

ABSTRACT
In humans, associative memories are more susceptible to age-related cognitive decline (ARCD) than are recognition memories. Reduced cAMP/cGMP signaling in the hippocampus may contribute to ARCD. Here, we found that both aging and traumatic brain injury-associated dementia increased expression of the cAMP/cGMP-degrading enzyme phosphodiesterase 11A (PDE11A) in the human hippocampus. Further, age-related increases in hippocampal PDE11A4 mRNA and protein were conserved in mice, as was the increased vulnerability of associative versus recognition memories to ARCD. Interestingly, mouse PDE11A4 protein in the aged ventral hippocampus (VHIPP) ectopically accumulated in the membrane fraction and filamentous structures we term “ghost axons”. These age-related increases in expression were driven by reduced exoribonuclease-mediated degradation of PDE11A mRNA and increased PDE11A4-pS117/pS124, the latter of which also drove the punctate accumulation of PDE11A4. In contrast, PDE11A4-pS162 caused dispersal. Importantly, preventing age-related increases in PDE11 expression via genetic deletion protected mice from ARCD of short-term and remote long-term associative memory (aLTM) in the social transmission of food preference assay, albeit at the expense of recent aLTM. Further, mimicking age-related overexpression of PDE11A4 in CA1 of old KO mice caused aging-like impairments in CREB function and remote social—but not non-social—LTMs. RNA sequencing and phosphoproteomic analyses of VHIPP identified cGMP-PKG—as opposed to cAMP-PKA—as well as circadian entrainment, glutamatergic/cholinergic synapses, calcium signaling, oxytocin, and retrograde endocannabinoid signaling as mechanisms by which PDE11A deletion protects against ARCD. Together, these data suggest that PDE11A4 proteinopathies acutely impair signaling in the aged brain and contribute to ARCD of social memories.
INTRODUCTION
After the age of 60, nearly all individuals experience some form of cognitive decline—particularly memory deficits—and no drugs prevent or reverse this loss [10, 11]. Even in absence of dementia, age-related cognitive decline (ARCD) increases health care costs and risk for disability [12]. ARCD is not a uniform process, with variability in symptom severity observed across individuals and cognitive domains [10]. For example, associative memories are more susceptible to ARCD in humans than are recognition memories [13-19] for reasons that are not clear but may be related to deficient activation of anterior hippocampus [20, 21] (a.k.a. ventral hippocampal formation in rodents, VHIPP). This lack of knowledge slows therapeutic development.
3’,5’-cyclic adenosine monophosphate (cAMP) and 3’,5’-cyclic guanosine monophosphate (cGMP) signaling are decreased in the aged and demented hippocampus (rodents and humans), particularly when there is a history of traumatic brain injury (TBI) [22-24]. Interestingly, cyclic nucleotide signaling deficits associated with neuropsychiatric and age-related diseases of the brain can be more prominent in one subcellular compartment versus another [23, 25-29]. Together, these findings suggest that select enzymes that generate (i.e., cyclases) and/or break down cAMP and cGMP (i.e., 3’,5’-cyclic nucleotide phosphodiesterases, PDEs) not only change in expression and/or activity, but may also become ectopically localized [30, 31]. Such an ectopic localization could prove exceptionally deleterious since the discrete localization of these enzymes within specific subcellular domains allows a single cell to recognize and respond uniquely to multiple stimuli [30, 31]. In other words, where a cyclase or PDE is localized is just as important to its overall function as is its catalytic activity [32].
Age-related changes in cyclic nucleotide signaling may worsen cognition by promoting proteinopathies (a.k.a. proteopathies)—abnormalities in protein synthesis, post-translational modification, folding, or deposition that occur with normal aging and age-related diseases [33-36]. For example, reductions in hippocampal adenylyl cyclase correlate with the accumulation of amyloid plaques [37], and increasing cyclic nucleotide signaling through the cAMP-PKA and cGMP-PKG pathways helps clear aggregated proteins that cause neurodegeneration [38-40]. On the other hand, aberrant phosphorylation driven by cAMP/cGMP-regulated kinases can drive the accumulation of tau and TDP-43 in many neurodegenerative diseases [41, 42]. Therefore, it is of great interest to determine how the dysregulation of cyclic nucleotides within specific brain regions and subcellular compartments may drive proteinopathies associated with aging and how that dysregulation may affect ARCD.
The age-related decreases in hippocampal cyclic nucleotides described above are consistent with our observation that expression of PDE11A4 increases with age in both the mouse and rat hippocampus [7, 8]. The PDE11A family is comprised of a single gene that is spliced into 4 isoforms, PDE11A1-4. The longest isoform, PDE11A4, is the isoform that is expressed in brain and is ∼95% homologous across mouse, rat and human [43, 44]. All 4 isoforms are considered dual-specific, in that they degrade both cAMP and cGMP; however, enzyme assays suggest PDE11A4 may hydrolyze cAMP with a higher Km and Vmax than cGMP [45, 46]. PDE11A single nucleotide polymorphisms are associated with major depressive disorder (MDD) [47-49], suicide risk [50], sleep quality [51], antidepressant response in patients with MDD [47, 48] (but see [49, 52]), and lithium response in patients with bipolar disorder [53-55]. It is, then, interesting to note that both MDD and BPD have been conceptualized as diseases of accelerated aging [56, 57]. PDE11A4 mRNA is strongly expressed in neurons of CA1, the subiculum, and the adjacently connected amygdalohippocampal area of the VHIPP [7, 9], with moderate expression in dorsal hippocampus (DHIPP) and little to no expression in other brain regions [7, 9, 58-60]. Thus, PDE11A4 molecularly defines an exceptionally discrete neuronal population within a brain region key to associative memory, making it ripe for study in the context of ARCD.
Consistent with its enrichment in the VHIPP, PDE11A4 regulates preferences for social interactions as well as the consolidation of social memories [1, 2, 6, 7, 9]. The effect of PDE11A deletion on social memory in young adult mice is quite unique in that it triggers a transient amnesia that ultimately produces a stronger remote long-term memory (LTM; i.e, intact short-term memory, impaired recent LTM, improved remote LTM) [6]. Further, we have shown PDE11A regulates signals important for social memory consolidation, including oxytocin signaling, glutamatergic signaling, calcium/calmodulin-dependent kinase II signaling, CREB function, and protein synthesis [7-9, 61, 62]. As such, here we test the hypothesis that age-related increases in hippocampal PDE11A4 are conserved across species, occur in an ectopic subcellular compartment, and impair social associative memories.
METHODS
Human expression data
To assess age-related changes in PDE11A, PDE5A, XRN2, and NONO mRNA, RNA sequencing data from human hippocampi were mined from the 2014 Allen Institute for Brain Science Brainspan database, as we previously published [63, 64]. Data were downloaded by navigating to the following website and using the enzyme names listed above in the search box: https://www.brainspan.org/rnaseq/search/index.html. Methods used for this RNA sequencing study have been previously published [65] and can be found here: http://help.brain-map.org/display/devhumanbrain/Documentation. Data were also mined from the 2016 Allen Institute for Brain Science Aging, Dementia, and traumatic brain injury (TBI) study, a study conducted in a cohort of adults 75+ years old. Data for the entire study were downloaded from http://aging.brain-map.org/download/index, and the methods for this RNA sequencing study can be found here: http://help.brain-map.org/display/aging/Documentation. Subjects from the Brainspan database were grouped into the following age categories based on our previous studies [63, 64]: prenatal, childhood (2 months-17 years old), and adulthood (18-40 years old). Subjects from the Aging, Dementia, and TBI database were divided into four groups based on whether they had a history of TBI and/or had been diagnosed with Alzheimer’s disease or a related dementia (ADRD).
Mouse Subjects
Mating trios (1 male x 2 females) of C57BL/6J and BALB/cJ mice were originally obtained from Jackson laboratory and then bred onsite either at the University of South Carolina School of Medicine or the University of Maryland School of Medicine. Old 129S6/SvEv were originally obtained from Taconic at approximately 2 months of age and were then aged onsite at the University of South Carolina School of Medicine, with young mice ordered from Taconic 2 weeks prior to tissue harvest for comparison. As previously published (e.g., [6, 9, 62]), the Pde11a mouse line was originally obtained from Deltagen (San Mateo, CA) and then maintained on a mixed C57BL6 background (99.8% multiple C57BL/6 substrains and 0.2% 129P2/OlaHsd [62]) as well as a 98.8% BALB/cJ background [62]. Note that knockouts on the BALB/cJ background were only used as negative controls in immunofluorescent staining experiments using PDE11A antibodies; no behavior was conducted in the BALB/cJ line. Pde11a mice were bred onsite in heterozygous (HT) x HT trio matings, with 3-5 same-sex wild-type (WT), heterozygous (HT), and knockout (KO) littermates (defined as any mice born at the same time to either dam) weaned together. As previously described [62, 66], litter effects are unlikely driving findings described herein as each dataset reflects either 1) a combination of 2+ subcohorts born and tested at different times or 2) is fully replicated in a separate cohort of mice. Further, any given litter of Pde11a mice typically contributes n=1-2 mice/genotype and, at most, parents contribute 2 litters to a cohort (i.e., a total of 2-4 mice/genotype). Males and females were used throughout each experiment, but not necessarily in sufficient numbers to power an analysis of sex effects (see figure legends for specific n’s/sex/group/experiment). For experiments herein, young was defined as 2-6 months old, middle aged was defined as 10-15 months, and old was defined as 18-22 months. As previously described [66], we do not conduct gross pathology, however, mice are generally healthy at the time of testing. Mice are routinely assessed by husbandry, veterinary and laboratory staff and mice with palpable tumors >1 cm, lethargy, altered gait, signs of malnutrition or dehydration are removed from study and euthanized. Mice demonstrated no evidence of a striking anatomical abnormality of the brain upon dissection (e.g., a pituitary tumor) [66]. Animals were housed on a 12:12 light:dark cycle and allowed ad lib access to food and water. Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Pub 85-23, revised 1996) and were fully approved by the Institutional Animal Care and Use Committee of the University of South Carolina and the University of Maryland, Baltimore.
Tissue Collection
As previously described [66], mice were euthanized during the light cycle via cervical dislocation. Mouse brains were harvested fresh, dissected on wet ice, and stored at -80 °C for further processing.
In situ hybridization
Brains were stored at −80 °C until being cryosectioned at −19 to -17 °C in the sagittal plane at 20 μm, with sections thaw mounted on superfrost slides. As previously described [8, 67], slides were pretreated in a series of 4% paraformaldehyde, 1M Triethanolamine/0.9% NaCl, ethanol (70-100%), chloroform, ethanol (100-70%), and sterile water and were then allowed to air dry before storing at -80 °C. In situ hybridization for PDE11A4 was also conducted as previously described [8, 67] using an S35-labelled antisense probe (5′-ccaccagttcctgttttccttttcgcatcaagtaatc-3′). The probe was end labeled with 35S-dATPα using terminal deoxytransferase and extracted using Illustra Microspin G-25 columns (GE Healthcare, North America). Each slide was then hybridized with 600,000 cpms of labeled probe in 125 μl of hybridization buffer and slides incubated overnight in a humid chamber warmed to 37° C. The following day, slides were washed in a series of saline-sodium citrate (SSC) buffers with 0.1 M dithiothreitol (2X SSC room temperature then 0.2X SSC at 55° C). Slides were dehydrated in 70% ethanol and allowed to air dry. Once dry, slides were exposed to Kodak BioMax MR film for 14 days, generating optical densities that were within the linear range of the film. Note, antisense probes yielded consistent expression patterns (see Figure 1) while sense probes yielded no signal as previously published [8, 67].

