

Abstract
The neonatal central nervous system (CNS) in mammals is known to support greater regeneration than the adult CNS. Established models of neonatal CNS injuries cause significant disruption to the microenvironments needed for axon growth and guidance. This limits their ability to investigate long-distance axon growth ability. We established a novel microsurgical approach to transect developing corticospinal neuron (CSN) axons, without producing an overt spinal lesion and investigated long-distance growth ability of the developing corticospinal tract (CST) in neonatal mice. We identified, rather surprisingly, that this ability is not equivalently lost at distinct spinal levels. At postnatal day 4 (P4), while this ability is robustly maintained at thoracic T11, it is completely abolished at cervical C2. We further identify that thoraco-lumbar-projecting CSN lose growth ability at cervical C2 even while extending axons to thoraco-lumbar segments. Further, long-distance regrowth is possible even when CSN axons are displaced from their “normal” location in the dorsal funiculus to the dorsolateral funiculus, where only a minority of the CST normally traverses. The differential loss of long-distance growth at distinct spinal levels does not appear to be due to segmental differences in astrocytic or microglial activation. Our results suggest that the initial control over long-distance axon growth is differentially governed across the length of an axon by context-specific mechanisms.
Highlights Long-distance axon growth ability is not lost equally across the entire length of a developing axon.
Corticospinal tract (CST) axonal regenerative ability in the cervical spinal cord is lost even during the period of CST growth to thoracic and lumbar segments.
The segmentally distinct loss of long-distance CST regeneration during development is not due to segmental differences in astrocytic or microglial activation.
Introduction
A central scientific tenet regarding axon regeneration in the mammalian central nervous system (CNS) is that long-distance axon regenerative ability remains high during development and this ability declines with adulthood (reviewed in [1]). It is believed that this developmental decline occurs due to a reduction in the intrinsic growth capacity of adult neurons [2-5] and developmental changes in the CNS environment [1, 6]. These environmental differences include differential responses of neonatal versus adult cell types to injury that result in diminished capacity for axon growth after lesions to the adult CNS [7, 8], albeit adult CNS lesions result in multiple distinct forms of axonal responses [9, 10]. Most long-distance axon growth in the CNS occurs during embryonic life, which makes experimental manipulation difficult during this time. It therefore remains unclear whether long-distance axon growth ability is robustly maintained throughout the developmental period of axon growth and if so, whether this ability is uniformly maintained across the entire trajectory of an axon as it extends to its eventual targets. The rodent corticospinal tract (CST) extends into the spinal cord entirely postnatally and therefore provides the unique advantage of relatively easy experimental perturbation to investigate long-distance growth competence during normal developmental period of axon extension. Numerous investigations have identified that lesions to the developing CST in neonates can result in greater capacity for anatomical plasticity and also better functional outcomes than similar lesions to the adult CST [11-19].
Seminal work in rodents identified that CST lesions in neonates elicit plasticity in the trajectory of the CST, where axons are re-routed and exhibit some, albeit limited, growth ability. In all these instances, long-distance CST growth is still largely abolished or severely diminished which suggested that this long-distance axon growth ability is already lost during this time [15, 18]. However, a critical limitation of these lesion paradigms (e.g., compression, over hemisection, transection) is that they cause significant disruption to the spinal cord neuronal, glial, and vascular microenvironments, which normally play a critical role in developmental axon growth and guidance[20]. For instance, over hemisection at thoracic T8-10, even prior to the arrival of CST axons, resulted in few axons extending into the lesion and no long-distance growth [18], indicating that the normal process of developmental axon extension is largely disrupted. This therefore precludes the ability of such lesions to ask fundamental questions of whether developmentally regulated regenerative decline is underpinned by intrinsic changes in neuronal biology, by modifications in the spinal environment, or by both. To overcome this issue and better address these questions, we have established a new approach to axotomize the developing CST without causing overt spinal damage. This approach enabled precise delineation of the spatial and temporal trajectory of the loss of long-distance CST growth ability through development into maturity in the absence of measurable changes in the cellular environment of the spinal cord. Here, we show that long-distance CST growth ability is lost at distinct times at distinct spinal levels and that the developmental trajectory of this loss across the spinal cord closely parallels the normal developmental trajectory of CST growth into the spinal cord. This loss at distinct spinal segments does not correlate with differences in levels of astrocytic or microglial reactivity. Our results indicate that long-distance axon regenerative ability appears to be initially controlled by “context-specific mechanisms” that operate at precise developmental times in the appropriate location, which are either similar or identical to mechanisms controlling normal axon guidance in development. The work presented here identifies that such context-specific mechanisms represent an initial control mechanism over long-distance axon regenerative ability during normal development.
Results
Newly established microsurgical lesions enable CST axotomy with minimal damage to the spinal environment
Established experimental models of neonatal spinal injuries in rodents cause significant damage to the spinal cord, which disrupts the guidance cues that normally direct CST axons in development. This limits their ability to interrogate the competence of the CNS to support long-distance CST growth. To investigate long-distance growth competence of the CST, we therefore needed a different approach whereby we could axotomize the developing CST while leaving the spinal environment largely unperturbed. For this, we established a novel microsurgical approach, in which the developing CST is axotomized via a beveled glass micropipette vibrating at ultrasonic frequency under visual guidance provided by ultrasound-guided backscatter microscopy (Fig. 1a-e, Extended Data Video 1). These microsurgical lesions (hereby referred to as “microlesions’’) precisely cut through the dorsal funiculus and cause minimal overt damage to the surrounding spinal environment (whole mount view of the spinal cord at P35 after a P4 microlesion is shown in Fig. 1f). Immunohistochemistry for glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule 1 (Iba1) shows minimal astrocytic and microglial reactivity, respectively at the microlesion (Fig. 1g -g’’) (we perform more in-depth analyses of astrocytic and microglial reactivity; these results are described in further detail in Extended Data Figs. 3 and 4). Together, these indicate that microlesions cause minimal damage to the spinal cord.

a. Beveled micropipette used for microlesion. b. The same micropipette is seen inserted into a neonatal spinal cord ex vivo. c-d. Ultrasound images show the beveled glass micropipette prior to insertion into the spinal cord (c), after insertion into the spinal cord (d), and after the vibrating apparatus has been activated to produce the microlesion (e). f. Cervical cord from a P35 mouse after microlesion at P4 (arrow indicates microlesion site). g-g’’. Horizontal section of the same cord stained for GFAP (astrocytes) (g’) and Iba1 (microglia) (g’’) shows minimal astrocytic and microglial activation at the microlesion. Scale bars: (a, b): 250 μm; (f, g): 500 μm.
