-yellow regions, which seemed to become lighter (Figs. 4D and S12). When observed under UV irradiation in the rn knockdown wings, the UV fluorescence was observed in the pale-yellow spots (Figs. 4D and S12), where UV fluorescence is not observed usually in the mimetic form. From these observations, we consider that rn plays an important role in the pigment synthesis characteristic of the mimetic phenotype.
We next examined the expression levels of genes inside and flanking the HDR (dsx-H, dsx-h, UXT, U3X, prospero, sir2, rad51) upon knockdown of dsx-H, UXT, and U3X. These genes were not included in the DEGs described above, but because the transcriptome sequence information used for screening was incomplete for the genes inside and around the HDR (especially for dsx, incomplete transcripts containing dsx fragments and male isoforms are included.), we manually constructed transcript sequence data and mapped them again for detailed examination. UXT, U3X, prospero, sir2 and rad51 were mapped to the full length of mRNA, and dsx-H and dsx-h were mapped to the open reading frame (ORF) sequences of female isoform. When dsx-H was knocked down, the expression of dsx-H was significantly decreased, but no significant expression changes were observed in other genes (Fig. 4E). Similarly, when UXT was knocked down, the expression of UXT tended to decrease (not statistically significant), but there was no significant effect on the expression of other genes (Fig. 4E). On the other hand, notably, when U3X was knocked down, in addition to the downward trend of U3X expression (not statistically significant), the expression of dsx-H was significantly decreased and the expression of UXT was significantly increased (Fig. 4E).
Functional analysis of prospero and sir2, two proximal genes outside the HDR-H
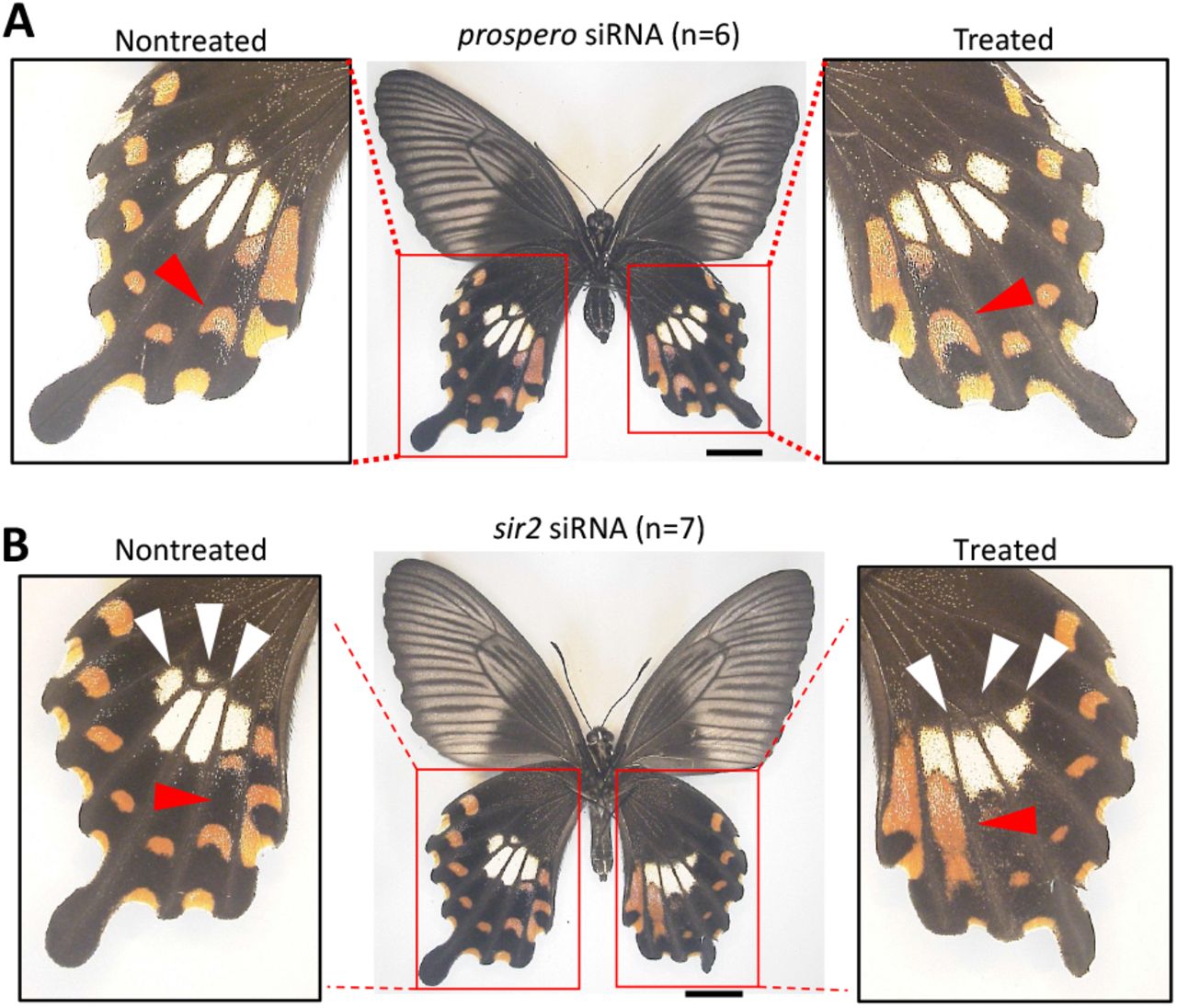
Next, the functional roles of prospero and sir2 which locate in close proximity to the HDR region but outside the inversion, were analyzed in mimetic female hindwings by in vivo electroporation mediated RNAi. In the prospero siRNA injected hindwing, there were no obvious changes, but the red spots characteristic of the mimetic form were subtly enlarged (Figs. 5A and S13). In the case of sir2 RNAi, the pale-yellow spots were flattened like non-mimetic phenotype, and the red spots were enlarged under the innermost pale-yellow spots (Figs. 5B and S14). The decreases in prospero and sir2 expressions by RNAi were confirmed by RT-qPCR, and although not statistically significant, there was a tendency for those expressions to decrease in the knockdown side (Fig. S15). These results suggest that multiple adjacent genes outside the HDR are involved in the formation of mimetic patterns.

siRNA was injected into the left pupal hindwing immediately after pupation and electroporated into the ventral side. Red and white arrowheads represent the changed red and pale-yellow regions, respectively. Scale bars, 1cm. Supplementary Figs. S13, 14 show other replicates. Photo Credit: Souta Shinozaki, The University of Tokyo.