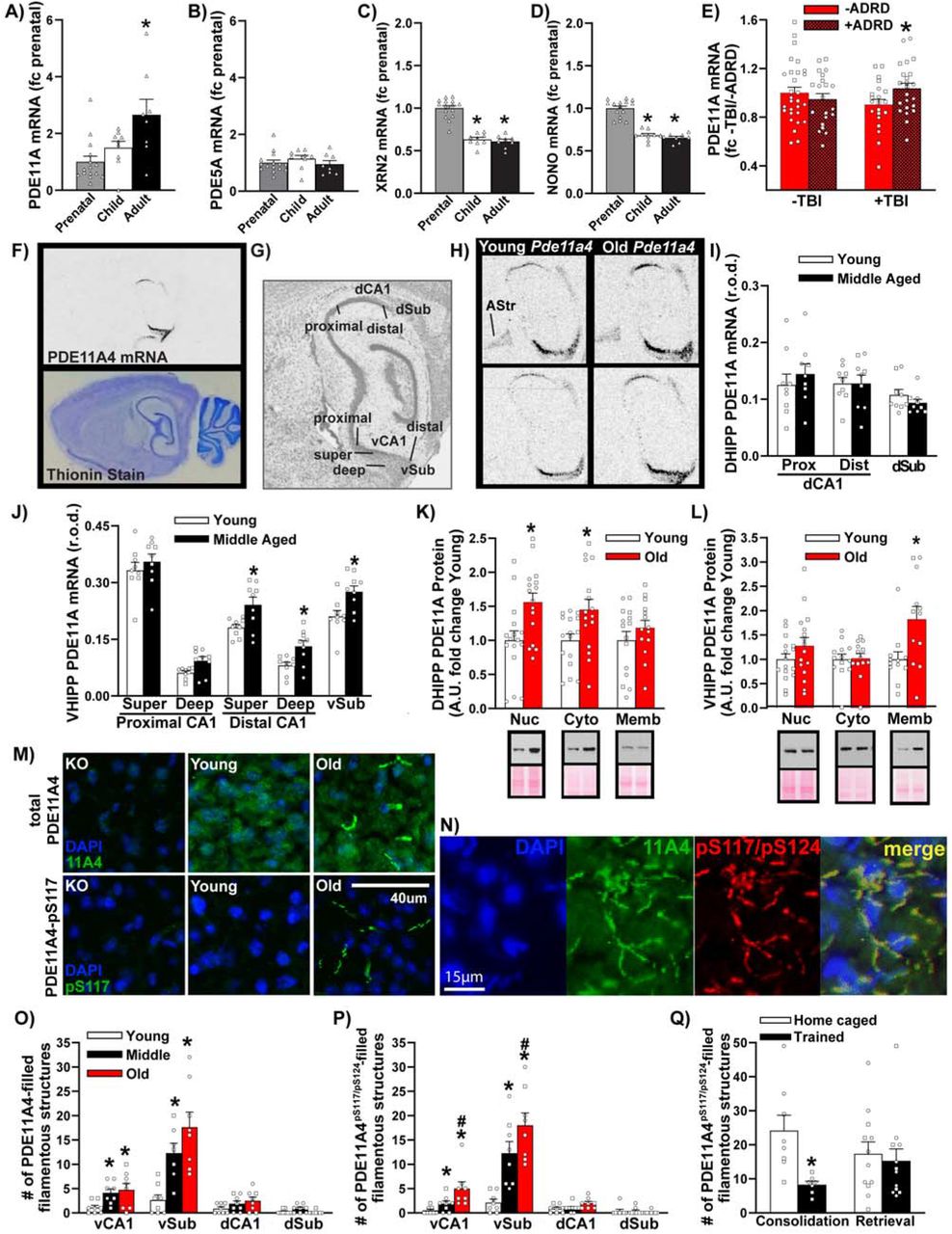
A) Data mined from the Allen Institute for Brain Sciences Brainspan database and expressed as a fold change (fc) of prenatal levels shows PDE11A mRNA expression in the human hippocampus robustly increases from the prenatal period (n=15; sex not specified), to childhood (n=9), through to adulthood (n=8; H(2)=9.48, P=0.009; Post hoc prenatal vs adult, P=0.008). B) In contrast, mRNA for PDE5A—the next closest related PDE—shows no change in expression (F(2,29)=0.72, P=0.496). This age-related increase in PDE11A mRNA expression correlates with age-related decreases in mRNA expression of C) the nuclear protein p54nrb/NONO (F(2,29)=66.29, P<0.001; Post hoc, each vs prenatal, P<0.001) and D) the exoribonuclease XRN2 (H(2)=23.35, P<0.001; Post hoc, each vs. prenatal, P<0.001), both of which work together to target PDE11A4 mRNA for degradation [3]. E) Data mined from the Aging, Dementia, and Traumatic Brain Injury (TBI) database (expressed as fc -TBI/-ADRD) shows that among elderly humans with a history of TBI, Pde11a mRNA is significantly higher in those that developed Alzheimer’s disease or a related dementia (n=10M,13F) versus those that did not (n=15M,6F; effect of TBI x dementia: F(1,90)=4.07, P=0.047; Post hoc +TBI/-dementia vs. +TBI/+dementia, P=0.049). F) To delineate where within the hippocampus age-related increases in Pde11a4 mRNA expression may be occurring, we turned to rodent studies. In situ hybridization shows Pde11a4 mRNA in the brain is restricted to the extended hippocampal formation. G) Robust expression is observed in all mice in CA1, the subiculum, and the amygdalohippocampal area. H) To our surprise, a subset of male and female young and old mice also showed Pde11a4 mRNA expression in the amygdalar-striatal transition area (AStr; top) while others did not (bottom)—AStr staining had not been previously noted in the mouse brain with weaker probes [7-9]. I) Dorsal hippocampal subfields show no effect of age on PDE11A mRNA expression (n=4M,5F/age. J) In contrast, select subfields of the ventral hippocampus show increased PDE11A4 mRNA expression in middle-aged versus young mice (2-Way RM ANOVA on all ventral subfields failed normality), in particular the superficial and deep subfields within distal CA1 (effect of age: F(1,16)=8.96, FDR-P=0.024) and the ventral subiculum (Sub; t(16)=2.97, FDR-P=0.024). K) Despite showing no age-related changes in PDE11A mRNA expression, the DHIPP does exhibit an age-related increase in PDE11A4 protein expression in the nuclear (Young, n=9M,7F; Old, n=11M,5F; t(30)=-2.91, P=0.0068) and cytosolic fractions (Young, n=9M,7F; Old, n=11M,5F; t(30)=-2.54, P=0.017) but not membrane fraction (Young, n=9M,6F; Old, n=9M,5F; t(27)=--1.07, P=0.296). L) In contrast, the ventral hippocampus shows no changes in PDE11A4 protein expression in the nuclear (Young, n=9M,7F; Old, n=11M,5F; t(30)=-1.34, P=0.189) and cytosolic compartment (Young, n=6M,7F; Old, n=8M,5F; failed normality; Rank Sum test for effect of age: T(13,13)=171.0, P=0.837) but does show an age-related increase in the membrane compartment (Young, n=6M,6F; Old, n=7M,5F; t(22)=-2.674, P=0.014). M) Immunofluorescence also revealed age-related increases in VHIPP PDE11A4 protein are compartment specific, with PDE11A4 accumulating in filamentous structures we term ghost axons (see Figure 2). PDE11A4 phosphorylated at serines 117 and N) 124 appears to be found exclusively in these ghost axons, and all PDE11A4 found in these ghost axons appears to be phosphorylated at S117/S124 as indicated by a complete overlap in staining patterns of the total and phospho-specific antibodies. O) Quantification (n=4/sex/age) shows significant age-related increases of PDE11A4-filled structures in vCA1 (F(2,14)=5.05, P=0.022; Post hoc vs Y: M P=0.028, O P=0.026) and vSub (F(2,13)=13.55, P<0.001; Post hoc vs Y: M P=0.005, O P<0.001) as well as N) PDE11A4-pS117/pS124-filled structures in vCA1 (fails normality; ANOVA on Ranks for effect of age: H(2)=12.62, P=0.001; Post hoc vs Young, each P<0.05) and vSub (F(2,14)=29.60, P<0.001; Post hoc vs Young, each P<0.001). Q) Increased presence of these pS117/pS124-PDE11A4-filled ghost axons may be deleterious since normally memory consolidation following STFP training is associated with a reduction in their presence (Home caged, n=3M,5F; trained, n=3M,4F; t(13)=3.16, P=0.007) although 24-hour memory retrieval is not (home caged, n=4M,8F; trained, n=6M,6F; failed normality; Rank Sum test for effect of age: T(12,12)=162.0, P=0.506). *vs prenatal, -ADRD, or young, P<0.05-0.001; #vs. middle-age, P<0.05. Data plotted as individual points (females as circles, males as squares) and expressed as mean ±SEM. Histogram stretch, brightness, and/or contrast of images adjusted for graphical clarity. A.U.—arbitrary units; r.o.d.—relative optical density.
Biochemical Fractionation and Western Blotting
Biochemical fractionation was performed as previously described [59, 64, 66]. In short, cells or tissue were sonicated in ice cold fractionation buffer (20 mM Tris-HCl, pH 7.5; 2 mM MgCl2; Thermo Pierce Scientific phosphatase tablet #A32959 and protease inhibitor 3 #P0044). For those samples processed by biochemical fractionation, successive rounds of differential centrifugation were conducted. Following a low-speed spin (1000 x g) to isolate the nuclear fraction pellet, the supernatant was transferred to a new tube and subjected to a high-speed spin (89,000 x g) to isolate the cytosolic proteins in the supernatant and the membrane proteins in the pellet. The pellet was then resuspended in fractionation buffer with 0.5% Triton-X 100 to solubilize the protein, with a subsequent high-speed spin (60,000 x g) to isolate the supernatant containing soluble membrane proteins. Note that the nuclear and membrane pellets were “rinsed” prior to the resuspensions described above. That is, these pellets were resuspended in the previous buffer and respun with resulting supernatant discarded. Protein concentrations were determined by DC Protein Assay kit (Bio-Rad; Hercules, CA, USA) according to manufacturer protocol, and were subsequently equalized across samples. Samples were stored at -80°C until further processing. For western blotting, 10μg of total protein for cells, 30μg of total protein for tissue, or 20μg for membrane, cytosolic, or nuclear fractions from tissue were loaded onto 12% NuPAGE Bis-Tris gels (Invitrogen, Waltham MA) and electrophoresed for one hour at 180 volts. Previously verified hippocampal tissue from a PDE11A KO mouse was included in all PDE11A4 blots as a negative control. Protein was transferred onto a 0.45μm nitrocellulose membrane for two hours at 100 mA. Membranes were then washed twice in tris-buffered saline with 0.1% tween20 (TBS-T) before staining with Ponceau S to determine total protein loading. Stained membranes were imaged and then rinsed in TBS-T to remove the stain. Blots to be probed with the #1 antibody were blocked in 5% milk while all others were blocked in Superblock Blocking Buffer (ThermoFisher, Cat#37515), each with 0.1% Tween 20. Overnight primary antibody incubation at 4°C was completed for the following targets: PDE11A (Aves custom PDE11#1 at 1:10,000; Fabgennix PD11A-112 at 1:500), CREB (Cell Signaling #4820 at 1:10,000) as well as synaptophysin (BD Transduction #611880 at 1:200,000) and histone 3 (Millipore 05-928 at 1:5000) or actin (Sigma A2066 at 1:10,000). The next day, membranes were washed four times with TBS-T for ten minutes each. Secondary antibody incubation was completed for one hour at room temperature. The following Jackson Immunoresearch secondary antibodies were used: anti-rabbit (Jackson Immunoresearch, 111-035-144; 1:10 000), anti-chicken (Jackson Immunoresearch, 103-035-155; 1:40 000), and anti-mouse (Jackson Immunoresearch, 115-035-003; 1:10 000). Three final washes in TBS-T were done for fifteen minutes each. The membranes were immersed in SuperSignal West Pico Chemilumiscent Substrate (ThermoScientific, Waltham MA) and then exposed to film. Multiple film exposures were taken to ensure signals were within the linear range, and Ponceau stain and western blot optical densities were quantified using Image J. To account for gel-gel variances in differences in film exposure, how well antibodies incorporated onto each membrane, how well chemiluminescence reacted between blots, etc., Western blot data were normalized to a control condition (e.g. EmGFP-mPDE11A4WT) on each blot, as previously described (e.g., [8, 64, 66].