CST long-distance growth is lost at distinct times at distinct spinal levels
Using this newly established approach, we first asked whether CSN axons that are lesioned during the developmental period of axon growth into the cord are still able to maintain their regenerative competence. The CST in mice grows into the spinal cord postnatally during the first postnatal week and at postnatal day 4 (P4), pioneer CSN axons are traversing the caudal most thoracic spinal segments [21-23], which suggested that long-distance growth ability would be maintained in the caudal thoracic cord. We therefore first investigated CST extension following P4 microlesions at thoracic T11. We injected AAV-TdTomato into the cortex to anterogradely label CSN axons 1 day prior to the microlesion (Fig. 2f) and analyzed mice at P35. We quantified CSN axon extension past the microlesion in both, the dorsal funiculus (DF) (normal location for majority of CSN axons in rodents), and the dorsolateral funiculus (dLF) (where normally only a minority of CSN axons transverse) (Fig. 2g, Extended Data Fig. 1). Following a P4 microlesion at T11, we find long-distance CSN axon growth in the DF is indistinguishable from non-lesioned controls. In non-lesioned mice, 10 ± 3% of CSN axons present in the ventral medulla reach lumbar L2. Similarly, following a P4 microlesion at T11, 9 ± 2% of CSN axons reach L2 (Fig. 2 h-j). This indicates that the CST maintains robust long-distance growth competence at thoracic T11 during the period of normal axon growth at this segmental level. Further, our results also indicate that microlesions do not significantly disrupt the spinal environment and that the minimal astrocytic and microglial reactivity induced by the microlesion (further shown in Fig. 3 and Extended Data Figs. 3 and 4) does not interfere with normal developmental processes of axon growth and guidance. This finding is in contrast to previous investigations that used much more severe lesions of the postnatal thoracic cord. These previous lesions were performed prior to arrival of CSN axons, and yet the uninjured CSN axons only exhibited limited growth into the lesion and did not extend much further [18]. Our new results indicate that this limited growth ability by non-lesioned axons was largely due to disruption to spinal environments that are normally required for such growth.
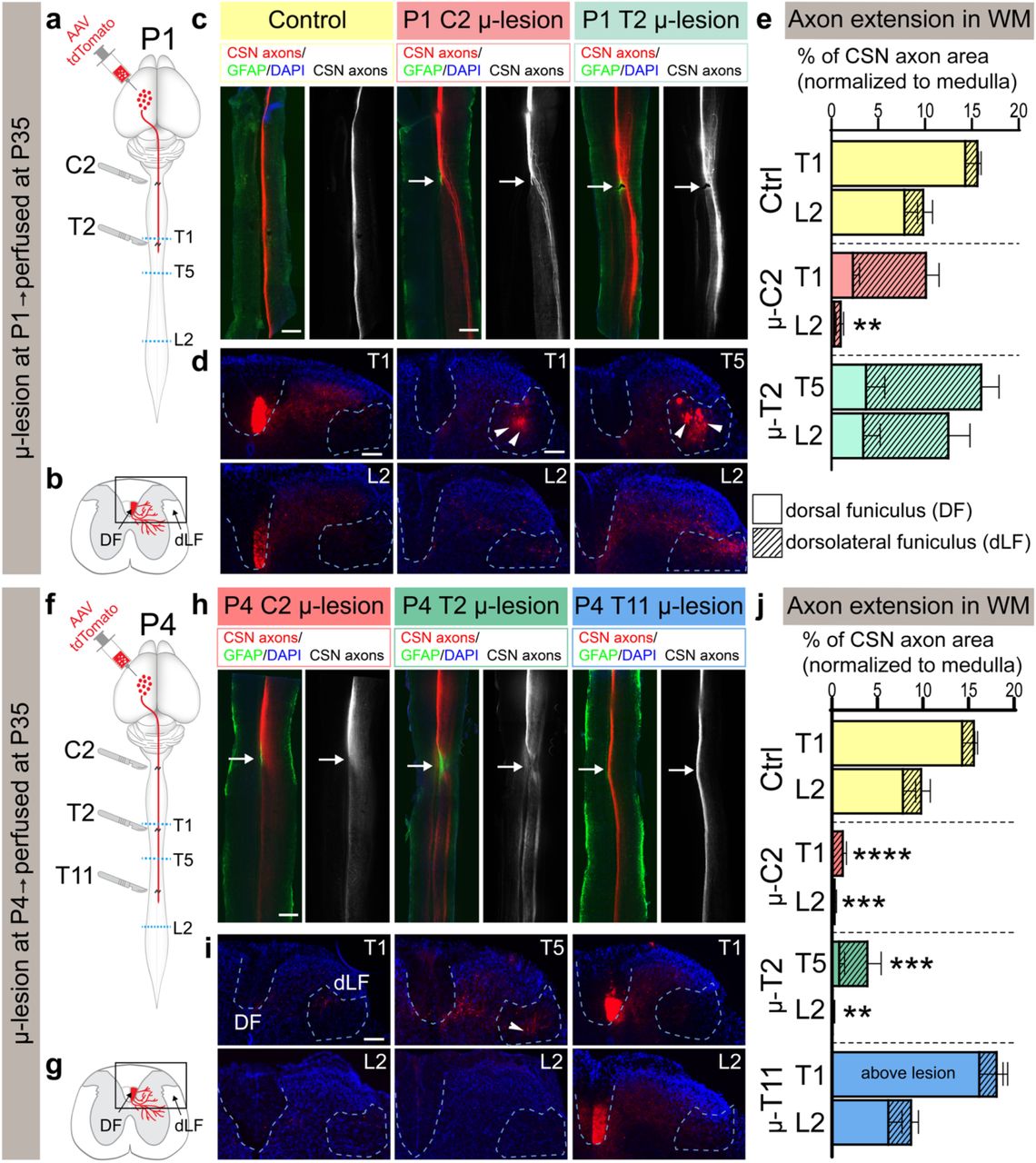
a, b, f, g. Schematics outlining experimental protocol and analysis. a, f. AAV-TdTomato was delivered into caudomedial cortex and mice underwent either a P1 or P4 microlesion at one of three distinct spinal segmental levels (C2, T2, or T11 as indicated). The segmental level where growing CST axon terminals are present at the time of the microlesion is indicated by the level of the red line in schematics. Mice were analyzed at P35. Blue dotted lines indicate the levels at which axial sections of the spinal cords were analyzed to investigate CSN axon extension. b, g. Schematics of spinal cord axial sections showing the location of dorsal funiculus (DF) and dorsolateral funiculus (dLF) where axon extension was quantified after both P1 and P4 microlesions. c, h. Horizontal spinal sections at P35 after P1 (c) and P4 (h) microlesions at distinct spinal segmental levels (as indicated). GFAP+ astrocytes (green) delineate microlesion (white arrow). CSN axons are in red (and in monochrome). d, i. Axial sections of the same spinal cords caudal to microlesion showing CSN axons either in dorsal (DF) or dorsolateral funiculus (dLF). Dotted outlines of the DF and dLF, where axon extension was quantified. Arrowheads indicate the diverted CSN axons in the dLF after P1 microlesions at C1 and T2, as well as after P4 microlesions at C2. e, j. Quantification of CSN axonal area at thoracic T1/T5 and lumbar L2. Total (DF + dLF) area of CSN axonal area is plotted; hatched bars indicate axon area quantified in dLF. For individual quantification of DF and dLF, see Extended Data Fig. 1. ** p<0.01, *** p<0.001, **** p<0.0001 control DF+dLF vs. microlesioned DF+dLF. Data are shown as mean + s.e.m.. Scalebars: c, h :500μm; d, i:100μm
