

ABSTRACT
Despite advancement in treatment, prostate cancer (PCa) remains the second leading cause of death among men. Neuroendocrine prostate cancer (NEPC) represents one of the most lethal forms of PCa and lacks life-prolonging treatment. Here we identified histone lysine demethylase KDM4A as a drive in NEPC progression and an effective therapeutic target. We found that KDM4A mRNA and protein are overexpressed in human and mouse NEPC compared to adenocarcinoma. Also, we showed that knockdown or knockout of KDM4A in NEPC cell lines suppressed cancer cell growth in vitro and in vivo. Importantly, inactivation of Kdm4a in a genetically engineered mouse model of prostate cancer led to reduced tumor burden, reduced incidence of NEPC, and prolonged overall survival. Mechanistically, we found that KDM4A KD led to suppression of MYC signaling through direct transcriptional regulation of MYC. Importantly, MYC signaling is hyper-activated in human and mouse NEPC. Furthermore, a potent pan-KDM4 inhibitor QC6352 significantly reduced NEPC cell growth in vitro and in vivo. Taken together, we demonstrated that KDM4A drives NEPC progression through regulation of MYC and targeting KDM4A can potentially be an effective therapeutic strategy for NEPC.
INTRODUCTION
Neuroendocrine prostate cancer (NEPC) is a highly lethal subtype of castration-resistant prostate cancer (CRPC) with a median survival of seven months after initial diagnosis,1-3 as compared to a median survival of 13 to 31 months in castration-resistant prostate adenocarcinoma, the more common subtype of CRPC.4 NEPC is characterized by attenuated androgen receptor (AR) signaling, the expression of neuroendocrine lineage markers (e.g., synaptophysin), uncontrolled hyperproliferation, and widespread metastasis (bone, liver, and lung). De novo NEPCs are rare (2%-5%); the majority arise as a mechanism of resistance from prostate adenocarcinoma treated with potent AR pathway inhibitors (ARPIs).5 The widespread use of ARPIs in non-metastatic CRPC and hormone-sensitive metastatic tumors has led to an increase in the incidence NEPC. Due to the lack of life-prolonging systemic therapies, there is an urgent need to better understand the mechanisms underlying the pathogenesis of NEPC.
Recent evidence suggests that epigenetic dysregulation is a hallmark of NEPC.5 Aberrantly expressed transcription factors (TFs) and regulators of DNA methylation and histone modification were found in NEPC preclinical models and patients.5 Extensive studies have led to the discovery of key regulators driving the transition from adenocarcinoma to NEPC.5 Among these epigenetic aberrations, histone lysine methylation, which is balanced by writers (histone lysine methyltransferase [KMT]) and erasers (histone lysine demethylases [KDM]), plays an important role in development and cancer, including prostate.6 Multiple lysine methylation modifiers (e.g., EZH2, WHSC1, DOT1L, LSD1/KDM1A, KDM4A, KDM4B)7-11 have been implicated in prostate cancer progression. However, their roles in NEPC are just started to emerge. For example, EZH2 has been recently implicated as a key player in NEPC.5,12-17Whether KDMs play any role in NEPC progression is unknown.
KDM4A regulates cell cycle, replication, and DNA damage response in cancers and has been found to be amplified or overexpressed in multiple cancer types, including breast, lung, and prostate.18 In prostate cancer, KDM4A overexpression (OE) is not sufficient to drive the formation of prostate adenocarcinoma in a GEMM.19 However, KDM4A cooperates with loss of tumor suppressor PTEN and OE of oncogenic ETV1 to enhance the development of primary prostate cancer. Although OE or shRNA KD of KDM4A affects prostate cancer cell proliferation,19 whether KDM4A plays a role in NEPC is unknown. Here we identified histone lysine demethylase KDM4A as a potential driver of NEPC progression and establish alternative epigenetic pathways driving NEPC progression. We also showed that KDM4A is a promising therapeutic target, which will contribute to therapies to treat NEPC.