Discussion
In this paper, using the in vivo electroporation mediated RNAi method, we show that not only dsx, but also UXT and U3X in the inversion region for the H-allele, furthermore even outside flanking genes prospero and sir2 are involved in the mimetic wing pattern formation in P. polytes (Figs. 3, 5 and 6A). The transcription factor dsx has been thought to function as a mimicry supergene as a single gene because it induces downstream genes to form the mimetic trait (20, 30). However, the present experiments indicate that multiple genes are involved in pattern formation. These genes are not downstream genes of dsx-H (Fig. S10), but are likely to function as members of the supergene. On the other hand, it is also important to note that we found that the expression of U3X may induce the expression of dsx-H (Fig. 4E). U3X is a long non-coding RNA not found in the h-allele and other genomic regions and is thought to have arisen specifically in HDR-H during evolution. U3X is located upstream of the transcription start site of dsx-H, and U3X may cis-regulates dsx-H expression. In Daphnia magna, long non-coding RNAs are also present upstream of dsx and regulate dsx function (31), which indicates that further investigating more details of the regulatory mechanism of U3X expression are necessary. The knockdown of U3X did not necessarily cause a change from the mimetic to the non-mimetic patterns, but the RNA-seq results showed that U3X also repressed the expression of UXT, suggesting that the knockdown of U3X may have had the effect of increasing the gene expression of UXT (Figs. 3C and 4E). Knockdown of UXT switches the pattern to resemble the non-mimetic phenotype, including flattening of the upper part of the pale-yellow spots in the center of the hindwing (Fig. 3A). Importantly, the mosaic knockout of UXT in Crispr/Cas9 resulted in a similar phenotypic change (Fig. 3B), and the results were consistent between the two completely different experimental methods.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) The mimicry highly diversified region (HDR) on chromosome 25 contains three genes, dsx, U3X, and UXT, and neighboring genes such as prospero, Nach-like, and sir2. It is hypothesized that dsx works as mimicry (switching) gene and that UXT, sir2 and probably prospero functions as modifier genes. U3X upregulates the dsx-H expression and represses the UXT expression, and that there may be cis-regulatory element (CRE) which influences expressions of supergene members. (B) Dsx-h switches from male to non-mimetic female and Dsx-H switches from non-mimetic to mimetic female, and genes such as UXT and sir2 may act as modifier genes to make the phenotype of the mimetic female more similar to the toxic model species. UXT and others may be involved in shaping the mimetic type of pale-yellow spots, while sir2 and others may be responsible for removing excess red spots. These modifier genes make the mimicry more like the model.