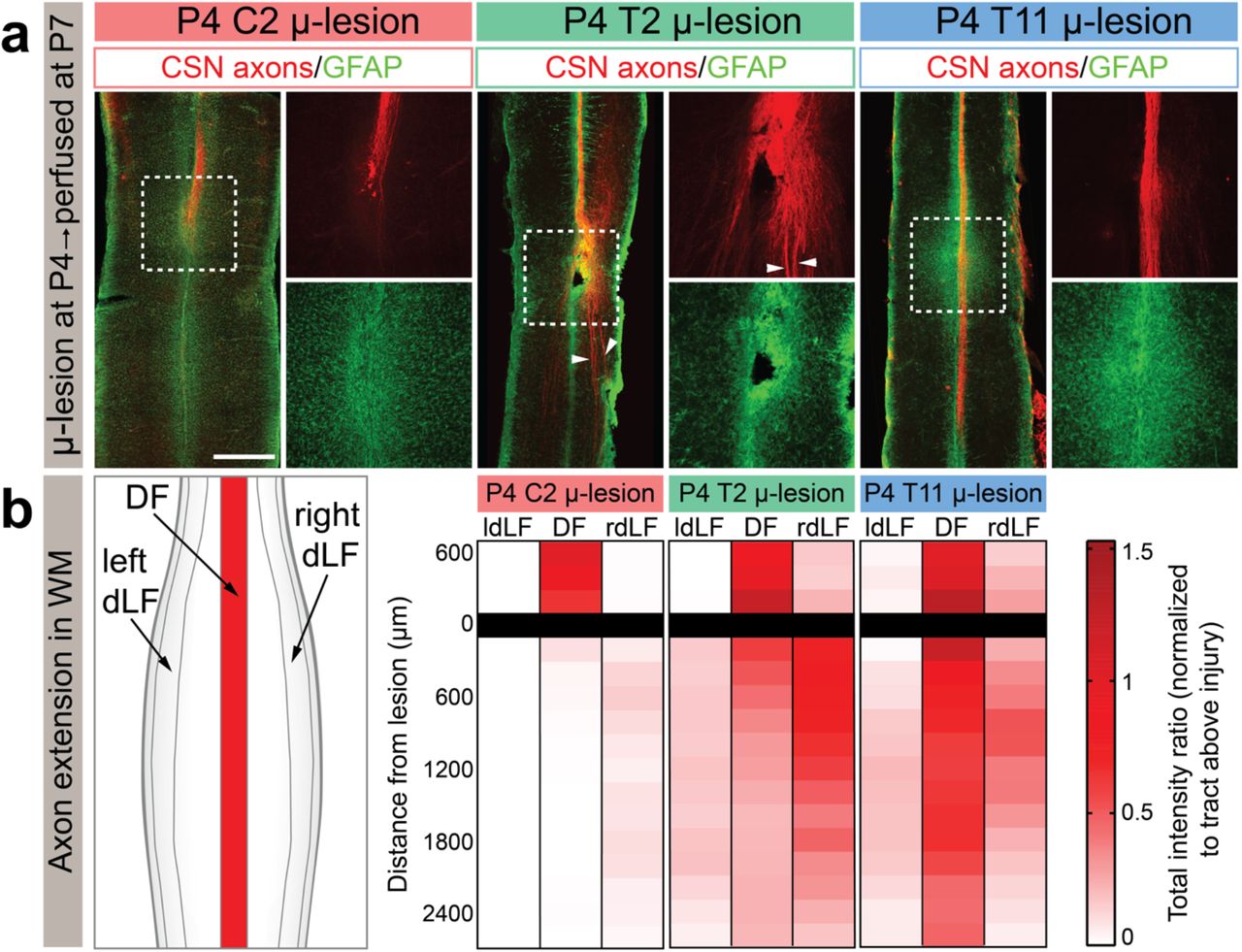
a. Horizontal sections of P7 mouse spinal cords after P4 microlesions at either C2, T2, or T11, analyzed 72 hours after microlesion. CSN axons are in red and GFAP+ astrocytes in green. Note that the segmentally distinct axon growth responses at distinct spinal levels are already evident at this acute time following the microlesions. CSN axons do not extend past C2 microlesion, but do extend past T11 microlesion. CSN axons can be seen diverted to the dLF after a T2 microlesion (arrowheads). b. Axon extension was quantified individually in either the dorsal funiculus (DF), as well as left/right dorsolateral funiculus (rdLF, ldLF, respectively). Scalebar: 500 μm.
We next performed P4 microlesions at 2 distinct spinal segments (cervical C2, and thoracic T2) and similarly analyzed CSN axon growth at P35 (Fig. 2f). These microlesion sites are located closer to the CSN soma but farther from the growing ends of the CST, with the microlesion at C2 located farther than the microlesion at T2. Given that at least some CSN are in a state of active axon growth to caudal thoracic and lumbar segments, we expected to find that the long-distance axon growth competence at these more rostral spinal segmental levels would be similarly maintained as observed at thoracic T11. Surprisingly, we find that this is not the case. After a P4 microlesion at cervical C2, there is almost complete loss of long-distance axon growth. In non-lesioned mice, 16 ± 2% of CSN axons present at the ventral medulla extend to thoracic T1 with 10 ± 3% extending to lumbar L2. In striking contrast, following a P4 microlesion at C2, only 1 ± 0.3% of CSN axons in the ventral medulla reach T1 (DF + dLF combined), and 0 ± 0.1% reach L2 (Fig. 2 h-j). This indicates a significant decline in long-distance axon growth ability, even during the period of active CST growth. When the P4 microlesion was performed at T2, i.e. closer to the growing axonal ends (the CST is growing toward caudal thoracic segments at P4 [21-23]), we observed limited long-distance axon growth (4 ± 2% of axons at ventral medulla extend to T5, but almost none extend to L2, Fig. 2 h-j); however, these CSN axons are diverted from DF to dLF (Fig. 2 h-j, Extended Data Fig. 1). This suggests that some CSN axons might maintain long-distance axon growth even when they are not extending in their normal location in the DF, although it remains possible that this minority is what would have normally extended in the dLF and were not lesioned by the microlesion. Regardless, these results indicate that long-distance CSN axon growth ability is not lost uniformly across the spinal cord. This ability also does not appear to depend on proximity to the CSN soma. Instead, growth ability appears to correlate with the normal trajectory of CST extension in the cord. When CSN axons are axotomized closer to the growing/leading edge of the developing CST, there is more robust long-distance regenerative ability as compared to when the axotomy occurs farther away from it. This suggested that the relative distance of the axotomy site from the growing ends of the CST predicts whether long-distance CSN axon growth ability remains intact.