RESULTS
KDM4A is overexpressed in neuroendocrine prostate cancer (NEPC)
To examine whether histone lysine demethylases play a role in NEPC, we analyzed a well-annotated RNA-seq dataset,20 which includes five pathologically defined subtypes based on a proposed classification approach:21 adenocarcinoma (n=34), NEPC (n=7), mixed NEPC-adenocarcinoma (N=2), small cell prostate carcinoma (SCPC) (n=4), and mixed SCPC-adenocarcinoma (n=2). Due to the small sample size of the non-adenocarcinoma samples, we combined NEPC and mixed NEPC-adenocarcinoma as one group (NEPC), and SCPC and mixed SCPC-adenocarcinoma as another group (SCPC). We compared the genome-wide gene expression of NEPC (NEPC and mixed NEPC-adenocarcinoma) to adenocarcinoma and SCPC (SCPC and mixed SCPC-adenocarcinoma), respectively, to identify differentially expressed genes (DEGs). We identified 4669 and 1834 upregulated genes in NEPC compared to adenocarcinoma and SCPC, respectively (Supple. Table 1-2). Also, we identified 1525 and 33 downregulated genes in NEPC compared to adenocarcinoma and SCPC, respectively (Supple. Table 1-2). KDM4A and KDM5D were uniquely upregulated in NEPC compared to adeno-CRPC and SCPC among the histone lysine demethylases (Figure 1A-B). Because KDM4A, but not KDM5D22, was shown to play an oncogenic role in prostate adenocarcinoma,19 we decided to focus on the functions of KDM4A in NEPC progression. To determine whether KDM4A mRNA is also upregulated in mouse NEPC, we took advantage of a publicly available RNA-seq dataset from Pb-Cre+;Ptenf/f;NMYC/NMYC, Rb1f/f NEPC model (referred to as PNR hereafter), we found that KDM4A, but not KDM5D, is upregulated in advanced tumors from older mice (16 weeks) compared to tumors from younger mice (12-13 weeks) (Figure 1C), suggesting KDM4A overexpression may play a role in NEPC progression. Also, KDM4A is overexpressed in the more aggressive NEPC from PNR mice compared to Pb-Cre+;Ptenf/f;NMYC/NMYC (PN) and Pb-Cre+;Ptenf/f;NMC/+;Rb1f/+ (PNhet Rhet) with comparable age (16-17 weeks) (Figure 1C), which develop prostate adenocarcinoma.

(A) Venn Diagram showing KDM4A and KDM5D as the two histone lysine demethylases that are overexpressed in NEPC using the Beltran et al. RNA-seq dataset. (B) The expression of KDM4A and KDM5D in NEPC compared to adeno-CRPC and SCPC. (C) Kdm4a mRNA is upregulated in prostate tumors from older PNR mice than tumors from young PNR mice, PN mice and PNhetRhet mice. (D) KDM4A IHC in primary NEPC and adenocarcinoma-CRPC. (E) KDM4A IHC staining in prostate cancer PDXs. (F) KDM4A IHC in primary tumors from PS, PTR, PSTR and TRAMP mice. (G) Correlation of KDM4A mRNA expression with NE markers CHGA and CHGB in the Beltran et al. RNA-seq dataset.
To confirm the findings from the human and mouse RNA-seq data, we performed IHC staining of KDM4A in human and mouse primary CRPC-adeno and NEPC. We found that KDM4A is overexpressed in human NEPC but not adeno-CRPC (Figure 1D). Also, we examined KDM4A protein expression in a well characterized prostate cancer patient-derived xenografts (PDX) tissue microarray (TMA).23 We found that KDM4A protein is overexpressed in NEPC PDXs compared to adenocarcinoma PDXs (Figure 1E). To determine the expression of KDM4A protein in murine NEPC, we examined its expression in three genetically engineered mouse models (GEMMs) that generate NEPC including: TRAMP24, prostate specific conditional knockout of Pten/Trp53/Rb1 (PTR),13 and a recently generated prostate specific cKO of Pten/Smad4/Trp53/Rb1 (PSTR). We found that KDM4A protein is highly expressed in the primary tumors from all three NEPC GEMMs compared to two well-established prostate adenocarcinoma models, i.e., the prostate specific cKO of Pten and Pten/Smad4 (PS model) (Figure 1F & Suppl. Figure 1A), consistent with our findings in human prostate tumor tissues. Of note, the specificity of KDM4A antibodies was validated using Kdm4a-KO cells using cell lines derived from PSTR primary tumors (Suppl. Figure 1B). We also determined whether KDM4A overexpression is associated with neuroendocrine markers. Using the Beltran et al. RNA-seq dataset,20 we observed a strong positive correlation between KDM4A mRNA and multiple neuroendocrine (NE) markers, including CHGA, CHGB, ENO2, and DLL3 (Figure 1G & Suppl. Figure 1C). We also examined the expression of KDM4A in the Abida et al. RNA-seq data set,25 which also contains a small subset of NEPC. We found that KDM4A mRNA is significantly higher in mCRPC tumor samples with neuroendocrine features compared to those without NE features (Suppl. Figure 1D). Collectively, our data strongly suggested that KDM4A may play a role in NEPC progression.