Immunofluorescence (IF)/Immunohistochemistry (IHC)
For most experiments, fresh-harvested, flash-frozen brains were embedded in matrix, cryosectioned in the sagittal plane at 20 μM, and thaw-mounted two sections per slide. One exception was that perfused tissue was sectioned with a vibratome at 50 μm for labeling using the AC3 antibodies described below. As previously described [7], 4% paraformaldehyde in PBS for 20 minute was used to fix frozen tissue. After fixation, the tissue was washed three times for ten minutes each in PBS with bovine serum albumin and triton X-100 (PBT). To reduce non-specific labeling, antibodies hosted in rodent species were pretreated with anti-Mouse FabFragments (0.15mg/ml; Jackson Immunoresearch # 715-007-003) in PBS for 2 hours. The FabFragments were then washed off by 3×10 minute washes in PBT prior to adding primary antibody. Overnight incubation with antibodies to the following targets was done at 4°C. Antibodies were mixed in PBT at optimized dilutions: PDE11A (FabGennix PD11-112 at 1:100; Aves custom PDE11A4 #1 at 1:10,000; Fabgennix PPD11A-140AP at 1:1000; Fabgennix PPD11A-150AP at 1:500), NF-L (Cell Signaling #2837 at 1:10,000), β-tubulin (Covance PRB-435P at 1:100,000), MBP (Aves MBP at 1:5000), MAP2 (Neuromics CH22103 at 1:2000), AC3 (PhosphoSolutions 85-AC3 at 1:500 or Thermofisher Scientific PA5-35382 at 1:500 produced equivalent results), α-internexin (Phosphosolutions 100-AIN at 1:5000), peripherin (Invitrogen PA1-10018, 1:1000), Ankryin-G (UC Davis/NIH Neuromab Facility Incorporated 75-146 at 1:100), lectin (Sigma, L1516 at 1:500), IBA-1 (Wako 019-19741 at 1:1000), and GFAP (Cell Signaling 12389P at 1:10,000). The following day, sections were washed 4x in PBT and incubated for 90 min in secondary antibody, with co-labeling studies prioritizing secondaries recognizing the PDE11A antibodies first (Alexafluor 488 AffiniPure Donkey Anti-Chicken, 1:1000, Jackson Immunoresearch #703-545-155 or Alexafluor 488 AffiniPure Donkey Anti-Rabbit, 1:1000, Jackson Immunoresearch # 711-545-152), followed by 3×10 min PBT washes, then the secondaries recognizing the co-labeling marker antibody (Alexafluor 594 AffiniPure species-specific, 1:1000, Jackson Immunoresearch). After the secondary antibody incubation(s), slides were washed in PBT three times for ten minutes each. The slides were then rinsed ten times in PBS. For IF, slides were then mounted with DAPI fluoromount (Southern Biotech, #0100-20). In the case of immunohistochemistry, slides were processed as previously described [61]. Slides were treated with ABC solution (Vector Laboratories, PK-6100) prepared in PBT for 90 minutes. More specifically, after washing 3×10 washes with PBS sections were subjected to a DAB reaction (Vector Laboratories, SK-4100) until fully developed. Slides were placed in PBS once the reaction was complete. After being dried at room temperature, slides were mounted with Permount mounting media (Fisher, SP15-100) and a coverslip. Slides were kept covered and refrigerated until viewed and imaged. For quantification of PDE11A-filled structures and some co-labeling studies, images were taken using a CX9000 camera and Neurolucida imaging software (MBF Bioscience; Williston VT) mounted on a Nikon Eclipse 80i bright-field microscope that is equipped with a dry 20X objective. For co-labeling studies, images were captured using a LASX software and a Leica SP8 confocal microscope with oil immersion objectives 20X-63X. The number of structures was quantified by an experimenter blind to treatment.
Social Transmission of Food Preference
As previously described [6, 7], mice were food restricted such that access to food was limited to 1 hour per day for 2 days prior to the start of training. 24 hours prior to training, all mice were first habituated to the powdered food by being given access to it in a clean home cage. The next day, a mouse designated as the “demonstrator” was moved to a clean home cage alone and fed powdered food laced with a household spice for one hour (e.g. basil vs. ginger, thyme vs turmeric, mint vs cardamom, orange vs anise, basil vs thyme). Note that some cohorts of mice were trained and tested at a given retrieval time point with one spice combination and then trained and tested at a different retrieval time point with a different spice combination in order to reduce the total number of mice used herein. For example, data shown in Figure S6 are all from the same cohort of middle-aged mice that were trained/tested at different times, with a different spice combination used at each retrieval time point. We have extensively detailed elsewhere that such an approach does not confound interpretation of our data [6]. The demonstrator is then returned to the home cage and their “observer” cage mates interact with the demonstrator for 15 minutes, during which time the observers make an association between the non-social odor (household spice) and the social pheromones in the breath of the demonstrator. 15 minutes, 24 hours, or 7 days after training, the observer mice are then tested for 60 minutes by being placed in a clean home cage by themselves and given access to 2 powdered foods--one laced with the spice that the demonstrator was given and the other a novel spice. The amount of food eaten and the amount of time spent eating the food (scored for 1 minute out of every 10 minutes) was calculated by an experimenter blind to group. All mice met minimum inclusion criteria of eating at least 0.25 gm of food and being observed to eat for at least 5 seconds. Memory formation/retrieval is defined as the observer mice eating more of the food laced with the familiar spice that was on their demonstrator’s breath versus the novel spiced food (preference ratio: familiar-novel/familiar+novel).
Odor Recognition
As previously described [6, 7], 1’’ round wooden beads (Woodworks) were placed in the subjects’ home cages for at least 7 days prior to testing to allow for habituation and saturation of the beads with social odors. Training involved a habituation trial with 3 beads from the subject’s home cage, followed by 2 subsequent trials that included 2 home-cage beads and 1 novel-scented bead. For social odor recognition (SOR), the novel-scented bead came from a cage of sex-matched mice of a different strain (e.g., C57BL/6J Jax #000664, BALB/cJ Jax #000651, 129S6/ SvEv Taconic #129SVE, C3H/HeJ #000659, A/J #000646). For non-social odor recognition (NSOR), the novel-scented bead came from a baggie of bedding saturated with a household spice (e.g., basil, ginger, marjoram, cumin, etc.). Memory was then tested 1 hour (short-term memory, STM), 24 hours (recent long-term memory, LTM) or 7 days later (remote LTM). For SOR testing, mice were presented with one home cage bead, one bead from the familiar donor strain used during training, and one bead from a second novel donor strain. For NSOR, mice were presented with 1 bead scented with the training spice and 1 bead with a novel household spice. The donor/spice assigned as “novel” within a given set of scents was counterbalanced across subjects. Mice were allowed to investigate the beads for 2 minutes and time spent investigating beads was manually scored by an experimenter blind to the conditions. All mice met minimum inclusion criteria of spending at least 3 seconds sniffing. Memory is defined as spending more time investigating the novel vs familiar scent (preference ratio: novel-familiar/novel+familiar).
Plasmid generation
Plasmids were generated as previously described [59]. Briefly, Genscript (Piscataway, NJ) generated constructs expressing either EmGFP alone containing an A206Y mutation to prevent EmGFP dimerization [68] or the mouse Pde11a4 (NM_001081033) sequence fused at the N-terminal with EmGFP. These constructs were initially generated on a pUC57 backbone and then subcloned into a pcDNA3.1+ mammalian expression vector (Life Technologies; Walthan, MA). Pde11a4 mutations (i.e., S117A, S124A, S117D, S124D, S162A, S162D) were introduced using the QuickChange procedure and products as per manufacturer’s instructions (Agilent Technologies; Santa Clara, CA). Oligonucleotide primers used in the QuickChange procedure were generated by Integrated DNA Technologies (Coralville, IA) and mutated DNA sequences were subsequently verified by Functional Biosciences (Madison, WI).
Cell culture and transfections
Cell culture and transfection were conducted as previously described [59], with cultures periodically confirmed to be mycoplasma negative by the laboratory of Dr. Mythreye Karthikeyan at the University of South Carolina. Briefly, COS-1 (male monkey fibroblast cell line as per web.expasy.org), HEK293T (female human embryonic kidney cell line as per web.expasy.org), and HT-22 cells (undefined-sex mouse hippocampal cell line) were grown in t-75 flasks containing Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO; Gaithersburg, MD, USA) with added 10% fetal bovine serum (FBS) (GE Healthcare Life Sciences; Logan, UT, USA) and 1% Penicillin/Streptomycin (P/S) (Mediatech, a Corning subsidary; Manassas, VA, USA). Cells were incubated at 37°C/5% CO2 and passaged once 70-90% confluent, using TrypLE Express (GIBCO; Gaithersburg, MD,USA) as a dissociation agent. One day prior to transfection, cells were plated in either 24-well plates or 6-well plates with cover slips for imaging experiments or 100 mm dishes for biochemistry, all of which contained DMEM+FBS+P/S. The following day, media was replaced with Optimem (GIBCO), and cells were transfected with the pcDNA3.1+ vectors using Lipofectamine 2000 (Invitrogen; Carlsbad, CA) according to manufacturer protocol using a ratio of 3.75μg DNA plus 10uL lipofectamine per 10 mLs of media. ∼19 hours post-transfection (PT), Optimem + Lipofectamine was replaced with DMEM+FBS+P/S. For most studies, cells were allowed to grow for five hours before sample processing (e.g. fixation for imaging, sonication for Western blot, etc.) and, thus, were harvested ∼24 hours following transfection. The one exception was in the case of the COS1 experiment that tracked protein expression changes between the S117A/S124A versus S117D/S124D mutants from 24 hours to 72 hours following transfection. For imaging experiments, cells were fixed with 4% paraformaldehyde in PBS for fifteen minutes and then kept in PBS for imaging using a CoolSNAP EZ CCD camera (Photometrics, Tuscon AZ) that was mounted on an inverted Leica (Wetzlar DE) DMIL microscope with a Fluotar 10X/0.3 ∞/ 1.2 objective. The images were captured using NIS-Elements BR-2.3 (Nikon,Tokyo Japan) software. Images were then systematically hand scored by an experimenter blind to condition. This systematic process involved placing a 4×5 grid over the image in powerpoint to help keep track of position and using the same digital zoom for all images within an experiment, typically between 250% to 400%, depending on the experimenter. The cells within each box of the grid were then classified as exhibiting cytosolic-only labeling versus punctate labeling, and data were then entered for that grid into a grid-shaped template. Once all grids for an image were evaluated, data were typed into an .xls template that calculated the total number of cells that were labelled and the percent of labeled cells that exhibited cytosolic versus punctate labeling (data graphed as the latter).