To test this idea, we next performed microlesions at P1, at a time when growing CSN axons are present in the rostral thoracic cord [21]. We performed P1 microlesions at two distinct spinal segments (C2 and T2) and similarly analyzed axon extension at P35 (Fig. 2a). Our results indicate that after P1 microlesions at thoracic T2, there is robust long-distance CSN axon growth (16 ± 2% of axons reaching T5, and 13 ± 3 % reaching L2), which is statistically indistinguishable from non-lesioned controls. After P1 microlesions at cervical C2, 2/3 of CSN axons exhibit long-distance growth, which is distinct from P4 microlesions at this level; however, this growth does not fully extend to normal targets– 10 ± 2% of axons at the ventral medulla reach T1, which is slightly reduced from 16 ± 2% in controls, but only 1 ± 0.3% reach L2) (Fig. 2 c-e). Therefore, similar to P4 microlesions, when the axotomy is performed close to the leading edge of the CST, there is more robust axon growth. However, in both these P1 microlesion groups, the majority of CSN axons are diverted from DF to dLF (Fig. 2 c-e, Extended Data Fig. 1). This further indicates that long-distance CSN axon growth does not require CSN axons to traverse in their normal location in the DF. CSN axons that get diverted to the dLF, where only a minority of CSN axons normally traverse, are still able to fully maintain long-distance growth. However, even this “diverted” growth at C2 is abolished by P4. Together, our findings indicate that the long-distance growth ability of CSN axons is lost significantly early in development (at least by P4 at cervical C2) and this ability is not lost uniformly across the spinal cord. Further, long-distance CSN axon growth ability appears to follow the normal developmental trajectory of CST extension in the spinal cord.
Segmentally distinct CSN axonal responses are seen as early as 3 days post microlesions
The previous results were obtained at P35, i.e. more than one month after the microlesions. Therefore, there still remained the possibility that CSN axons might have initiated a growth response following a P4 microlesion at C2 that was subsequently aborted (before P35). This would give the appearance that there was no long-distance axon growth at this spinal level at P4. We therefore investigated CSN axons acutely after P4 microlesions at distinct spinal levels. We labeled CSN axons and performed P4 microlesions at the three distinct spinal levels (C2, T2, and T11) as described above, and analyzed the spinal cords 72 hours later. We find that the distinct effects on long-distance CSN axon growth at these distinct spinal levels observed at P35, are already evident at P7, 3 days after the microlesions. While axons have already fully elongated past the microlesion at T11, CSN axons have not extended past the microlesion at C2. Further, growing axons are already seen diverted to the dLF after the microlesion at T2. Notably, CSN axons can be seen growing even through a site of GFAP activation at thoracic T11 (Fig. 3a). Note that the intensity of GFAP immunoreactivity is not different between microlesions at these distinct spinal levels (Fig. 3a). We quantified axon extension in DF and dLF, and find that the observed differential effects on long-distance axon growth occur very early (within the first 72 h) after microlesions (Fig. 3b). Further, the absence of long-distance axon growth at cervical C2 that was observed at P35 following P4 microlesions is not due to an initial regenerative attempt that is then aborted.
Segmentally distinct loss of long-distance axon extension even by CSN that are in a state of active axon growth
The complete lack of long-distance CSN axon growth following P4 microlesion at cervical C2 is surprising, especially because axons from thoraco-lumbar projecting CSN (CSNTL) are still extending toward thoracic and lumbar spinal segments at P4. CSNTL are located in medial sensorimotor cortex where they reside interdigitated with cervical-projecting CSN [22]. Therefore, with AAV-mediated anterograde labeling, both cervical- and thoraco-lumbar projecting CSN are labeled. At P4, while CSNTL axons are extending to caudal levels of the spinal cord, cervical-projecting CSN are already collateralizing into the cervical spinal gray matter. Since the initiation of synapse formation is known to cause a decline in regenerative ability [24, 25], this could account for some reduction in long-distance CST growth by P4 at cervical C2. It therefore remained theoretically possible, although unlikely, that if only CSNTL axons were selectively analyzed, which are still actively extending to their targets at P4, these axons might exhibit a greater ability for long-distance growth after a P4 microlesion at C2. To address this, we utilized intersectional genetic reporter mice: Crim1CreERT2;Emx1FLPo;Ai65 (CERai65 mice, [22]). Crim1 expression prospectively identifies CSNTL [22]. In these intersectional genetic reporter mice, CSNTL axons can be visualized in the spinal cord via TdTomato expression (Fig. 4a) [22]. We used these mice to specifically investigate whether CSNTL axons exhibit greater long-distance growth ability when compared to the overall population of CSN axons arising from medial sensorimotor cortex. We performed P4 microlesions in CERai65 mice at both cervical C2 and thoracic T11 and analyzed axon growth as described above. Interestingly, while long-distance CSNTL axon growth is completely intact following a P4 microlesion at thoracic T11; however, long-distance CSNTL axon growth is completely absent after P4 microlesion at cervical C2 (Fig. 4 b-c). These results are nearly identical to our previous findings using AAV-mediated anterograde labeling. Thus, CSNTL axons lose their long-distance growth ability at cervical C2 even when they are extending toward caudal thoracic and lumbar spinal segments; this growth ability remains fully intact at thoracic T11.

a. In Crim1CreERT2;Emx1FlpO;Ai65 triple transgenic (CERai65) mice, only Crim1+ cortical neurons are labeled via TdTomato expression. Crim1 expression identifies thoraco-lumbar projecting CSN (CSNTL)[22]. P4 microlesions were performed at either C2 or T11 (as indicated by scalpels in the schematics). b. Horizontal spinal sections after P4 microlesion at either cervical C2 or thoracic T11 at P4. CSNTL axons are in red (and in monochrome). White arrow indicates the microlesion site. Minimal astrocytic activation is seen at the microlesion (GFAP, green). c. Quantification of CSNTL axonal area extending to thoracic T1 and lumbar L2, normalized to axonal area in the ventral medulla. DF – dorsal funiculus, dLF – dorsolateral funiculus. Data are shown as mean + s.e.m.. ** p<0.01, control DF+dLF vs. microlesioned DF+dLF. Scalebar: 500 μm.
Segmentally distinct loss of long-distance axon extension is not due to segmental differences in astrocytic and microglial activation or in levels of myelin
We investigated whether these segmentally distinct responses of long-distance CSN axon growth at distinct spinal levels are due to segmental differences in numbers of astrocytes, oligodendrocytes, or microglia in the spinal white matter. We therefore analyzed P4 spinal cords in non-lesioned mice at cervical C2, thoracic T2, and thoracic T11, using immunohistochemistry— GFAP (astrocytes), MBP (oligodendrocytes), and Iba1 (microglia). We find no overt differences between these distinct spinal segments in the dorsal funiculus (Extended Data Fig. 2). We next investigated whether the segmentally distinct responses could be accounted for by differences in levels of astrocytic or microglial reactivity in response to microlesions. Although microlesions do not result in the formation of a lesion core, there is still some, albeit minimal, astrocyte reactivity. It therefore remained possible that there might be segmental differences in levels of astrocytic activation that could cause the distinct effects on long-distance axon growth at distinct spinal levels. We therefore quantified the astrocytic and microglial response after microlesions at distinct spinal levels using GFAP and Iba1 immunohistochemistry, respectively. Our results show that the intensity of the GFAP at the microlesions does not differ between the different groups at P35 (Extended Data Fig. 3 a,e). We also measured the area of increased GFAP+ reactivity in the spinal cord at P35. These data showed that area of GFAP reactivity at microlesions does not differ between all the microlesioned groups (P1 C2, P1 T2, P4 C2, P4 T2, and P4 T11 microlesions); the increased GFAP reactivity extends ∼800 μm rostrocaudally and ∼300 μm mediolaterally across all groups (Extended Data Fig. 3 c-d). Further, there is also no difference in the volume of the microlesions (3D volume of increased GFAP+ immunoreactivity as compared to baseline; detailed in Methods) between all the microlesioned groups (Extended Data Fig. 3f). 3D volumetric reconstructions clearly highlight that the volume of increased GFAP immunoreactivity is equivalent across all P4 microlesion groups, even though CSN axons show segmentally distinct growth responses– there is robust long-distance growth through the P4 T11 microlesion, partial, diverted growth through the P4 T2 microlesion, and no growth through the P4 C2 microlesion (Extended Data Fig. 3g). Similarly, microglial reactivity, identified using Iba1 immunohistochemistry, was not different between the different groups at P35 (Extended Data Fig. 4).