KDM4A KD and KO impaired the growth of mouse NEPC proliferation in vitro
To examine the functions of KDM4A in NEPC progression, we used shRNA and siRNA to generate KDM4A knockdown (KD) and knockout (KO) cells. Mouse prostate cancer cell lines derived from PSTR and PTR primary tumors, which were confirmed to have upregulated expression of NE markers such as Ncam1, Chga, Syp, Foxa2, Prox1, and Sox2 compared to Myc-CaP adenocarcinoma using qRT-PCR (Suppl. Figure 2A-B), were used in this study. We confirmed the efficient KD of Kdm4a using Western blot in PSTR cells (Figure 2A). Given the highly proliferative nature of NEPC cells, we first examined whether KDM4A is required for the proliferation of NEPC cell lines using Kdm4a-KD cells and control cells. We found that Kdm4a KD using shRNA or siRNA significantly reduced cell proliferation in PSTR cells as shown by foci-formation assay (Figure 2B & Suppl. Figure 2C). Kdm4a KD in PTR cells also reduced cell proliferation (Figure 2C-D). To confirm the findings using shRNA, we also used CRISPR/Cas9 system to generate Kdm4a KO and control PSTR cells. We confirmed the complete KO of Kdm4a using Western blot (Figure 2E). We found that Kdm4a KO in PSTR cells dramatically reduced cell proliferation (Figure 2F). To determine the effect of KDM4A KD on human NEPC cells, we generated KDM4A KD and control human NEPC cells using the 144-13 and LASCPC-01 models (Figure 2G & Suppl. Figure 2D). Human NEPC cell line 144-13 was derived from a NEPC PDX MDA PCa 144-13)26 and LASCPC-01 was derived from Myr-AKT/N-MYC transformed PCa.16 We found that KDM4A KD in 144-13 and LASCPC-1 cells also significantly reduced cell proliferation (Figure 2H & Suppl. Figure 2E), consistent with the findings using mouse cell lines. Together, our data suggest that KDM4A is required the growth of NEPC cells in vitro.

(A-B) Kdm4a KD in PSTR cells, as confirmed by Western blot (A), led to significant reduced growth in foci-forming assay in vitro (B). (C-D). Kdm4a KD in PTR cells, as confirmed by Western blot (C), led to significant reduced growth in vitro (D). (E-F) Kdm4a KO in PTR cells, as confirmed by Western blot (E), led to significant reduced growth in foci-forming assay in vitro (F). (G-H) KDM4A KD in 144-13 cells, as confirmed by Western blot (G), led to significant reduced cell number (H). (I-J) KDM4A KD in 144-13 cells (I) or Kdm4a KO in PSTR cells (J) led to reduced number of colonies and reduced colony sizes using soft agar colony formation assay.
We also tested the impact of KDM4A KD and KO in NEPC cells on the anchorage independence growth, an assay that measured the tumorigenicity of cancer cells in vitro, using soft agar colony formation assay. We found that KDM4A KD in 144-13 cells led to reduced colony number and size compared to control siRNA (Figure 2I). Similar results were observed in PSTR Kdm4a-KO cells (Figure 2J). Together, our data suggest that KDM4A is required the growth of NEPC cells in vitro.