PDE assay
As previously described [2], cells were harvested in buffer containing 20 mM Tris-HCl and 10 mM MgCl2 and kept on ice until ready to use. [3H]cGMP (Perkin Elmer, NET337) or [3H]cAMP (Perkin Elmer, NET275) were prepared at 4× 104 disintegrations per minute in the same solution in which the cells were harvested. In a tube containing 50 uL of cell lysates, 50 uL of [3H]cGMP or [3H]cAMP was added and the reaction took place for 10 minutes. The reaction was then quenched with 0.1 M HCl and neutralized with 0.1 M Tris base. To convert the product of PDE reaction (5’AMP or 5’GMP) to the corresponding nucleoside and phosphate, 3.75 mg/mL of snake venom (Crotalus atrox, Sigma V-7000) was added to the samples for 10 minutes at 37°C. Samples were then loaded into 5’polystyrene chromatography columns with coarse filters (Evergreen, 208-3383-060) containing DEAE Sephadex A-25 resin (VWR, 95055-928). Once the sample completely flowed through the column, the columns were eluted with 4 washes of low salt buffer containing 20 mM Tris-HCl and 0.1% sodium azide. Once all 4 washes were complete, 4 mL of Ultima Gold XR scintillation fluid (Fisher, 50-905-0519) was added directly to the samples and vortexed vigorously. The samples were then run through a scintillation counter (Beckman LS 6000) to obtain the counts per minute. Data were then normalized to the total amount of protein per sample.
Stereotaxic Surgery
As previously described [6], the NeuroStar motorized stereotaxic and drill and injection robot (Tubingen, Germany) was used to conduct stereotaxic surgery. Mice were anesthetized with a mixture of oxygen and isoflurane at an induction rate of 3%. Upon initial administration, lack of reflexes was verified and isoflurane administration was maintained at 1.5-2%. A robotic drill was used to drill small holes at the following coordinates relative to Bregma (dCA1 AP, 1.7, dCA1 ML, +/ 1.6, vCA1 AP, 3.5, vCA1 ML, +/3.0). Once the needle reached the appropriate depth (Bregma: dCA1 DV, - 1.3, vCA1 DV, 4.4), the injection robot was used to inject 2 μl of lentivirus (titers: eGFP-only, 10×106 TU/ml; eGFP-PDE11A4 7.4×106) suspended in 0.2M sucrose/42 mM NaCl/0.84 mM KCl/2.5 mM Na2HPO4/0.46 mM KH2PO4/0.35 mM EDTA at a rate of 0.167 μl/minute. The syringe was left in place for 3 minutes following injection before withdrawing the needle. Mice were allowed to recover for at least 2 weeks prior to behavioral testing.
RNA sequencing
As previously described [1], the Epigenomics Core at Weill Cornell Medicine performed the RNA and library preparation, sequencing, and post-processing of the raw data. The Functional Genomics Core of the University of South Carolina then performed the alignment and generation of the gene list of log-fold changes with their raw and FDR-corrected P-values. A cryoPREP TM Impactor was used to dry fracture tissue that had been stored at -80°C in a tissueTUBE device, using manufacturer recommendations (Covaris, Woburn, MA). Each pulverized sample was resuspended in 350uls of Qiagen RNeasy Plus Mini RTL lysis buffer and RNA extracted as per manufacturer recommendations (Qiagen, Valencia, CA). The Lab Chip GX (Perkin Elmer, Waltham, MA) assessed RNA integrity, with samples having a quality score >8.0. Established Illumina methods were used to prepare TruSeq RNA libraries (Illumina, San Diego, CA, Part #RS-122-2001). Samples were sequenced across 3 lanes, with each library being made with one of the TruSeq barcode index sequences. The pools were clustered at 6.5pM on a pair end read flow cell and sequenced for 100 cycles on an Illumina HiSeq 2500. Illumina’s Real Time Analysis software (RTA) conducted primary processing of sequencing images. Image capture, base calling and demultiplexing was conducted using CASAVA 1.8.2 software. STAR v2.3.1 [69] was used to align sequences to the mouse mm10 genome (http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/). Reads, which were summarized at exon, transcript, or gene level were, counted using the featureCounts function of the Subreads package [70] in R (https://www.R-project.org/) using Gencode M6 GTF (http://www.gencodegenes.org/mouse_stats/archive.html). Only those reads mapping uniquely to the genome were used. A minimum threshold of 10 was used for mapping quality (MAPQ). The edgeR package within R [71] was used for differential expression analysis. The average read depth for the samples was 64 million reads, and only genes with at least 1 count per million average depth were considered for differential expression analysis. The trimmed mean of m-values (TMM) method was used to normalize raw counts. The estimateGLMRobustDisp function [72] was then used to calculate the dispersion estimates. The function glmFit [73] was then used to fit normalized read counts to a generalized linear model. The function glmLRT was then used to perform gene-wise tests for significant differential expression. Benjamini-Hochburg’s FDR [74] was then used to correct the raw P-value for multiple testing. Genes that differed between WT versus KO mice with aLogFC >+/-0.5 and an FDR-P<0.01) were included in pathway analyses using String v11.0 (string-db.org) [75] (accessed 7/27/21).
2-dimensional difference in gel electrophoresis (2-D DIGE) and mass spectroscopy (MS)
Phosphoproteomics studies were conducted by Applied Biomics (Hayward, CA; https://www.appliedbiomics.com/2d-dige/phosphoproteomics/). Ventral hippocampi from 4 cohorts of Pde11a WT and KO mice were harvested and homogenized as described above for Western blotting. Lysates for all WTs within a cohort (Cohort 1: 2F + 3M; Cohort 2: 3M; Cohort 3: 4F; Cohort 4: 6M), and all KOs within a cohort were pooled (Cohort 1: 2F + 3M; Cohort 2: 3M; Cohort 3: 4F; Cohort 4: 6M) to allow for sufficient starting material. Protein sample buffer was exchanged with 2D lysis buffer (30 mM Tris-HCl, pH 8.8, containing 7 M urea, 2 M thiourea and 4% CHAPS), with subsequent protein concentrations determined by Bio-Rad Protein Assay Kit II #500-0002 according to manufacturer’s protocol and all sample concentrations adjusted to 6 mg/mL. These 4 sample sets were first characterized by a proteomic 2-D DIGE study, with total protein expression differences identified by MALDI-TOF MS and TOF/TOF tandem MS/MS reported elsewhere [61]. Subsequently, samples from Cohort 1 and 2 were combined as were samples from Cohort 3 and 4, to ultimately yield 2 WT samples and 2 KO samples with sufficient starting material for a phosphoproteomics study. For each sample, 30μg of protein was mixed with the sample buffer (8 M urea, 4% CHAPS, 20 mg/ml DTT, 2% pharmalytes and trace amount of bromophenol blue) and 100 ul destreak solution and rehydration buffer (7 M urea, 2 M thiourea, 4% CHAPS, 20 mg/ml DTT, 1% pharmalytes and trace amount of bromophenol blue) for a total volume of 250 ul. Labeled samples were run with IEF (pH3-10 Linear) following the protocol provided by Amersham BioSciences, with IPG strips subsequently incubated for 15 minutes in freshly made equilibration buffer-1 (50 mM Tris-HCl, pH 8.8, containing 6 M urea, 30% glycerol, 2% SDS, trace amount of bromophenol blue and 10 mg/ml DTT) and rinsed for 10 minutes in freshly made equilibration buffer-2 (50 mM Tris-HCl, pH 8.8, containing 6 M urea, 30% glycerol, 2% SDS, trace amount of bromophenol blue and 45 mg/ml DTT). After rinsing in the SDS-gel running buffer, IPG strips were transferred into 12% SDS-gels that were run at 15°C until the dye front ran out of the gels. Gel images were scanned immediately following the SDS-PAGE using Typhoon TRIO (GE Healthcare), with the gels subsequently stained using Pro-Q ® Diamond Phosphoprotein Gel Stain following manufacturer’s protocol (Invitrogen) and scanned again. Gels were analyzed using DeCyder v6.0 (GE Healthcare), along with manual inspection, to select differentially phosphorylated spots. Spots were then mapped in DeCyder and pixel counts exported for calculation of the phospho/total protein ratios. Spots with a KO/WT ratio ≤0.6 (with neither sample >0.7) or a ratio ≥1.58 (with neither sample <1.5) were subsequently identified by MALDI-TOF MS and TOF/TOF tandem MS/MS on an AB SCIEX TOF/TOF™ 5800 System (AB SCIEX). MALDI-TOF mass spectra were acquired in reflectron positive ion mode, averaging 4000 laser shots per spectrum. TOF/TOF tandem MS fragmentation spectra were acquired for each sample, averaging 4000 laser shots per fragmentation spectrum on each of the 10 most abundant ions present in each sample (excluding trypsin autolytic peptides and other known background ions). Peptide mass and the associated fragmentation spectra were then submitted to a GPS Explorer workstation equipped with the MASCOT search engine (Matrix science) to search the database of National Center for Biotechnology Information non-redundant. Signals with protein scores/C.I.% and total ion scores/C.I.% >100 were entered into String v11.0 (string-db.org; accessed 07/27/2021) for pathway analyses.
Data Analysis
Data were collected blind to treatment and experiments were designed to counterbalance technical variables across biological variables. Data points greater than two standard deviations away from the mean were removed as outliers prior to analyses, as previously described (e.g., [6, 8, 76, 77]). Outliers removed/total n: Figure 1O, 1/24; Figure 3A, 4/89; Figure 3B, 3/73; Figure 3C, 7/110; Figure 3I, 4/73; Figure 3L, 2/43; Figure 3M, 5/4; Figure 4L, 2/48; Figure S6F, 2/38; Figure S7C, 5/108. Data were analyzed for effect of genotype, age, behavioral parameter (e.g., bead, food, etc), and sex for experiments with greater than 6/sex/genotype [6-9]). Where datasets met assumptions of normality (Shapiro-Wilk test) and equal variance (Levene’s test), the following parametric statistical analyses were run on Sigmaplot 11.2 (San Jose, CA, USA): ANOVA (F), Student’s t-test (t), one-sample t-test (t; Table 1). In the case of one-sample t-tests (determining whether or not a given group demonstrated memory), a false-rate discovery (FDR) correction was applied to all P-values within an experiment to mitigate the risk of Type I error associated with multiple comparisons. Where normality and/or equal variance assumptions failed, the following non-parametrical statistical analyses were run: Kruskal-Wallis ANOVA (H), Mann-Whitney rank sum test (T), or Wilcoxon Signed Rank Test (Z). Repeated measures analyses were used where appropriate (e.g., in analysis of behavior across multiple trials). Post hoc analyses were performed according to the Student-Newman-Keuls or Dunn’s method and significance was defined as P<0.05.