The above analyses were performed ∼5 weeks after microlesions (at P35). However, the segmental differences in long-distance axon growth are evident by 72 h after microlesions. We therefore also performed unbiased transcriptomics of the dorsal spinal cord 72 h after microlesions at two distinct spinal levels - cervical C2 and thoracic T11. We chose these segmental levels since CSN axons exhibit a diametrically opposite response following microlesions at these two levels. We extracted RNA from the dorsal spinal cord 3 days after P4 microlesions at cervical C2 and thoracic T11 and performed RNA-sequencing; non-lesioned P7 littermates served as controls (Fig. 5a). Multi-dimensional scaling plots highlight that molecular differences between the spinal segmental levels (including non-lesioned and micro-lesioned mice) appear to be greater than the molecular effects of the microlesions (Fig. 5b). Hox genes are expressed at the appropriate spinal levels—HoxA5 and HoxC5 highly expressed at C2 vs. T11, while HoxA9 and HoxC9 are specifically expressed at T11 and not C2 [26-28]. This segment level-specific expression is also maintained after microlesions suggesting preservation of spinal microenvironments (Fig. 5c). We also find no difference in levels of GFAP (astrocytes), MBP (myelin basic protein), myelin associated glycoprotein (Mag), or TMEM119 (microglia) between the two segments in non-lesioned mice (Fig. 5d), confirming and extending our earlier immunohistochemical findings at P4 (Extended Data Fig. 2). Further, a comparison of non-lesioned and microlesioned mice shows that there is a similar fold-increase in levels of GFAP (∼30%), and TMEM 119 (∼20%) at both C2 and T11 in microlesioned mice compared to non-lesioned controls. This further indicates comparable astrocytic and microglial activation following microlesions at these two segments. We also analyzed microglial genes that are now known to promote scar-free healing in the neonatal spinal cord [7] to investigate whether these genes are differentially activated following microlesions at C2 vs. T11. We similarly find no segmental difference in the levels of any of these genes (Fig. 5e). These data further indicate that the distinct responses of long-distance CSN axon growth ability are not due to differences in astrocytic or microglial activation or segmental differences in levels of myelination.

a. After P4 microlesion at C2, or T11, the dorsal spinal cord was dissected at P7 and used for RNA-Seq; P7 non-lesioned mice served as controls. b. Multi-dimensional scaling (MDS) plot shows that segmental differences vs. response to microlesions can be dissociated across different principal components – PC1 distinguishes lesion responses while PC2 discerns segmental differences. Note that the microlesioned groups cluster closer to their segmentally matched non-lesioned counterparts. c. Hox gene expression in non-lesioned (yellow) vs. lesioned groups at C2 (pink) and T11 levels (blue) confirm spinal segment specificity. Segmental specificity of appropriate Hox gene expression is unaltered after microlesions. d. Expression of Gfap (reactive astrocytes), Tmem119 (microglia), Mbp (myelin basic protein) and Mag (myelin associated glycoprotein). A slight increase in Gfap and TMEM119 after microlesions at both levels indicates some astrocytic and microglial activation. However, there are no differences between the two spinal segments in either non-lesioned mice or after microlesions. e. Expression of microglial genes previously identified by Li, et. al., Nature 2020 [7] after P4 microlesions at C2 vs T11. Cd68 – activated macrophages or microglia, Spp1 – activated microglia, P2ry12 – homeostatic microglia, Ms4a7 – embryonic microglia. All expression data are FPKM and presented as mean ± s.e.m..
Discussion
Regenerative ability in the nervous system has been lost several times throughout vertebrate evolution [29]. Regeneration, when applied to an injured axon refers to re-growth of a transected axon, which would be an essential way to restore function after injuries to the adult CNS[10]. True and complete regeneration would require a transected axon to regrow after injury to its previous target(s) to reestablish original circuitry prior to injury. In this regard, long-distance axon growth of an injured axon would be critical to re-establish such connectivity[30]. Prior work in the field had identified that lesions to the neonatal mammalian CNS resulted in some anatomical plasticity whereby some injured axons are either re-routed to a different trajectory or exhibit some regrowth into the lesion; in all instances, there is very limited long-distance regrowth. However, these previous investigations used lesion paradigms that disrupt mechanisms that are expressed in an appropriate context to direct normal long-distance axon growth in development. Therefore, these previous investigations were not able to precisely delineate long-distance axon growth ability. The microlesions used in this work do not produce measurable changes in the spinal microenvironment. As a result, these microlesions enable more precise delineation of long-distance axon growth ability during development.
It was previously understood that regenerative ability declines when developmental axon growth is complete and the expression of axon growth genes that are normally highly expressed in development is downregulated in adult neurons. Misexpression of these developmental genes in adult neurons, without manipulation of the lesion environment, has been shown to increase regenerative ability of adult CSN (e.g., mTOR [31-33], Klf7 [34], Sox11 [35]). Further, the termination of axon growth program is followed by the formation of synapses, which also leads to a decline in regenerative ability [24]. However, to our surprise, we find that at P4, when at least a subset of CSN (CSNTL) are still in the developmental phase of axon extension to the thoraco-lumbar spinal cord, they are still unable to mount long-distance axonal regrowth after microlesions at cervical C2. Known developmental axon growth genes (e.g., Gap43, Cap23, Scg10, Atf3, and c-jun) are still highly expressed by CSN at this time; high expression levels by CSN are maintained at least until P14 [36]. This indicates that high-level expression of these genes is still not sufficient to direct long-distance regrowth of lesioned axons in the spinal white matter. In contrast, P4 microlesions at T11 showed robust long-distance axon growth to spinal levels caudal to the microlesion. At P4, only pioneer CSNTL axons have arrived at thoracic T11, with ∼20% of CSN axons at thoracic T1 extending to this caudal thoracic level [23]. We do not find any reduction in axon extension to the spinal levels caudal to the P4 microlesion at T11, suggesting that these pioneer axons were able to re-grow after the microlesion. In addition, this group of animals clearly showed that the environment at the microlesion, including activated astrocytes, did not prevent late-arriving, non-lesioned axons from growing through the microlesion in their appropriate position in the spinal white matter (Fig. 2). This is in striking contrast to previous models of neonatal spinal injuries. For instance, over hemisections of the rat thoracic spinal cord resulted in minimal CSN axon growth into the lesion and not much further [18]. Since these lesions were performed even before the arrival of the CST at this spinal level, this indicates that the lack of long-distance growth in these instances was largely because of disruption of the normal guidance mechanisms in the spinal cord. Our results therefore strongly indicate that in contrast to these previous neonatal injury paradigms, microlesions sufficiently preserve the spinal environment such that it is not inhibitory or growth-limiting to these late-arriving, non-lesioned axons. Astrocytic activation after more disruptive CNS lesions are known to be required for reestablishment of tissue homeostasis [37, 38]. Given the minimal disruption caused by the microlesions, it appears consistent that we observe a very minimal astrocytic response, which is diminished even when compared to other models of neonatal spinal cord lesions [7].