By functional analysis of dsx-h, which has been considered to have no specific function, it was shown that dsx-h induced non-mimetic patterns in females (Fig. 2A). This clearly indicates that the non-mimetic pattern in males is a default trait, and if dsx-h is expressed there, it becomes non-mimetic female, and if dsx-H is expressed there, it becomes mimetic female (Fig. 6B). This result suggests that dsx-h was originally involved in the regulation of sexual dimorphism in wing pattern, and the recombination was suppressed by an inversion, resulting in the differentiation of dsx-H and the evolution of female-limited Batesian mimicry. In butterflies, the evolution of female-limited polymorphism based on sexual dimorphism has been frequently hypothesized from evolutionary studies (30, 32). Furthermore, functional analysis suggests that only isoform 3 of dsx-H, induces a mimetic pattern among the three female isoforms in this study (Fig. 2, D–F). The expression levels of each isoform were not significantly different (Fig. 2C), suggesting that the function of isoform 3 as a protein is important for the induction of mimetic traits, rather than the regulation of expression. Dsx is a transcription factor involved in sexual differentiation, and each isoform binds to a different response element, suggesting that the downstream gene network may change among three isoforms. Iijima et al. (29) explored the downstream gene network of dsx-H for all isoforms, and it may be necessary to explore the downstream genes specific to isoform 3 of dsx-H for clarifying the mimicry mechanism in the future.
In recent years, many examples have been reported of supergenes in which complex adaptive phenotypes showing intraspecific polymorphism are regulated throughout a certain region of the chromosome (33, 34), but this study is the first to investigate the functions of multiple genes in and flanking the HDR and to show that the gene cluster adjacent to dsx work as a supergene (Fig. 6A). dsx-H is thought to switch the phenotype from a non-mimetic to a mimic phenotype, and genes such as UXT and sir2 are thought to make the mimetic phenotype more similar to the model (Fig. 6B). Genes such as dsx-H are called the mimicry gene, while those such as UXT and sir2 are called modifier genes that are fine-tuned to improve mimicry (35–38). It is predicted that the mimicry gene evolved first, and modifier genes evolved later (35–38). We hypothesize about the evolution of the mimicry supergene in P. polytes as follows. First, inversion occurred around dsx, and then dsx-H and U3X originated, and mimicry females evolved, then U3X and cis-regulatory elements in the HDR may establish a regulatory mechanism for the expression of surrounding genes, and these genes may come to act as modifier genes (Fig. 6).
On the other hand, the results of expression analysis of each gene do not clearly indicate the regulatory relationships among genes in and flanking the mimicry HDR, and whether each gene is involved in the control of mimicry pattern formation (Fig. 1, C and D). In this study, all mRNA samples were prepared from the entire hindwing, and thus if a gene is expressed in a specific region (e.g., red spot region), it may not be possible to clearly judge the functional involvement of the gene in a mimetic pattern from the expression level. The only way to solve this problem is to compare the expression of each gene by in situ hybridization. In addition, we here compared gene expression levels at only three developmental timing: W (the first stage of the prepupa), P2, and P5. In order to obtain clear results, it is necessary to continuously compare gene expression levels at a wider range of time points. Furthermore, due to technical limitations, electroporation-mediated RNAi (siRNA injection) in the wing can only be performed immediately after pupation, which may not necessarily correspond to the time when each gene is functioning. In the case of dsx-H knockdown, it is noteworthy that the mimetic pattern is switched to the non-mimetic pattern even if RNAi is performed immediately after pupation (21), suggesting that the fate of pattern formation is carried over at least to the early pupal stage. If RNAi can be applied to other stages of development, the functional role of each gene can be more clarified.