To determine whether or not a given group exhibited a signifciant memory in a given task at a given time point, preference ratios for each group were analyzed by one-sample t-test to determine if their performance significantly differed from chance (i.e., 0), with resulting P-values corrected for multiple comparisons within an experiment using false discovery rate correctio (FDR).
RESULTS
Age-related increases in PDE11A mRNA are conserved
We previously showed that PDE11A4 mRNA and protein expression in the hippocampus increase across the lifespan in rodents [7, 8]. Here we assessed age-related changes in expression in human HIPP by mining data from the Allen Institute for Brain Sciences Brainspan database. As in rodents, hippocampal PDE11A mRNA expression in humans increased across the lifespan (Figure 1A). The large 2.66-fold increase in expression between the prenatal and adulthood period (18-44 years of age) is unique to PDE11A, as PDE5A—the closest related PDE—shows no such increase in hippocampus (Figure 1B). Further, our previous work on the next closest-related PDEs shows only a modest 0.36-fold increase in PDE2A mRNA along with a 0.42-fold and 0.54-fold decrease in PDE9A and PDE10A mRNA, respectively [63, 64]. Next we determined if this age-related increase in PDE11A mRNA expression might be related to increased transcript stability by examining expression of p54nrb/NONO and the exoribonuclease XRN2, both of which form a complex with PDE11A4 mRNA to target it for degradation [3]. In concert with the age-related increases in PDE11A mRNA expression, we see that p54nrb/NONO and XRN2 mRNA decrease (Figure 1C-D). Given evidence that TBI can exacerbate age-related decreases in hippocampal cAMP levels [24], we next mined data from the Aging, Dementia, and Traumatic Brain Injury database that includes data from a group of 75+ year-old patients characterized with regard to their history of TBI and dementia. Among elderly humans with a history of TBI, hippocampal PDE11A mRNA expression was significantly higher in those that developed dementia versus those that did not (Figure 1E). In contrast, there was no difference between TBI patients with vs. without dementia in terms of PDE2A (-dementia, 0.95 ±0.04; +dementia 0.90 ±0.03), PDE5A (-dementia, 0.96 ±0.02; +dementia, 0.97 ±0.03), or PDE10A expression (-dementia, 0.83 ±0.03; +dementia 0.86 ±0.04). Together, these data suggest that PDE11A expression uniquely increases with age in the human hippocampus in part due to increases in transcript stability and that these age-related increases can be exacerbated in those with dementia and a history of TBI.
Age-related increases in PDE11A4 protein expression occur in a compartment-specific manner
As noted above, PDEs are discretely localized to specific brain regions and subcellular domains [30, 31]. At the regional level, PDE11A4 mRNA is restricted to the extended hippocampal formation, with enrichment in VHIPP versus DHIPP. At the subcellular level, PDE11A4 is enriched in the cytosol vs. membrane and nucleus, which stands in contrast to PDE2A and PDE10A—the two most closely related PDEs expressed in hippocampus—both of which are greatly enriched in the membrane versus cytosol [78]. As such, we determined if age-related increases in PDE11A4 were uniformly distributed or were restricted to discrete, potentially ectopic, compartments. In situ hybridization shows that PDE11A4 mRNA in the middle-aged mouse brain remains largely restricted to the extended hippocampal formation (Figure 1F), including CA1, subiculum, the AHi, and—in a subset of male and female mice—the amygdalar-striatal transition area (AStr) that sits just anterior to the hippocampus (Figure1H). Note that expression in the AStr had not been noted with previous weaker probes [7-9]. In DHIPP, there was no effect of age on PDE11A4 mRNA expression (Figure1I); however, protein expression was increased in the nuclear and cytosolic compartments of old versus young mice (Figure 1K; see Figure S1A for validation of biochemical fractionations). In contrast, select subfields of the VHIPP did show increased PDE11A mRNA expression in middle-aged versus young mice (i.e., the superficial and deep subfields within distal CA1 and the ventral subiculum; Figure 1J), and PDE11A4 protein was increased only in the membrane compartment of old versus young mice (Figure 1L). Native gels suggest that PDE11A4 is not associated with the membrane directly (e.g., via palmitoylation), but rather via protein-protein interactions (Figure S1B). Phosphorylation is a post-translational modification known to alter PDE protein-protein interactions [32]. As such, we validated antibodies that recognize phosphorylation of PDE11A4 serines 117 and 124 (Figure S1C). Immunofluorescence with antibodies that detect all PDE11A4 (Figure 1M-N), PDE11A4-pS117 (not shown but see Source Data), or PDE11A4-pS117/pS124 (Figure 1M-N; see Figure S1 for antibody validation) confirmed the differential compartment-specific localizations of PDE11A4 protein in the aged DHIPP versus VHIPP, with PDE11A4 in VHIPP accumulating in filamentous structures that are rarely seen in young C57BL/6J mice (Figure 1O-P). This increased accumulation of PDE11A4 in the aged VHIPP is also observed in BALB/cJ (Figure S2A, S2C) and 129S6/SvEv mice (Figure S2D). We term these PDE11A4-filled structures ghost axons (see Figure 2) and find they are enriched for PDE11A4 that is phosphorylated at serine 117 and 124 (pS117/pS124; Figure 1M bottom and 1N). This age-related increase in PDE11A4-pS117/pS124-filled structures may be deleterious since associative memory consolidation in the social transmission of food preference assay—but not memory retrieval—normally triggers a reduction in their presence (Figure 1Q).
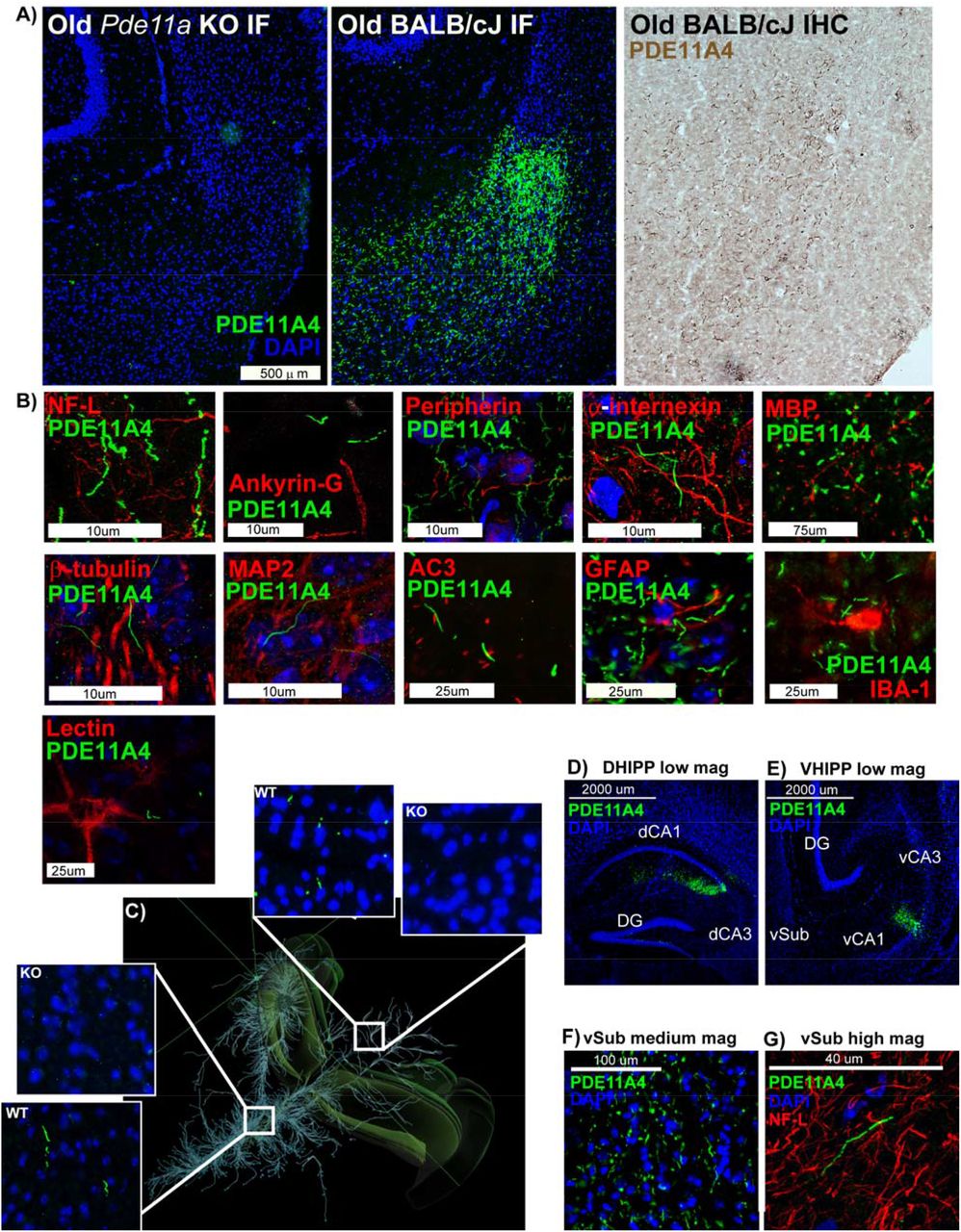
A) Immunofluorescence with the PDE11A4-pS117/pS124 antibody (green; nuclear marker DAPI shown in blue) shows no signal in the ventral subiculum (vSub) of a Pde11a KO mouse on a BALB/cJ background, but does show the accumulation of PDE11A4-filled filamentous structures throughout the ventral subiculum (vSub) of a Pde11a WT mouse on a BALB/cJ background. The structures can also be visualized in a WT mouse using immunohistochemistry. B) Staining for PDE11A4-filled structures appears very much like that of intermediate filaments in terms of diameter and tortuosity; however, PDE11A4-filled structures fail to colocalize with markers for axons (NF-L, Ankryin-G, peripherin, internexin, MBP), neurons (tubulin), dendrites (MAP2), cilia (AC3), glia (IBA-1--microglia, GFAP--astrocytes) or perineuronal nets (lectin). C) Given these structures look very much like axonal markers, coupled with the fact that several age-related diseases are associated with the aberrant accumulation of proteins in axons [4, 5] we determined if such PDE11A4-filled structures could be found in other terminal fields of vCA1 projections that themselves do not express PDE11A4 mRNA (terminal fields defined by Allen Institute for Brain Science’s Mouse Brain Connectivity; projections shown from top left perspective of mouse brain). Indeed, PDE11A4-filled structures can be found in such terminal fields (shown: hypothalamic nuclei in left insets, colliculi in right insets), strongly suggesting PDE11A4 is accumulating in terminal regions of axons projecting from CA1. D) To confirm this, we injected GFP-tagged mPDE11A4 into the dorsal and ventral CA1 (dCA1, vCA1) of Pde11a KO mice on the C57BL/6J background. Despite the fact that PDE11A4 expression was only being generated in CA1, E) PDE11A4-filled structures emerged in vSub consistent with an accumulation of the protein within axons projecting from CA1 to vSub. Like endogenous PDE11A4-filled structures, recombinant PDE11A4-filled structures appear identical in size and shape to NF-L, but fail to colocalize with NF-L. These studies, along with electron microscopy results (Figure S3), suggest high levels of PDE11A4 expression leads to an accumulation of PDE11A4 in axons that either occludes co-localization of other axonal protein or possibly leads to the degeneration of the surrounding axon as occurs with Tau ghost tangles [4, 5], hence the adoption of the term ghost axons herein. NF-L, Neurofilament light chain; MBP, myelin basic protein; MAP2, microtubule associated protein; AC3, adenylyl cyclase 3; IBA-1, ionized calcium binding adaptor molecule 1; GFAP, glial fibrillary acidic protein. Histogram stretch, brightness, and/or contrast of images adjusted for graphical clarity.