Kdm4a KO or KD impairs NEPC progression in vivo
To examine the effect of Kdm4a KO on the NEPC cell growth in vivo, we implanted Kdm4a-KO PSTR cells and control cells subcutaneously in nude mice. We found that Kdm4a KO significantly reduced the tumor growth in vivo (Figure 3A). To further establish the role of KDM4A in NEPC progression in vivo, we crossed the Kdm4aloxP/loxP mice27 to Probasin-Cre4 transgenic mice to generate the prostate-specific Kdm4a KO mice, which were further crossed with TRAMP mice in C57BL/6 background, a well characterized prostate cancer model that recapitulate the multi-stage tumor progression in prostate cancer patients and develop both prostate adenocarcinoma and neuroendocrine prostate cancer.24,28-31 Since the tumor progression in the TRAMP mice is affected by the copy number of the transgene (tg), we generated TRAMPtg/tg homozygous and Pb-Cre;Kdm4aloxP/loxP;TRAMPtg/tg compound mutant mice (aka KT mice) for comparison. We confirmed the loss of KDM4A protein expression by IHC staining in the KT mice (Figure 3B). We found that Kdm4a KO reduced the tumor weight in age matched KT and TRAMP mice (Figure 3C). We found that prostate tumors from KT mice were largely PIN with focal invasion whereas the prostate tumors from age-matched TRAMP mice are largely invasive (Figure 3D). Also, Kdm4a KO led to a significant increase in overall survival in KT mice compared to TRAMP mice (Figure 3E). To characterize the impact of Kdm4a KO on the cancer cell proliferation in vivo, we performed IHC staining of Ki67 in tumors from age-matched TRAMP and KT mice. We found that Kdm4a-deficient tumors have reduced Ki67 staining (Figure 3F-G). We observed that a subset of TRAMP mice developed aggressive tumors that required euthanasia before 30-week-old (Figure 3E). The majority of these prostate tumors (9 out of 10) displayed a NEPC-predominant phenotype as shown by IHC staining of SYP (Figure 3H-I). For TRAMP mice die after 30-week-old, most of these prostate tumors (7 out of 8) displayed an adenocarcinoma-predominant phenotype (Figure 3H-I). These findings are consistent with previous reports that TRAMP mice develop both NEPC and prostate adenocarcinoma and the aggressive nature of NEPC. In contrast, only 1 out 16 KT mice rarely required euthanasia due to big tumor burden before 30-week-old and most of KT mice died around 40-41 weeks (Figure 3H). For KT mice died after 30-week-old, the majority of these prostate tumors (10 out of 11) displayed an adenocarcinoma-predominant phenotype (Figure 3H-I). Collectively, our data suggest that KDM4A plays an important role in NEPC progression in vivo.

(A) Kdm4a KO delayed the growth of PSTR cells in subQ implantation model. (B) IHC staining confirmed the loss of KDM4A expression in tumors from KT compound mutant mice compared to tumors from TRAMP mice. (C) Reduced tumor weights in KT mice (20-28 weeks) compared to tumors from age-matched TRAMP mice. (D) H & E staining of prostate tumors from 20-week-old TRAMP and KT mice. (E) Kaplan Meier survival analysis comparing KT mice to TRAMP mice. (F-G) Ki67 IHC staining in prostate tumors from TRAMP and KT mice. (H) The incidences of prostate adenocarcinoma and NEPC in TRAMP and KT mice. (I) IHC staining of SYP in prostate tumors from >30-week-old and <30-week-old TRAMP mice and KT mice.
KDM4A regulates MYC expression to promote tumor progression in NEPC cells
To determine the mechanisms by which KDM4A promotes NEPC progression, we performed RNA-seq of Kdm4a-KD and -control PTR cells. We identified 1359 genes that are downregulated and 2608 genes that are upregulated in Kdm4a-KD cells compared to control PTR cells (FDR≤0.05, fold change≥1.5) (Suppl. Table 3). We performed gene set enrichment analysis (GSEA) to identify pathway that are activated or suppressed in Kdm4a KD cells. We found that Kdm4a KD led to reduced activation of E2F signaling (Suppl. Figure 3A), which is consistent with previous findings that KDM4A interacts with E2F1 to regulates transcription.32 Interestingly, we identified MYC signatures (HALLMARK MYC TARGETS V1 and HALLMARK MYC TARGETS V2) as the top downregulated pathways in Kdm4a KD cells (Figure 4A & Suppl. Table 5). Multiple MYC target genes (e.g., Ldha, Nme1, and Srsf2, etc.) were significantly downregulated in Kmd4a-KD cells (Figure 4B). Interestingly, MYC has been implicated to play a functional role in NEPC, as overexpression of MYC, AKT1, BCL2, or RB1-KD/p53-KD transformed normal prostate epithelial cells into NEPC.33 We also found that MYC is the top hallmark pathways activated in both human and mouse NEPC as shown by GSEA analyses of publicly available RNA-seq data of adenocarcinoma and NEPC from patients20 and mice13 (Figure 4C-D). These data suggest that KDM4A may promote NEPC progression through regulation of MYC signaling.