The segmental differences in long-distance axon growth ability likely reflect a combination of mechanisms. Prior work using in vitro cultures of crushed neonatal spinal cords in the opossum indicates that there are segmental differences between cervical and lumbar segments that correlate with a rostral to caudal gradient of spinal cord development [39]. These differences in regeneration were attributed to differences in levels of environmental inhibitors in the spinal cord that are known to increase with maturation [13]. Our in vivo experiments, however, indicate that these distinct effects on long-distance axon growth at distinct spinal levels are not due to segmental differences in levels of astrocytic, microglial activation, or due to differences in myelin levels. This is also corroborated by both immunohistochemical and transcriptomic analyses. While it remains possible that there might be distinct responses by spinal cell types to microlesions at distinct spinal levels, RNA-seq analyses indicate that molecular differences between distinct spinal segments, even in non-lesioned mice, are greater than the effects of the microlesions themselves (Fig. 5b). These data further corroborate that microlesions cause minimal effects on the spinal environment. The molecular differences between C2 and T11 in non-lesioned mice likely represent axon guidance mechanisms that are responsible for guiding CSN axons to their appropriate segmental targets. These mechanisms likely also control long-distance regrowth ability. Manipulating such guidance mechanisms might help elucidate the contribution of these pathways to the initial control over long-distance regenerative ability.
An implicit corollary to this idea would be that CSN-intrinsic mechanisms that normally control CSN axon growth at distinct spinal levels might also control the responses of axotomized CSN axons at distinct spinal levels. CSN projecting to distinct spinal levels express distinct genes that control their segmentally distinct axon extension [22, 23]. It is tempting to speculate that the molecules that direct CSN axon extension to appropriate spinal segments would also control regrowth ability at these distinct levels. In line with this idea, we find that when CSN axons are lesioned close to the spinal level where they are normally growing (T11 at P4, T2 at P1), we find robust long-distance growth ability in the white matter. However, when the axons are lesioned at a distant site from the growing ends (P4 microlesion at C2), they show complete loss of long-distance axon regrowth. i.e. when the segmentally-appropriate axon growth mechanisms are “in sync” with where the microlesion occurs, there is robust axon regrowth. In contrast, microlesions that disrupt this developmental synchrony result in no long-distance regrowth. There is prior evidence for such “chronicity” of neuronal intrinsic mechanisms controlling regenerative ability in the developing chick CNS [40]. In these experiments, heterochronicity of neuronal transplants show that intrinsic developmental changes in hindbrain projection neurons are more critical than environmental changes in the spinal cord in controlling the developmental decline in regenerative ability of these projection neurons. Therefore, normal control mechanisms that function to direct axon growth and guidance during development might also control long-distance regenerative ability. These “context appropriate mechanisms” will include developmental stage-specific gene expression in CSN soma, appropriate and specific molecules being localized to CSN axon growth cones at the correct developmental time, and expression of specific guidance cues under spatial and temporal control in the spinal cord.
It is also possible that microlesions at distinct spinal levels elicit distinct molecular responses in CSN depending on the segmental level, which could underlie the distinct responses of the lesioned axons. It is likely that a combination of all such mechanisms will control the developmental loss of long-distance regenerative ability and future investigations will be required to delineate their respective contributions.
Following P1 microlesions at thoracic T2, we find CSN axons maintain robust long-distance growth ability, albeit in the dorso-lateral funiculus. This indicates that the signals in the spinal cord that direct long-distance CSN axon growth are likely expressed more broadly than a confined location in the dorsal funiculus. This means that the dorsolateral funiculus is, in fact, capable of supporting long-distance growth of the majority of CSN axons at least beyond thoracic T2. This suggests largely overlapping or shared molecular mechanisms for long-distance axon growth in dorsal versus dorsolateral funiculus. This finding appears consistent with the fact that CSN that normally extend axons within the dorsal funiculus versus those that extend outside it, share similar cortical locations [41]. It remains unclear, however, whether these redirected axons after a thoracic T2 microlesion still form appropriate connectivity with their targets in the caudal thoracic and lumbar cord. Future investigations will be needed to elucidate this question.
Interestingly, even though long-distance axon growth does not occur following a P4 microlesion at cervical C2, we do observe extensive axonal sprouting into the spinal gray matter that extend significantly further caudally into the cervical cord and largely do not extend into thoracic segments. Regenerative sprouting is applied to growth arising from a lesioned axon, which does appear to apply in this case. It is tempting to speculate that the absence of context-appropriate growth mechanisms results in loss of long-distance axon growth, but that the axon growth mechanisms discussed above, might be sufficient to produce such sprouting.
Finally, in this work, we specifically analyzed the long-distance regenerative ability of the CST, but it is tempting to speculate that similar, segmentally-distinct mechanisms might also function to regulate long-distance growth of other descending, spinal-projecting pathways, e.g., rubrospinal and reticulospinal pathways. This will require experimental manipulation at earlier developmental times since their developmental axon growth into the spinal cord occurs in utero in rodents.
Together, our data delineate a precise timeline of the decline in long-distance CST regenerative ability during development. Our results identify that this decline can occur even when neurons are in a state of developmental axon growth, and even prior to synapse formation. This suggests that there are additional steps that control the decline of long-distance regeneration through development into maturity in a context-specific manner. Future investigations into the mechanistic underpinnings of this initial control over regenerative ability are likely to identify novel molecular substrates for corticospinal regeneration and repair following adult injury.
Author Contributions
C.R., and V.S. designed research; C.R., J.K., and P.P. performed research; C.R., J.K., and F.S. analyzed data; and C.R., and V.S. wrote and edited the manuscript.
Methods
Mice
All mouse studies were approved by the Weill Cornell Medical College Institutional animal care and use committee, and were performed in accordance with institutional and federal guidelines. Wild-type mice on a CD1 background were obtained from Charles River Laboratories (Wilmington, MA). The day of birth was designated as P0. We obtained Crim1CreERT2:Emx1-IRES-Flpo:ai65 (CERai65) triple transgenic mice from Prof. Jeffrey D. Macklis where they were previously generated by crossing Crim1GCE/+, Emx1-IRES-FlpO/+, and ai65 (RCFLtdT)/+ mice [22]. CreERT2 activity was induced as previously described whereby P3 mouse pups were injected intraperitoneally with 100 μl of tamoxifen (Sigma, T5648) solution (3.5 mg/ml) dissolved in corn oil (Sigma, C8267).