Preventing age-related increases in PDE11A is sufficient to prevent cognitive decline of social associative memory
Human studies show that associative memories involving verbal and/or non-verbal stimuli (e.g., faces and names) are more susceptible to age-related cognitive decline than are recognition memories for individual items [13-19]. We were able to recapitulate a differential sensitivity of aLTMs vs recognition LTMs (rLTMs) in C57BL/6J mice using social memory tasks (Figure S4). To test the hypothesis that ARCD of aLTMs is driven by the age-related increases in PDE11A4 expression described above, we compared male and female young vs. old Pde11a WT and KO mice in social transmission of food preference (STFP). [Note that female Pde11a WT and KO mice are indistinguishable in terms of cycle length or reproductive senescence (Figure S5), consistent with our previous report showing no gross peripheral pathology in middle-aged KO mice [9].] Our initial focus on social aLTM stems from the fact that in young adult mice, PDE11A4 regulates social behaviors and social memory, but not non-social memory [1, 2, 6, 7, 9]. To assess the specificity of effects for aLTMs, we also tested these mice for social odor recognition memory (SOR) and non-social odor recognition memory (NSOR). In all assays, we tested short-term memory (STM), recent LTM 24 hours after training, and remote LTM 7 days after training given that we previously showed deletion of PDE11A produces transient amnesia for social memories in adolescent and young adult mice (e.g., intact STM, impaired recent LTM, and intact/improved remote LTM). Old Pde11a WT mice exhibited STM for STFP but it was weaker than that of young WT mice (Figure 3A, S6A; Table 1). This deficit worsened as time progressed with old WT mice failing to demonstrate any aLTM for STFP at the 24-hour and 7-day timepoints (Figure 3B-C, S6B-C). Despite the fact that both young and old Pde11a KO mice demonstrate a transient amnesia for recent aLTM 24 hours after STFP training (Figure 3B, S6B), old KO mice show stronger STM and remote aLTM 7 days after STFP training relative to old WT mice (Figure 3A-C, S6A-C). Middle-aged Pde11a HT and KO mice similarly show enhanced remote aLTM relative to middle-aged WT mice 7 days after STFP training (Figure S6F), despite middle-aged KO mice exhibiting no recent aLTM (Figure S6B). Consistent with our observations in the C57BL/6J mice (Figure S4), there was no ARCD of SOR or NSOR in any genotype at any time point; however, both young and old Pde11a KO mice exhibited a transient amnesia for SOR in line with our previous work in young mice (Figure 3D-F) [6]. Importantly, the effect of genotype on these memory tasks cannot be explained by performance aspects of these assays. Although old mice consistently ate less food during STFP and spent less time investigating during SOR than young mice, and females ate more food during STFP and spent more time investigating during SOR than males as we previously described [6], there were no main effects or interactions with the effect of genotype (Table S1). This suggests the effects of age and genotype on memory versus food consumption and total time investigating social odors are dissociable. Thus, old Pde11a KO mice exhibit transient amnesia for social but not non-social memories much like young KO mice, and the ability of Pde11a deletion to prevent ARCD of STFP aLTMs is related to the associative nature of the memory as opposed to age-related changes in recognizing the social or non-social components.

A) WT-Y (n=16M,7F), KO-Y (n=18M,6F), WT-O (n=13M,7F), and KO-O mice (n=15M,3F) each showed STFP STM when memory was defined by the amount of food eaten (top graphs) or the time spent eating the food (bottom graphs); however, old WT mice in particular demonstrated weaker memory than young mice (food effect of age: F(1,81)=6.59, p=0.012; time effect of age: F(1,81)=5.02, p=0.028; time effect of genotype: F(1,81)=5.02, P=0.028). B) 24 hours after training, only WT-Y mice (n=7M,8F) showed STFP memory, with KO-Y (n=8M,6F), KO-O (n=8M,12F), and WT-O mice (n=8M,13F) performing significantly worse (food effect of genotype x age: F(1,62)=4.31, P=0.042; Post hoc vs. WT-Y: WT-O P=0.028, KO-Y P=0.002, KO-O P=0.029; time effect of genotype x age: F(1,60)=3.83, P=0.055). C) 7 days after STFP training, WT-Y (n=14M,12F), KO-Y (n=18M,10F), and KO-O mice (n=14M,12F) exhibit stronger remote aLTM than WT-O mice (n=13M,11F; food effect of genotype x age: F(1,96)=11.68, P=0.0009; Post hoc: WT-O vs each group, P<0.001; time effect of genotype x age: F(1,95)=13.34, P=0.0004; Post hoc: WT-O vs each group, P=0.0001). D) All groups demonstrated strong STM for SOR, including WT-Y (n=8M,7F), KO-Y (n=7M,7F), WT-O (n=8M,7F), and KO-O (n=8M,7F) with no difference among groups. E) 24 hours after SOR training, WT-Y (n=4M,5F) and WT-O mice (n=7M,6F) exhibit significantly stronger rLTM than KO-Y (n=5M,4F) and KO-O mice (n=6/sex; effect of genotype: F(1,38)=5.67, P=0.022). F) 7 days after SOR training, however, KO-Y (n=10M,8F) and KO-O mice (n=10M,8F) exhibit stronger rLTM than WT-Y (n=9/sex) and WT-O mice(n=11M,7F; (F(1,67)=5.25, P=0.025) consistent with our previous observation in young KO mice [6]. G) Each group showed strong NSOR STM, with old mice slightly outperforming young mice (n=3/sex/age/genotype; effect of age: F(1,20)=5.08, P=0.036). H) There were no differences among groups, however, for NSOR recent rLTM (WT-Y n=4M,5F; KO-Y n=5/4F; WT-O n=6M,9F; KO-O n=6M,9F) or I) NSOR remote rLTM (WT-Y n=7M,9F; KO-Y n=9M,7F; WT-O n=9M,6F; KO-O n=13M,9F). J) Next, we determined if overexpressing PDE11A4 in dorsal and ventral CA1 old KO mice (see Figure 2D for expression pattern observed 2 weeks following injection and Figure S8A for map of expression pattern observed >2 months injections) would be sufficient to eliminate their protective phenotype by infusing a lentivirus expressing either GFP alone or a GFP-tagged mPDE11A4 [6] that exhibits robust catalytic activity (n=8 biological replicates of HT-22 cells/group; cGMP: (t(14)=-16.58, P<0.001; cAMP: t(14)=-14.61, P<0.001). K) Relative to infusion of GFP only, infusion of PDE11A4 significantly impaired remote aLTM for STFP (KO-GFP n=2M,3F; 11A4 n=4M/3F; t(10)=-2.24, P=0.049) and L) SOR (KO-11A4 n=10M,12F; KO-GFP n=8M,11F; effect of virus: F(1,37)=11.47, P=0.002), M) but not NSOR (KO-11A4 n=8M,11F; KO-GFP n=7M/10F; effect of virus: F(1,32)=.09, P=0.763). N) Overexpressing PDE11A4 in KOs is also sufficient to elicit aging-like biochemical phenotype in the VHIPP, namely decreased CREB in the nucleus (GFP n=6M,6F; 11A4 n=6M,5F; t(21)=2.11, P=0.047). *vs WT or GFP, P=0.049 to <0.001, ^vs. Young, P=0.028 to <0.001; #has memory (i.e., significantly >0; see Table 1 for individual statistics), P=0.018 to <0.001. Data plotted as individual points (females as circles, males as squares) and expressed as mean ±SEM. Brightness and/or contrast of images adjusted for graphical clarity. PS—Ponceau stain.
Mimicking age-related overexpression of PDE11A4 in Pde11a KO is sufficient to trigger aging-like deficits in biochemical and behavioral endpoints
To determine whether Pde11a KO mice are protected against age-related cognitive decline specifically due to the loss of PDE11A4 signaling in the aged brain or due to compensatory changes across the lifespan, we determined if overexpressing PDE11A4 in old KO mice would be sufficient to eliminate their protective phenotype. Infusions of a lentivirus expressing either GFP alone or a GFP-tagged mPDE11A4 [6] were targeted to dorsal and ventral CA1 of KO mice (KO-GFP vs. KO-11A4, respectively; Figure 2D and Figure S8A) since our previous work identified this as the hippocampal subfield within which PDE11A4 critically regulates social memory consolidation [6]. The recombinant GFP-tagged mPDE11A4 behaves similarly to endogenous mPDE11A4 in that it exhibits robust cAMP/cGMP-hydrolytic activity (Figure 1J), undergoes phosphorylation at S117/S124 (Figure S8B-C), and traffics to relevant compartments of the hippocampus (i.e, the cell body and dendritic layer of dorsal and ventral CA1, Figure 1D; ghost axons vSub, Figure 1E). As expected, KO-GFP mice demonstrated strong remote LTM for STFP, much like unsurgerized KO mice, but overexpression of PDE11A4 in the aged KO CA1 was sufficient to reverse this protective phenotype (Figure 3K). There was no difference between treatments in terms of the total amount of food eaten during the test (KO-GFP, 1.07 ±0.11 gm; KO-11A4, 1.01 ±0.14 gm; t(11)=-0.34, P=0.737). Although KO-GFP and KO-11A4 mice learned equally well during training for SOR (Figure S8D), KO-GFP mice demonstrated strong remote rLTM for SOR but KO-11A4 did not (Figure 3L) as previously reported for young adult mice [6]. In contrast, KO-GFP and KO-11A4 mice showed both strong learning (Figure S8E) and remote rLTM for NSOR (Figure 3M). Overexpressing PDE11A4 in KOs was also sufficient to elicit aging-like decreases in VHIPP CREB activity [29], as indicated by reduced nuclear localization of CREB (Figure 3N). Together, these data suggest the protective phenotype observed in old Pde11a KO mice is directly related to the absence of PDE11A4 signaling in the aged brain as opposed to a compensatory mechanism triggered by the chronic loss of PDE11A across the lifespan.