We would like to reconsider what is a supergene. Historically, it was assumed that multiple genes work together to produce more complex traits and to prevent recombination by placing genes adjacent to each other on the chromosome to avoid intermediate forms in the next generation, and such regions were defined as supergene (9– 13). In many of supergenes, chromosomal inversions are observed, and the structural diversity of multiple alleles is thought to be fixed by the inversions. And, Thompson and Jiggins (39) defined supergene as ‘a genetic architecture involving multiple linked functional genetic elements that allows switching between discrete, complex phenotypes maintained in a stable local polymorphism’. In the case of the female-limited polymorphic Batesian mimicry of P. polytes and its close relative, P. memnon, the whole genome sequence and GWAS showed that the causative region of the mimicry was a 150-kb region including dsx on chromosome 25 (25). Both species have two types of low homology sequences (HDRs) corresponding to mimetic and non-mimetic alleles, but there is an inversion between the two alleles in P. polytes, but not in P. memnon (25). It is not clear how sequence diversity arose and was maintained in P. memnon, but at least in P. memnon, the supergene cannot be defined in terms of the internal region of inversion. Then, it may be possible to define a supergene inside an HDR with low sequence homology, but is it possible to define a supergene including outer regions adjacent to an HDR with low sequence homology? This is an important question for understanding how we should think about the unit of the supergene and how the supergene has evolved.
Most supergene by an inversion contain more than a few dozen genes (some large supergenes contain more than 100 genes) (33, 34). However, there has been no evidence that multiple genes belonging to the supergene are involved in complex adaptive traits. The fact that the mimicry supergene of P. polytes is only 130 kb in size and contains only three genes in the inversion region makes it more suitable than other supergenes for answering the above questions. In addition, it is a great advantage to be able to discuss it in comparison with the supergene of a related species, P. memnon, which does not have an inversion. Further investigation of gene function around the HDR, using multiple closely related species, will reveal more details about the function and evolution of the supergene.
Our present results suggest that the unit of the mimicry supergene can be defined to include at least the external neighboring genes. The results of comparative transcriptome analysis after knockdown of dsx-H, U3X, and UXT did not show that the expressions of sir2 and prospero were not affected as downstream genes of the 3 genes, suggesting that sir2 and prospero expression is likely to be regulated by some cis-elements within HDR-H. The existence and location of cis-regulatory elements need to be investigated in the future, including possible epigenetic regulation of multiple genes in HDR-H. The significance of having related genes adjacent to each other on the chromosome should also be re-considered from the perspective of such expression regulation. For example, in the past, recombination sites may have been located further out and HDR dimorphism may have been more widespread, including adjacent sir2 and prospero. In the process of evolution, sir2 and prospero acquired functions involved in the pattern formation in addition to their original functions, and if these genes are able to work, they may not necessarily be in the recombination repression region. However, the regulation of their expressions may need to be affected by cis-regulatory elements inside the HDR-H.
Materials and Methods
Butterfly rearing
We purchased wild-caught P. polytes from Mr. Y. Irino (Okinawa, Japan) and Mr. I. Aoki (Okinawa, Japan), and obtained eggs and used for the experiment. The larvae were fed on the leaves of Citrus hassaku (Rutaceae) or on an artificial diet, and were kept at 25 °C under long-day conditions (light:dark = 16:8 h). Adults were fed on a sports drink (Calpis, Asahi. Japan).
Analysis of expression levels of genes in and flanking the HDR
In this study, we used the entire hindwing of P. polytes to analyze the expression levels of internal (U3X, UXT, dsx) and external flanking genes (Nach-like, sir2, prospero) in the HDR by RNA-seq and RT-qPCR. In addition to the published RNA-seq read data of P. polytes (BioProject ID: PRJDB2955) (21), newly sampled RNA was used for the analysis. The sample list used in the experiment is shown in Table S1. The newly added RNA-seq reads were obtained by the following procedure. The entire hindwing was sampled for RNA extraction on pupal day 2 (P2) and pupal day 5 (P5), and RNA extraction was performed using TRI reagent (Sigma) in the same manner as Nishikawa et al. (21) and Iijima et al. (29). The extracted and DNase I (TaKaRa, Japan) treated RNA was sent to Macrogen Japan Corporation for library preparation by TruSeq stranded mRNA (paired-end, 101 bp) and sequenced by Illumina platform. The obtained RNA-seq reads were quality-checked by FastQC (version 0.11.9) (40), mapped by Bowtie 2 (version 2.4.4) (41), and the number of reads was counted using SAMtools-(version 1.14) (42). Based on the number of reads, FPKM (Fragments Per Kilobase of transcript per Million mapped reads) was calculated (FPKM=number of mapped reads/gene length(bp)/total number of reads×109). For mapping, full-length mRNA sequences including UTRs were used for prospero, UXT-H, UXT-h, U3X, sir2, and Nach-like, and ORF region sequences were used for dsx-H and dsx-h. dsx-H and dsx-h were mapped to three female isoforms for female individuals and one male isoform for male individuals. Sequence information for each gene was obtained from Nishikawa et al. (21) and Iijima et al. (29).