Anterograde labeling of CST
For anterograde labeling of the CST, pAAV1-CAG-tdTomato (Addgene) particles (2× 10^13 GC/ml) were injected a day before the lesion (at P0 or P3) into the cortex as previously described [22, 23]. Briefly, pups were anesthetized using hypothermia for 2-3 minutes and AAV was injected unilaterally under the guidance of ultrasound backscatter microscopy (Vevo 770; VisualSonics, Toronto, Canada) via a pulled glass micropipette attached to a nanojector (Nanoject II, Drummond Scientific, Broomall, PA). For mice analyzed at P35, 7× 23 nl (total volume of 161 nl) was injected, while mice analyzed at 72 h post microlesion were injected with 21 × 23 nl (total volume 483 nl). After the injections, the pups were placed on a heating pad for recovery and returned to the dam soon after.
Microsurgical CST lesions
Pups were anesthetized by hypothermia at P1 or P4 for 2-3 minutes. For performing microlesions, a beveled (30°) glass micropipette (tip ∼150 μm) was used. This was attached to a high-frequency vibrating apparatus (Oral B PRO 1000 electronic toothbrush, 8800 oscillations/minute, 20000 pulsation/minute). We measured the maximal displacement of the micropipette when attached to this apparatus and identified it to be 400 μm. The pup was placed on its side, the dorsal side oriented towards the micropipette. The micropipette was inserted into the spinal cord at the correct segmental level (C2, T2, or T11) at the spinal midline until the central canal, under visual guidance via ultrasound-guided backscatter microscopy. Once the micropipette was in position, the vibration on the apparatus was turned on causing vibrations of the micropipette with a displacement of up to 400 μm. During the next 10 seconds, the pipette was slowly pulled out of the spinal cord with brief pauses every 2 seconds to completely axotomize the CST (ultrasound video shown in Extended Data Video 1). The 10 sec period was determined as the requisite minimum period to axotomize the CST (and largely similar to the 10 sec long crush lesions that were performed in neonatal opossums [13]. After the microlesion, pups were placed on a heating pad for recovery and returned to the dam. Since the lesion is barely visible on the pup’s body, the dams continued to take care of the pups without needing any additional care or intervention.
Tissue collection and sectioning
At the experimental endpoints (72 h after microlesion for acute time points and at P35 for chronic time points), mice were transcardially perfused with cold PBS, followed by 4% paraformaldehyde (PFA) in PBS. Brains and spinal cords were carefully dissected and post-fixed in 4% PFA overnight. Prior to processing, wholemount images of the brain and spinal cord were acquired using a fluorescence stereomicroscope (Nikon SMZ18). This was to ensure that the AAV injections in all the mice were well matched. The following blocks of tissue from each mouse were collected separately: 1) spinal cords containing the microlesion site (C1-C8 for C2 lesioned mice; C5-T4 for T2 lesioned mice; T6-L1 for T11 lesioned mice), 2) medulla for all groups, 3) tissue blocks for axial sections – T1 for C2 and T11-lesioned mice; T5 for T2-lesioned mice; L2 for all groups; as well as 3) brains. These samples were cryoprotected separately for each mouse in 30% sucrose in 1xPBS overnight, and frozen in OCT (Tissue Tek, Sakura Finetek). All tissue blocks were cut using a cryostat (Leica CM3050S) at 50 µm (brains and spinal cord axials) or 70 µm (spinal cord horizontals) thickness and collected as free-floating sections in 1xPBS. For horizontal sections of the spinal cord, each and every section was collected, stained, and mounted serially (dorsal to ventral). These were used to reconstruct the microlesion and analyze the placement of the microlesion site in the cord (described in greater detail below). Since serial sections were used, we were therefore able to reliably and reproducibly reconstruct the microlesion volume across the entire dorsoventral extent of the spinal cord.
Immunohistochemistry
Non-specific binding was blocked by incubating tissue sections in 0.3% BSA (Sigma, A3059-100G) in 1x PBS with 0.3% Triton X-100. Primary antibodies were incubated in this blocking solution overnight at 4°C. The following primary antibodies and dilutions were used: mouse anti-GFAP (1:500, Sigma-Aldrich), rabbit anti-Iba1 (1:500, Fujifilm Wako), rat anti-MBP (1:500, Millipore), and rabbit anti-RFP (1:500, Rockland Immunochemicals). Next, sections were incubated with appropriate Alexa Fluor secondary antibodies for 3 h at room temperature (1:250, Invitrogen). Sections were mounted on glass slides and coverslipped using DAPI-Fluoromount-G (VWR).
Exclusion of animals from analysis
Given the small size of microlesions, we undertook stringent analyses to ensure that the site of the microlesion occurred in the correct location in the dorsal funiculus. A proper site was identified using GFAP immunohistochemistry to visualize the astrocytic reaction. Mice where the microlesion site was either off the midline, too ventral, or more than a segment off from the intended segmental level, were excluded. In addition, mice where AAV injections were off from the target area (caudomedial cortex) or noticeably weaker than others, were also excluded since reliable CSN axon extension analysis in these animals was not possible.
Quantification of axon extension in the spinal cord
In mice analyzed at chronic timepoints (endpoint P35), axon extension in the spinal cord was quantified using 20X images of axial sections of the spinal cord taken on a Leica SP8 confocal microscope (Leica SP8) with 2 µm step z-stacks. Image analysis was done using NIH Fiji and a semi-automated macro. 3 sections were analyzed per mouse and the 3 brightest images from each z-stack were used for analysis. Briefly, an area containing the CST (tdTomato+) was selected as a region of interest (ROI) (pyramid in the medulla, ventral part of the dorsal funiculus, and dorsolateral funiculus in thoracic and lumbar axials), followed by manual thresholding of signal vs. background, and measurement of “Total Area of thresholded pixels” in these ROIs. Measurements at thoracic and lumbar levels were normalized to measurement at the ventral medulla and presented as a percentage for each mouse.
In mice analyzed at acute time points (72 h post-lesion), axon extension was quantified from 5x images of serial horizontal spinal cord sections containing the microlesion site, imaged using Zeiss Axioimager M2 epifluorescence microscope. All sections containing labeled CST axons were used to quantify the tract in the dorsal funiculus and the dorsolateral funiculus. All analyses were performed using NIH Fiji and a semi-automated macro. In brief, dorsal and dorsolateral funiculi were outlined on DAPI-images (DAPI signal distinguishes spinal gray from spinal white matter). These outlines were converted into masks and the TdTomato signal was analyzed within these masks. Every horizontal section of the cord containing the CST was then binned rostrocaudally into bins of 2000 × 200 um (W x H) extending ∼600 μm rostral, and ∼2600 μm caudal to the microlesion site. TdTomato+ pixels were manually thresholded to distinguish signal from the background, and the number of thresholded pixels was measured in each bin for each ROI (i.e. 1 value for the dorsal funiculus and one value for each dorsolateral funiculus). The thresholded pixels in the corresponding areas and bins from each horizontal section were summed to represent the total number of CST axons present in the spinal white matter both rostral and caudal to the microlesion site for each mouse. Axon intensity bin values for individual mice were then normalized to the average CST intensity for each mouse rostral to the lesion site. Data were then presented as heatmaps of averages for each group (as shown in Fig. 3b).