(A) GSEA analysis of RNA-seq on Kdm4a-KD and control PTR cells showed that MYC signaling is suppressed in Kdm4a-KD cells. (B) Fold changes in the expression of selective MYC-target genes comparing Kdm4a-KD cells to control cells. (C-D) GSEA analysis of Beltran et al. RNA-seq data (C) and Ku et al. RNA-seq data showed that MYC signaling is activated in NEPC compared to prostate adenocarcinoma. (E-F) Western blot showed that KDM4A KD led to reduced MYC expression in PSTR (E) and PTR (F) cells. (G) qRT-PCR showed that Kdm4a KD led to reduced Myc mRNA expression in PTR cells. (H) ChIP-seq analyses identified MYC as a KDM4A-target gene in 144-13 cells. (I) MYC KD led to reduced cell proliferation in 144-13 cells. (J) MYC inhibitor MYCi975 led to reduced cell proliferation in 144-13 cells.
To further delineate the mechanisms by which KDM4A regulates MYC signaling, we first determined whether KDM4A directly regulates MYC transcription, given the well characterized function of KDM4A in epigenetic regulation. We performed qRT-PCR and Western blot analyses to examine the effect of KDM4A KD on MYC expression in PSTR, PTR, and 144-13 cell lines. We found that KDM4A KD reduced MYC mRNA and protein expression in all three cell lines (Figure 4E-G & Suppl. Figure 3B). To determine whether MYC is a direct target gene of KDM4A, we performed ChIP-seq in 144-13 cells using anti-KDM4A, anti-H3K9me3, anti-H3K36me3, anti-H3K27ac, and anti-CTCF. We found that KDM4A binds directly to MYC promoter as shown by ChIP-seq analysis (Figure 4H), suggesting an active MYC transcription in NEPC cells. We also found that H3K9me3 is barely detectable at MYC locus whereas H3K36me3 and H3K27ac were present at MYC locus (Figure 4H), suggesting active transcription at MYC locus. As a positive control for anti-H3K9me3, ZNF114, ZNF333, ZNF175, genes that are constitutively silent due to the large H3K9me3 domain, showed significant H3K9me3 signal at its promoter region (Suppl. Figure 3C-E). Lastly, we determined the effect of MYC KD or MYC inhibitor on NEPC cell proliferation in 144-13 cells. We found that MYC KD or MYC inhibitor MYCi975 significantly reduced cell proliferation in 144-13 (Fig. 4I-J). Collectively, our data suggest that KDM4A promotes NEPC progression through direct regulation of MYC transcription.
KDM4 inhibitor suppressed cell proliferation and induced apoptosis in NEPC in vitro
Because our genetic data showed that KDM4A plays an important role in NEPC progression, we examined whether targeting KDM4A can be an effective therapeutic strategy for NEPC. Although there are no KDM4A-specific inhibitors, several pan-KDM4 inhibitors (KDM4i) [e.g.,QC6352,34,35 NSC636819,36 NCGC00244536,37 IOX1,38 SD7039] have been shown to suppress the growth of cancer cells. For example, QC6352 was shown to suppress the growth of colorectal cancer (CRC) and triple negative breast cancer (TNBC) and NCGC00244536 was shown to suppress the growth of prostate cancer cell lines PC3 and LNCaP. Among them, QC6352 is a highly selective inhibitor against the KDM4 family and rigorously characterized (e.g., cocrystal structure with KDM4A) with potent anti-tumor activities, favorable pharmacokinetics, and low toxicity in mice.34,35 We found that treatment of PSTR cells with QC6352 resulted in a significant increase in global level of H3K9me3 and H3K36me3 expression, two of KDM4’s substrates (Figure 5A). We then examined the effect of QC6352 on the growth of PSTR cells in vitro, which are largely resistant to AR inhibitor enzalutamide (ENZ) (Suppl. Figure 4A). We found that QC6352 treatment led to a dramatic decrease in cell proliferation in a dose-dependent manner (Figure 5B). QC6532 also induced apoptosis in PSTR cells as shown by Western blot analysis for cleaved caspase 3 and cleaved PARP1 (Figure 5C) and positivity of green fluorescent signal from IncuCyte Caspase-3/7 Green Apoptosis Assay Reagent (Figure 5D). Similarly, QC6352 treatment also dramatically suppressed cell proliferation and induced apoptosis in PTR cells (Figure 5E-F) and human NEPC cell lines (144-13) (Figure 5G & Suppl. Figure 4B). Similarly, NCGC00244536, another potent inhibitor for KDM4 histone lysine demethylase,37 also suppressed cell proliferation and induced apoptosis in PSTR and 144-13 cells (Figure 5G & Suppl. Figure 4C). In addition, we compared the response of Kdm4a-WT and -KO PSTR cells to QC6532 treatment. We found that KDM4A KO largely abrogated the response of PSTR to QC6532 (Figure 5H), supporting the notion that KDM4A is the critical target of QC6532 in NEPC cells. We also found that MYC mRNA and protein expression were downregulated in QC6352-treated NEPC cells compared to control cells (Suppl. Figure 4D), consistent with the role of KDM4A in regulating MYC expression.