The expression levels of U3X, UXT-H, UXT-h, dsx-H, dsx-h, sir2, and prospero were also analyzed by RT-qPCR. RNA obtained by the above method was subjected to cDNA synthesis using Verso cDNA synthesis kit (Thermo Fisher Scientific). The qPCR was performed using Power SYBR® Green Master Mix (Thermo Fisher Scientific) by QuantStudio 3 (ABI). The detailed method was followed by Iijima et al. (29). A total of 18 whole hindwing samples from 18 individuals were used, including three each of mimetic females (Hh), non-mimetic females (hh), and males (Hh) of P2 and P5. RpL3 was used as an internal standard and the primers used are shown in Table S2.
Knockout of UXT by Crispr/Cas9
A single guide RNA (sgRNA) was used to generate deletions and frameshifts within the prefoldin domain of UXT (Figure S6). A sgRNA was designed using CRISPRdirect (https://crispr.dbcls.jp), and the specificity of the sequence of sgRNA was assessed using BLAST to ensure that there were no multiple binding sites. The target sequence is shown in Figure S6a. The sgRNA template was generated by PCR amplification with forward primers encoding the T7 polymerase binding site and the sgRNA target site (Pp_UXT_F1modi, GAAATTAATACGACTCACTATAGGCCGACCAGAAGCTTCATCGTTTAAGAGCT ATGCTGGAAACAGCATAGC), and reverse primers encoding the remainder of the sgRNA sequence (sgRNA_Rmodi, AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATT TAAACTTGCTATGCTGTTTCCAGCATA), using Phusion DNA polymerase (M0530, New England Biolabs, Ipswich, MA, USA) (43). In vitro transcription was performed using the Megascript T7 Kit (Thermo Fisher Scientific) and sgRNA was purified with the MEGAclear Transcription Clean-Up Kit (AM1908, Thermo Fisher Scientific). To collect eggs for injection, host plants were provided to female butterflies and allowed to lay eggs for 1 hour. The obtained eggs were aligned on a glass slide and fixed with an instant glue Aron Alpha (Toagosei Company, Japan). The fixed eggs were disinfected with formalin for 3 min, the tip of the glass capillary was cut with a razor at an angle of 30–40°, perforated with a tungsten needle, and the capillary was injected with an injection mixture containing sgRNA (500 ng/ul) and Cas9 protein (CP-01, PNA Bio; 500 ng/ul) (injection pressure Pi 100 Pa, steady pressure Pc 40–80 Pa). Finally, the holes were sealed with Arone Alpha, placed in a Petri dish, and stored in a plastic case along with a well-moistened comfort towel. The hatched larvae were reared in the same manner as described above. The emerged adults were observed for phenotype, and parts of the head, abdomen and wings were taken for genotyping. DNA was extracted using a phenol-chloroform protocol and PCR amplified across the target sites (primers, Pp_UXT_cr_F1, ttcgtgttcaggatcaacag; Pp_UXT_cr_R1, tatttgttaactgcccgatg). PCR products were used to perform TA cloning, Sanger sequencing, and genotyping.
Functional analysis by RNAi using in vivo electroporation
siDirect (http://sidirect2.rnai.jp/) was used to design the siRNAs. The target sequences were blasted against the predicted gene sequence (BioProject: PRJDB2954) and the genome sequence (BioProject: PRJDB2954) in P. polytes to confirm that the sequences were highly specific, especially for the target genes. The designed siRNA was synthesized by FASMAC Co., Ltd. (Kanagawa, Japan). The RNA powder received was dissolved in Nuclease-Free Water (Thermo Fisher, Ambion), adjusted to 500 μM, and stored at -20°C. The sequence information of the siRNA used is listed in Table S3. A glass capillary (Narishige, GD-1 Model, Glass Capillary with Filament) was processed into a needle shape by heating it at HEATER LEVEL 66.6 using a puller (Narishige, PP-830 Model). The capillary was filled with siRNA. siRNA was adjusted to 250 μM when only one type of siRNA was used for one target gene (dsx-H, dsx-h&H, UXT, U3X, rn), and 500 μM siRNA solution was mixed in equal amounts when two types of siRNA were mixed for one target gene (prospero, sir2). The capillary was filled with siRNA and 4 μl of siRNA was injected into the left hindwing under a stereomicroscope using a microinjector (FemtoJet, eppendorf). Then, siRNA was introduced into only the positive pole side of the electrode by applying voltage (5 square pulses of 7.5 V, 280 ms width) using an electroporator (Cellproduce, electrical pulse generator CureGine). A PBS gel (20×PBS: NaCl 80g, Na2HPO4 11g, KCl 2g, K2HPO4 2g, DDW 500ml; 1% agarose) was placed on the dorsal side of the hindwing and a drop of PBS was placed on the ventral side of the hindwing. The detailed method follows that described in the previous paper (22). The pictures of all the individuals who performed the function analysis are described collectively as Supplementary figures.