Quantification of astrocytic and microglial reactivity
GFAP immunohistochemistry was used to identify astrocytic activation. All analyses quantifying GFAP were performed on 5x epifluorescence images of serial horizontal sections of the spinal cord taken using a Zeiss Axioimager M2 epifluorescence microscope. GFAP intensity at the microlesion was analyzed using NIH Fiji software. 3 sections containing the CST were chosen for each animal. The microlesion area, identifiable by increased GFAP immunoreactivity as compared to sites distant from the microlesion, was outlined and defined as the ROI. The raw intensity density (sum of the intensity values of each pixel) in each ROI was measured. We next measured GFAP intensity of an identical ROI on the same image rostral and caudal to the microlesion (baseline value), at a similar location along the midline. The intensity at the microlesion was then divided by the baseline value and results are presented as a normalized GFAP intensity ratio for each mouse analyzed (as shown in Extended Data Fig. 3e).
Next, GFAP volume was measured using Neurolucida software (MBF Bioscience, version 2010.1.3). The lesion area (identified by increased GFAP immunoreactivity) was outlined in every section of the spinal cord. The dorsoventral position of each horizontal section was assigned a z-value, with adjoining sections being separated by 70 μm. Since we collected serial sections for each mouse, we could then align all sections to generate a 3D model using Neurolucida (as shown in Extended Data Fig. 3g). Neurolucida Explorer was then used to measure the full volume of the lesion area (in µm3).
Lastly, the signal distribution of GFAP (activated astrocytes) and Iba1 (reactive microglia) relative to the distance from the microlesion was quantified using a semi-automated macro written for NIH Fiji. The same sections were analyzed that were used for the GFAP intensity analysis described above. 30 ROI bins of 200 µm x 3000 µm (H x W) were drawn horizontally (rostral to caudal), covering the full width of the spinal cord using NIH Fiji. Similarly, 16 bins of 100 µm x 2000 µm (W x H) were drawn vertically (from left to right) to cover the microlesion site. 3 spinal cord sections from each mouse that contained the CST were used for this analysis. In these sections, the spinal cord outline was traced to create a mask such that we only analyzed pixels that were located inside the section, excluding the background. Then, bins of ROIs (both horizontal and vertical) were centered at the microlesion and images were manually thresholded for both GFAP and Iba1 to distinguish signal from background. For each bin, the fraction of the total area that is occupied by positive, thresholded pixels was measured to generate distribution plots as shown in Extended Data Figs. 3 and 4.
Transcriptomics via RNA Sequencing
In CD1 mouse pups, the dorsal spinal cord (to include both dorsal and dorsolateral funiculus) was dissected from C2, and T11 spinal segments, from both non-lesioned (at P7) and lesioned (72 h after P4 C2, and P4 T11 microlesions) pups (schematized in Fig. 5a). RNA was extracted using Direct-zol RNA MicroPrep kit (Zymo). RNA concentration was measured on Qubit and quality was confirmed using Bioanalyzer 2100 (Agilent Technologies) Samples were treated with TURBO DNAse to remove genomic DNA contamination, and rRNA depletion was done using QIAGEN FastSelect rRNA HMR Kit (Genewiz). For library preparation, NEBNext Ultra II RNA Library Preparation Kit for Illumina was used (Genewiz). Next, samples were sequenced on Illumina HiSeq (2×150bp configuration, single index, per lane) by Genewiz. Raw sequence reads were mapped to mouse mm10 genome using STAR aligner (v2.7.5c). Read counts are determined using HT-seq (v0.11.1) with mouse mm10 refSeq gene model as reference. Raw count matrix was filtered for low expressed genes followed by VSD normalization.
Statistical analysis
For the comparison of multiple groups, we used one-way ANOVA followed by Tukey’s posthoc test where microlesion groups were compared to the control, non-lesioned mice. All statistical tests were performed and graphic presentations obtained using Graphpad Prism 9.2. A p-value of <0.05 was considered statistically significant. No statistical methods were used to pre-determine sample sizes.
Extended Data Figures and Videos

Quantification of CSN axon extension in the spinal white matter after distinct microlesions. Thresholded area of CSN axons at thoracic T1/T5 and lumbar L2 (normalized to total area in ventral medulla) is shown separately for the dorsal and dorsolateral funiculus. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 injured DF/dLF vs. control DF/dLF. Data are presented as mean + SEM. Each dot represents a separate mouse.

a. Schematic on the left showing the spinal locations of the transverse sections taken from P4 non-lesioned mice. b. Transverse sections were stained for astrocytes (GFAP, green), myelin (MBP, blue) and microglia (Iba1, red). Full transverse sections from C2, T2 and T11 are shown with the insets for dorsal funiculus closeup. No overt differences are seen between the segments. Scalebar: 200 μm.

a. Representative images of GFAP immunohistochemistry (astrocytic response) on horizontal sections of the mouse spinal cord at P35. Magnified views of the microlesion site are shown following either P1, or P4 microlesions at the distinct spinal levels indicated. b. Schematic showing the spinal cord locations for images shown in a. c-d. Quantification of GFAP signal distribution relative to the distance from the microlesion was quantified in 200 μm horizontal bins (rostral to caudal) (c) and 100 μm vertical bins (left to right) (d). Data are shown as mean ± SEM. There is no difference in the extent of GFAP activation between the multiple, distinct groups. e. Quantification of GFAP intensity at the microlesion site across the different microlesioned groups. f. Quantification of lesion site volume (3D volume of GFAP+ area) across the different microlesioned groups. g. 3D representations of the lesion site (GFAP in green, CSN axons in red) from the ventral view (upper row) and lateral view (lower row). There are no significant differences in either GFAP intensity or GFAP+ volume between the groups. Data are shown as mean ± SEM. Scalebars (a): 200 μm; (g): 500 μm.
Extended Data Fig. 4. Segmentally distinct loss of long-distance CSN axon extension is not due to segmental differences in microglial activation.
a. Representative images of P35 mouse spinal cord horizontal sections at the microlesion site following distinct P1 or P4 microlesions indicated. Sections are stained for Iba1 (microglial response). b. Schematic showing the spinal locations for images shown in a. c. Quantification of the signal distribution of Iba1+ area in 200 μm horizontal bins centered at the microlesion at specific distances from the microlesion (rostral to caudal). There is no significant difference between the multiple, distinct groups. Scalebar (a): 200 μm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A video taken under ultrasound-guided backscatter microscopy showing the microlesion procedure. An axial view of a P4 mouse pup on its side, dorsal side towards the micropipette (on the left). Spinal cord, vertebral bodies and CST are highlighted in the initial frames of the video for reference. In the video, the glass micropipette is first seen prior to insertion into the spinal cord. It is then inserted up to the central canal, and the high frequency vibrating apparatus is activated to axotomize the CST. After the activation of the vibration, the micropipette is withdrawn from the cord over a duration of 10 seconds (with brief 2s pauses during the withdrawal).
Acknowledgements
We thank Sargunvir Sondhi, Jake Lustig, and Aayushma Kunwar for technical assistance. This work was supported by funding from the National Institutes of Health (NIH; (NTRAIN/NICHD K12HD093427), a project grant from the Wings for Life Foundation, and a pilot grant from Craig H. Neilsen Foundation to V.S.. C.R. was partially supported by a postdoctoral fellowship from the NY State (NYS) Spinal cord injury research board (SCIRB). J.K. was partially supported by a postdoctoral fellowship from the Swiss National Science Foundation.
References