RNA sequencing and phosphoproteomics of the VHIPP implicate many of the same pathways in the protective effect of PDE11A deletion
To gain insight into how the absence of PDE11A4 in the adult hippocampus provides protection against ARCD, we conducted an RNA sequencing study in one cohort of mice followed by a confirmatory phosphoproteomic study in a second cohort of mice using VHIPP samples from aged Pde11a KO and WT mice. Importantly, changes in gene expression and protein phosphorylation were enriched in many of the same pathways (Table 2). Consistent with PDE11A4 regulating cyclic nucleotide signaling and social behaviors, both studies identified the cGMP-PKG signaling pathway and oxytocin signaling pathway. Interestingly, both studies also identified the calcium signaling, glutamatergic synapse, cholinergic synapse, and Alzheimer’s disease pathways, all of which have been implicated in ARCD and/or dementia (see Discussion). Given the age-related accumulation of PDE11A4 in ghost axons, it is also interesting to note that both studies identified pathways related to the axon and axon guidance, regulation of transport, chemical synaptic transmission, as well as the synapse, presynapse, and synaptic vesicle. An effect of PDE11A deletion on these latter pathways is consistent with PDE11A4 colocalizing with adaptin, a marker of intracellular transport vesicles (Figure S9C). Additional age-related pathways of note that were identified by one technique but not the other included longevity regulating pathway (RNA sequencing, strength 0.46, FDR-P=0.001), longevity regulating pathway-multiple species (RNA sequencing, strength 0.55, FDR-P<0.001) and cellular senescence (phosphoproteomics, strength 1.25, FDR-P=0.008).
Pathways identified as significantly enriched for both mRNA changes (a total of 1597 identified) and phosphoproteomic changes (a total of 22 identified) in VHIPP of old Pde11a KO vs WT mice.
Phosphorylation of serine 117 (S117) and serine 124 (S124), but not serine 162 (S162), are sufficient to increase PDE11A4 expression and drive PDE11A4 accumulation
Previous studies have suggested PDE11A4 regulates its own protein expression levels [1, 2]. As such we determined if the increased phosphorylation of PDE11A4 at S117 and S124 observed in the aged hippocampus (Figure 1M-P and Figure S2) was sufficient to cause aging-like increases in PDE11A4 protein expression and accumulation. To do so we tested the effect of phosphoresistant (alanine) versus phosphomimic mutations (aspartate, D) at these sites in vitro. We also assessed the specificity of these signals by similarly targeting S162, as it is also a PKA/PKG-regulated serine in the N-terminal of PDE11A4 [58, 78]. Across multiple experiments and cell lines (i.e., COS-1 and HT-22), preventing phosphorylation at S117/S124 or S124 alone was sufficient to decrease PDE11A4 protein expression while mimicking phosphorylation at S117/S124 or S124 alone increased expression (Figure 4A-C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Previous studies suggested PDE11A4 regulates its own protein expression levels [1, 2]. As such we determined if the increased phosphorylation of PDE11A4 at S117 and S124 observed in the aged hippocampus (Figure 1) was sufficient to cause age-related increases in PDE11A4 protein expression and accumulation. To do so we tested the effect of phosphoresistant (alanine, A) versus phosphomimic (aspartate, D) mutations. A) In 2 experiments using HT-22 cells harvested 24 hours following transfection (combined data shown here), preventing phosphorylation at S117/S124 (A/A) decreased PDE11A4 protein expression while mimicking phosphorylation (D/D) increased protein expression relative to wild-type (WT; n=12 biological replicates/group; F(2,33)=17.03, P<0.001; Post hoc: WT vs. A/A P=0.011, WT vs. D/D P=0.004, A/A vs. D/D P<0.001. B) This pattern was replicated in HT-22 cells when preventing vs. mimicking phosphorylation only at S124 (S124A vs S124D, respectively; n=4 biological replicates/group; F(2,9)=35.94, P<0.001; Post hoc: WT vs. S124A P=0.001, WT vs. S124D P=0.005, S124A vs. S124D P<0.001). C) COS1 cells were then harvested 24-72 hours following transfection and expression levels for each mutant were normalized to their own 24-hour baseline in order to determine if the differential expression noted 24 hours following transfection would escalate over time. Indeed, across 2 experiments (combined data shown) the difference in expression between S117A/S124A (A/A) vs. S117D/S124D (D/D) continues to grow over time (n=12 biological replicates/mutant/time; effect of mutation x time: F(4,44)=4.89, P=0.002; Post hoc A/A 60hr vs D/D 60hr P=0.003, A/A 72hr vs D/D 72hr P<0.001), with D/D showing significant increases in expression over its own 24hr baseline as early as 48 hours following transfection (Post hoc, D/D 24hr vs DD 48hr P=0.013) but A/A not showing a significant increase in expression over its own 24-hour baseline until 72 hours after transfection (A/A 24hr vs A/A 72hr P=0.011). D) Immunocytochemistry of transfected COS1 cells with an antibody that recognizes PDE11A4-pS117 on both mouse PDE11A4 (mPDE11A4) and E) human PDE11A4 (hPDE11A4) shows PDE11A4-pS117 is enriched in the accumulated pools of PDE11A4. The lack of pS117 signal in F) the phosphoresistant mutant (S117A) and the G) no primary antibody conditions argues the pS117 signal observed is specific. Next we determined if preventing/mimicking phosphorylation at S117 and S124 would be sufficient to reduce/increase trafficking of mPDE11A4 into puncta. Although S117A alone had no effect, both S124A and S117A/S124A reduced the presence of PDE11A4-filled puncta in H) COS-1 cells (n=15-18 biological replicates/group; F(3,62)=7.02, P<0.001; Post hoc vs WT: S124A P=0.004, S117A/S124A P=0.01), I) HEK293T cells (n=8 biological replicates/group; F(3,28)=16.45, P<0.001; Post hoc: WT vs. S124A and S117A/S124A, P<0.001), and J) HT-22 cells (n=11-12 biological replicates/group; F(3,42)=10.78, P<0.001; Post hoc vs. WT: S124A P=0.004, S117A/S124A P=0.005). In contrast, S117D and S124D alone or in combination increased the presence of PDE11A4-filled puncta in K) COS-1 cells (n=8 biological replicates/group; F(3,28)=13.17, P<0.001; Post hoc vs. WT: S117D P=0.008, S124D P=0.002, S117D/S124D P<0.001; Post hoc vs. S117D/S124D: S117D P=0.007, S124D P=0.023), L) HEK293T cells (n=7-8 biological replicates/group; F(3,26)=5.29, P=0.006; Post hoc vs WT: S117D P=0.033, S124D P=0.012, S117D/S124D P=0.004), and M) HT-22 cells (n=12 biological replicates/group; F(3,44)=5.39, P=0.003; Post hoc vs S117D/S124D: WT P=0.003, S117D P=0.008, S124D P=0.005). The ability of S117A/S124A to reduce the presence of PDE11A4-filled puncta does not require phosphorylation of S162 in Q) COS-1 cells, (n=12 biological replicates/group; F(2,33)=12.63, P<0.001; Post hoc: WT vs. S117A/S124A/S162A, P<0.001) or R) HT-22 cells (n=12 biological replicates/group; F(2,33)=22.89, P<0.001; Post hoc: WT vs. S117A/S124A/S162A, P<0.001). In contrast, phosphomimic mutation at S162 is able to prevent S117D/S124D-induced accumulation of PDE11A4 in S) COS-1 cells (7-8 biological replicates/group; F(2,20)=44.10, P<0.001; Post hoc: S162D vs. S117D/S124D/S162D P=0.885) and T) HT-22 cells (n=12 biological replicates/group; F(2,33)=447.17, P<0.001; Post hoc: S162D vs. S117D/S124D/S162D P=0.201). *vs. WT, P=0.011-<0.001; #vs. other mutant(s), P=0.023-<0.001; @greater than 24hr, P=0.011-<0.001; ^greater than 48hr, P=0.026-<0.001; &greater than 60hr, P=0.019. Data plotted as individual data points and mean ±SEM. Histogram stretch, brightness, and/or contrast of images adjusted for graphical clarity.
As is observed in the aging brain, mouse and human PDE11A4 accumulates in punctate structures in vitro and PDE11A4-pS117 is preferentially found in these puncta (Figure 4D). As such, we next determined if preventing/mimicking phosphorylation at S117 and S124 would be sufficient to reduce/increase trafficking of mPDE11A4 into puncta. Although S117A alone had no effect, both S124A and S117A/S124A reduced the presence of PDE11A4-filled puncta in COS-1, HEK293T and HT-22 cells (Figure 4J-L). In contrast, S117D and S124D alone or in combination increased the presence of PDE11A4-filled puncta in COS-1 and HEK293T cells (Figure 4M-N); however, only S117D/S124D in combination was sufficient to increase puncta in HT-22 cells (Figure 4O). The ability of phosphorylation to increase the accumulation of PDE11A4 in puncta appears to be selective for S117 and S124 in that a phosphomimic mutation at S162 (i.e., S162D) had the opposite effect of reducing the accumulation of PDE11A4 in COS-1, HEK293T, and HT-22 cells (Figure 4P-R). In COS-1 and HT-22 cells, the ability of S117A/S124A to reduce the presence of PDE11A4-filled puncta does not require phosphorylation of S162 (Figure 4S); however, a phosphomimic mutation of S162 is able to prevent S117D/S124D-induced accumulation of PDE11A4 (Figure 4T). Together, these data suggest that pS117/pS124 is a key molecular mechanism by which PDE11A4 expression and accumulation increase with age.
DISCUSSION
Here we show that age-related increases in hippocampal PDE11A4—but not PDE5A—are conserved across humans and rodents, as is the vulnerability of associative memories—but not recognition memories—to ARCD. Our work is consistent with previous studies showing cGMP and cAMP are decreased in the aged and demented hippocampus (rodents and humans), particularly when there is a history of TBI [22-24]. Age-related increases in PDE11A4 appear to be driven by factors influencing both transcript and protein stability, with age-related increases in PDE11A4 protein expression occurring in a subcellular compartment-specific manner. Strikingly, age-related increases in PDE11A4 in the VHIPP ectopically accumulate in filamentous structures we term ghost axons due to increased phosphorylation of PDE11A4 at S117/S124 (PDE11A4pS117/pS124; Figure 1M-N and Figure 4A-C), reminiscent of proteinopathies caused by hyperphosphorylation of tau (c.f., [29]). This age-related increase in PDE11A4-pS117/pS124 is thought to be deleterious since STFP memory consolidation is normally associated with reduced levels (Figure 1Q). Indeed, we show here that preventing age-related increases in PDE11A via genetic deletion is sufficient to protect against ARCD of social associative memories, while mimicking age-related overexpression of PDE11A4 in CA1 of old Pde11a KO mice is sufficient to cause aging-like impairments in memory (Figure 3K) and CREB function (Figure 3N). This suggests the protective effect of PDE11A deletion in old mice is due to the absence of PDE11A4 signaling in the aged brain and a subsequent increase in VHIPP CREB activity [2], as opposed to a compensatory mechanism triggered by the chronic loss of PDE11A across the lifespan (Figure 3K-M). RNA sequencing and phosphoproteomics analysis of the VHIPP from old Pde11a KO versus WT mice suggest PDE11A4 overexpression contributes to ARCD of social memories primarily via the cGMP-PKG pathway as opposed to the cAMP-PKA pathway, and confirm downstream effects on multiple pathways associated with ARCD and Alzheimer’s disease (Table 2). Together, these results suggest that increases in PDE11A expression that occur with age and TBI-associated dementia contribute to cognitive decline.