Regulatory relationship of dsx-H, UXT, and U3X expression by RNAi and downstream gene screening
After sampling the hindwings of individuals with dsx-H, UXT, and U3X knockdown by RNAi in the P2 stage with the siRNA-injected side as knockdown and the non-injected side as control, total RNA was extracted and DNase I treated RNA was sent to Macrogen Japan Corporation. Libraries were prepared using TruSeq stranded mRNA (paired-end, 101 bp) and sequenced using the Illumina platform. Sample and read information are shown in Table 4. dsx-H_Control_2 and dsx-H_knockdown_2 are the read data used in a previous study (29). We first performed quality check using FastQC (Version 0.11.9) (40), and the reads were mapped to the transcript sequences of P. polytes to calculate the expression levels. The transcript sequence was obtained from NCBI, GCF_000836215.1_Ppol_1.0_rna.fna (BioProjects: PRJNA291535, PRJDB2954). Because the transcript sequence information of the genes around H locus described in GCF_000836215.1_Ppol_1.0_rna.fna was incomplete (H and h derived transcripts of dsx and UXT were confused), read mapping to the genes around H locus (prospero, UXT, U3X, dsx-H, dsx-h, sir2, rad51) was performed separately: the full-length mRNA sequences including UTRs were used for prospero, UXT, U3X, sir2, and rad51, and the ORF region sequences for dsx-H and dsx-h were used. Mapping and calculation of FPKM value were performed as described above.
In addition, R software was used to extract genes with variable expression by statistical analysis of read data, and comparison between two groups with correspondence using Wald-test of DESeq2 (version 3.14) (44) was performed. The transcription factors and signaling factors were extracted using the GO terms of the top hit amino acid sequences by Blastx against the Uniprot protein database. “Transcription factor activity” [GO:0003700], “DNA-binding transcription factor activity, RNA polymerase II-specific” [GO:0000981] as transcription factors and “signaling receptor binding” [GO:0005102], “DNA-binding transcription factor activity” [GO:0003700] as signaling factor.
Statistical analysis
Statistical analysis of the data was performed with R software (45). In the analysis of gene expression levels (Figs. 1, C and D and 2C), we explored the effects of stage and/or genotype/sex using a generalized linear model (GLM) with a normal distribution. Tukey’s post hoc tests were used to detect differences between groups using the “glht” function in the R package multcomp (46); P<0.05 was considered statistically significant. For the analysis to examine the effects of gene knockdown (Figs. 2, G–I, 4A and S15), one-tailed paired t-test was used; P<0.025 was considered statistically significant.
Funding
This work was supported by Ministry of Education, Culture, Sports, Science and Technology/Japan Society for the Promotion of Science KAKENHI (20017007, 22128005, 15H05778, 18H04880, 20H04918, 20H00474 to H.F.; 19J00715 to S. K).
Author contributions
HF conceived the study; SK, SY, YK, SS and KT conducted experiments; SK and HF wrote the paper. H F supervised this project. All authors reviewed the manuscript.
Competing interests
Authors declare that they have no competing interests.
Data and materials availability
The raw sequence data were deposited in DNA data bank of Japan (DDBJ). Accession information: transcriptome sequence accession ID, SAMD00000018646, SAMD00018647, SAMD00018649–SAMD00018657, SAMD00128718 and SAMD00128715 (the new transcriptome sequence will be deposited upon manuscript acceptance.).
Acknowledgments
We thank Drs. T. Kojima and T. Iijima for helpful comments and experimental supports.
References