Age-related increases in PDE11A4 are driven by increased mRNA stability and phosphorylation of N-terminal residues S117 and S124
Parsimoniously, age-related increases in PDE11A protein expression might arise from increased transcription/translation and/or increased transcript/protein stability. With regard to PDE11A4 transcript stability, the nuclear protein p54nrb/NONO and the exoribonuclease XRN2 appear to be major players [3]. When p54nrb/NONO activity is compromised, XRN2 fails to bind PDE11A4 transcripts and PDE11A4 mRNA expression significantly increases, particularly in the nucleus [3]. Interestingly, cAMP stimulates PDE11A4 mRNA degradation via p54nrb/NONO [3], and cellular senescence has been associated with a loss of nuclear p54nrb/NONO function [79]. Together, these studies suggest that age-related decreases in cAMP signaling [29] lead to increased PDE11A mRNA expression, at least in part, by virtue of the age-related reductions in p54nrb/NONO-XRN2 expression measured here (Figure 1C-D). That said, the fact that mouse DHIPP exhibits age-related increases in PDE11A4 protein in absence of any upregulation of mRNA suggests there are additional factors at play regulating translation and/or protein stability.
Here, we identified PDE11A4-pS117/pS124 as a key intramolecular signal that increases both protein expression and accumulation of PDE11A4. Post-translational modifications have been shown to regulate the subcellular compartmentalization of other PDE families in vitro, including PDE2, PDE4, PDE5, and PDE10 (c.f., [32]). For example, phosphorylation of PDE10A blocks palmitoylation and prevents membrane insertion [80]. PDE11A4 most likely associates with the membrane by binding to a macromolecular complex as opposed to direct membrane insertion, given native gels of membrane fractions show PDE11A is localized to large macromolecular complexes and is not found as a dimer (Figure S1B). In contrast, PDE11A4 appears as a free dimer and as part of macromolecular complexes in the nuclear and cytosolic fractions. We demonstrate that pS117, pS124 and pS162 all regulate the subcellular localization of PDE11A4, with pS117/pS124 promoting the accumulation of PDE11A4 and pS162 causing dispersal. PKA and PKG phosphorylate S117 and S162 in vitro [44] and are predicted to phosphorylate S124 [58]. Thus, PDE11A4 may regulate its own function through a direct feedback/feedforward loop (PDE11A4→cAMP/cGMP→PKA/PKG→PDE11A4-pS117/pS124/pS162), as has been described for other PDE families [32].
Age-related increases in PDE11A occur in a subcellular compartment-specific manner
We find here that age-related increases in VHIPP PDE11A4 do not occur in a uniform/distributed manner, but rather are ectopically localized. Age-related increases in VHIPP PDE11A4 are restricted to the membrane fraction, a compartment that normally expresses very low levels of PDE11A4 (Figure S1B; [78]). These findings add to a growing body of literature showing that PDE11A4 in the VHIPP membrane is the pool more readily altered by genetic and environmental factors [1, 59, 61]. We also find that age-related increases in VHIPP PDE11A4 accumulate ectopically in filamentous structures we term “ghost axons” (Figure 2). Ectopic protein expression and accumulation is a hallmark of age-related brain pathologies [36]. For example in Alzheimer’s disease, the hyperphosphorylation of tau within neurons leads to neurofibrillary tangle that overwhelm the neuron to the point of cell death [81]. Similarly, we show that age-related increases in the phosphorylation of PDE11A4-S117/S124 drives PDE11A4 to accumulate in filamentous structures that strongly resemble axonal intermediate filaments in terms of diameter and tortuosity. Despite their physical appearance, PDE11A4-filled structures fail to colocalize with markers of axons, dendrites, cilia, or glia (Figure 2), even when injection of recombinant PDE11A4 into CA1 of a Pde11a KO mouse generates PDE11A4-filled structures in brain regions distal to the injection site (Figure 2D). We speculate that PDE11A4 accumulation within the axon overwhelms the system, occluding the co-localization of other axonal proteins or causing degeneration of the surrounding structure. Thus, we name these PDE11A4-filled structures “ghost axons” in homage to tau “ghost tangles” [4, 81]. Notably, mRNA expression and phosphoproteomic changes in VHIPP of old Pde11a WT vs KO mice were enriched for pathways related to Alzheimer’s disease, axon guidance, regulation of transport, chemical synaptic transmission, as well as the axon, synapse, presynapse, and synaptic vesicle (Table 2). It will be of interest to future studies to determine whether the lack of PDE11A4 co-localization with axon markers may reflect occlusion or axonal degeneration.
Although the age-related increases in PDE11A4 protein expression are clearly deleterious to aLTM, as demonstrated by the protective phenotype observed in Pde11a KO mice, it is possible that the accumulation of PDE11A4 in ghost axons reflects a protective mechanism attempting to sequester and/or inactivate the excess protein. Such sequestration is reported to occur with select PDE4A isoforms when they are both catalytically inhibited and conformationally altered [82]). If so, it would be important for a PDE11A4-targed therapeutic to not only reduce the accumulation of PDE11A4, but also to promote its clearance from the system. Disruption of PDE11A4 homodimerization may then represent a refined mechanism for therapeutically targeting age-related proteinopathies in PDE11A4 since in vitro studies show that disrupting PDE11A4 homodimerization both reduces the punctate accumulation of PDE11A4 and promotes proteolytic degradation of membrane-associated PDE11A4 [59].
Age-related increases in PDE11A4 are more deleterious in combination with brain injury
In elderly humans with a history of TBI, hippocampal PDE11A mRNA expression is significantly higher in those that developed Alzheimer’s disease or a related dementia versus those that did not (Figure 1E). This is particularly interesting given that pathway analysis of our previous proteomics study of VHIPP from Pde11a WT vs KO mice [61], along with pathway analyses of the RNA sequencing and phosphoproteomics study reported herein (Table 2), identified the Alzheimer’s disease pathway. This TBI-associated dementia-related increase in PDE expression is unique to PDE11A in that PDE2A, PDE5A, and PDE10A expression remained unchanged. That said, we do acknowledge that inhibition and/or genetic deletion of several PDE families has shown efficacy in models of cognitive impairment [83-85], suggesting a PDE itself does not have to change with age/disease in order for its targeting to elicit therapeutic effects. The fact that age-related increases in PDE11A are exacerbated in elderly patients with TBI-associated dementia is consistent with reports in humans that TBI worsens age-related decreases in hippocampal cAMP levels [24] as well as ARCD [86]. Further, TBI accelerates the onset of dementia [87-89], Alzheimer’s disease [90], and Parkinson’s disease [91]. Even surgical injury in aged humans contributes to post-operative cognitive dysfunction and increases risk of developing dementia [92]. This raises the interesting possibility that brain injury itself causes an aging-like dysfunction of PDE11A4, leading those with higher baseline levels or larger age-related increases in PDE11A4 to transition from simple ARCD to dementia. Alternatively, age-related dysfunction of PDE11A4 may interact with some other pathology induced by injury, such as inflammation [59, 61, 66] or altered proteostasis (c.f., [93]). Indeed, whereas unsurgerized aged mice with elevated PDE11A4 expression demonstrate intact remote rLTM for SOR (Figure 3F), hippocampally-surgerized aged mice with elevated PDE11A4 expression show deficits (Figure 3L). Importantly, all mice show intact remote rLTM for NSOR, pointing to a hippocampus-specific deficit. It will be of interest to future studies to determine how PDE11A4 may play a role in the pathological consequence of brain injury across the lifespan.
Deletion of PDE11A triggers changes in pathways related to ARCD and dementia
Pathway analyses from an RNA sequencing and confirmatory phosphoproteomics study comparing VHIPP from old Pde11a WT vs KO mice identified multiple mechanisms by which PDE11A4 may contribute to ARCD and/or dementia (Table 2). Of note, the cGMP-PKG signaling was identified but not the cAMP-PKA pathway, which is consistent with literature reporting age-related decreases in cGMP but not cAMP in the hippocampus [29, 94, 95]. Indeed, several studies show nootropic effects of cGMP-elevating compounding in the context of ARCD and neurodegenerative diseases [85, 96-100]. In addition to regulating learning and memory, the cGMP-PKG pathway is also required for regulating sleep and circadian rhythms [101-103]. Sleep quality reduces with age [104-106] and age-related diseases [107-109], and sleep disruption appears to negatively impact cognitive function more severely in aged versus young adults [110, 111]. As such, we find it highly interesting that circadian entrainment was one of the top PDE11A-regulated pathways identified in this study, particularly when a PDE11A mutation (Y727C) has been associated with sleep quality in humans [51]. It will be of interest to future studies to determine if the ability of PDE11A deletion to prevent age-related cognitive decline of social aLTMs is a direct consequence of ameliorating age-related deficits in circadian rhythms and/or quality of sleep.
Conclusions
In summary, proteinopathies in PDE11A4 develop with age, and age-related increases in PDE11A4 expression that are conserved across species are sufficient to cause ARCD of social associative memories. PDE11A4 is unique because in brain it is the only PDE to be expressed preferentially in the hippocampal formation, a structure critical for associative memories [8, 9]. This—along with the fact that PDE11A is a highly druggable enzyme [58]—makes PDE11A a very attractive therapeutic target because itstands to selectively restore aberrant cyclic nucleotide signaling in a brain region affected by age-related decline without directly affecting signaling in other brain regions or peripheral organs that might lead to unwanted side effects [58]. By gaining a further understanding of the intramolecular signals controlling PDE11A4 subcellular localization (e.g., N-terminal phosphorylation, homodimerization), we hope to develop even more sophisticated therapeutics that can target disease-specific PDE11A4 proteinopathies.
FUNDING
Research supported by a SPARC Fellowship from the University of South Carolina Office of the Vice President for Research (KP), an NSF Graduate Research Fellowship (KP), University of South Carolina Magellan Scholar Program (WRC), the ASPET SURF Program (WRC), a SURF grant from the University of South Carolina Honors College (WRC), a senior thesis grant from the University of South Carolina Honors College (WRC), a Research Starter Grant in Pharmacology & Toxicology from the PhRMA Foundation (MPK), an ASPIRE award from the Office of the Vice President for Research from the University of South Carolina (MPK), a Research Development Fund Award from the University of South Carolina School of Medicine (MPK), a NARSAD Young Investigator Award from the Brain & Behavior Research Foundation (MPK), R01MH101130 from NIMH (MPK), R01AG061200 from NIA (MPK), P20GM109091 from NIGMS (MPK), and start-up funds from the University of Maryland School of Medicine (MPK). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ACKNOWLEDGEMENTS
The authors would like to thank Shweta Hegde, Nicolous Poupore, Abi Smith, David Smith, and Alex Sougianis for technical assistance with biochemical or behavioral assays as well as Sophie Bruckmeier for technical and editorial assistance. The authors would also like to thank Dr. Christina Sigurdson for introducing us to the concept of “ghost axons”, Drs. Steven Wilson and Seungjin Shin of the University of South Carolina Viral Core for lentiviral preparations, Dr. Mythreye Karthikeyan for the LAMP1 antibody and mycoplasma testing, and John at Applied Biomics for advice on sample preparation and experimental design of the phosphoproteomics study. Finally, the authors would like to thank Drs. David Oliver and Misha Shtutman at the University of South Carolina Functional Genomics Core for performing the alignment of the raw data generated by the Weil Epigenomics core along with the list of significantly changed mRNAs.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.
- 27.
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.
- 36.↵
- 37.↵
- 38.↵
- 39.
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.
- 98.
- 99.
- 100.↵
- 101.↵
- 102.
- 103.↵
- 104.↵
- 105.
- 106.↵
- 107.↵
- 108.
- 109.↵
- 110.↵
- 111.↵