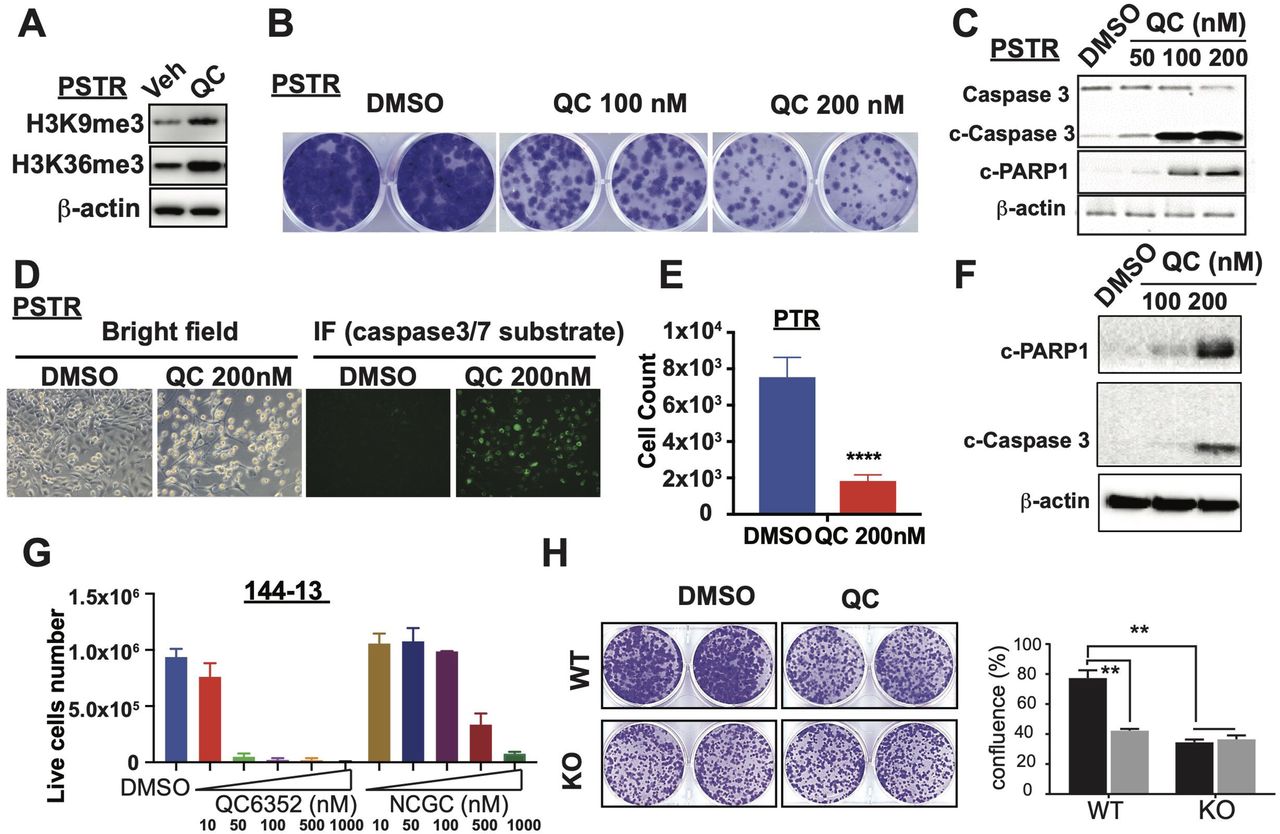
(A) KDM4 inhibitor QC6352 treatment led to increased total H3K9me3 and H3K36me3 in PSTR cells. (B) QC6352 suppressed PSTR growth in vitro as shown by foci-forming assay. (C-D) QC6352 treatment induced apoapsis in PSTR cells as shown by Western blot analysis of cleaved caspase 3 and cleaved PARP1 and fluorescence imaging of caspase 3/7 substrates. (E) QC6352 suppressed the growth of PTR cells. (F) QC6352 induced apoptosis in PTR cells as shown by WB analysis of cleaved caspase 3 and cleaved PARP1. (G) QC6352 and NCGC00244536 suppressed the growth of 144-13 cells in vitro. (H) Kdm4a-KO blunt the effect of QC6352 treatment on cell growth in vitro.
KDM4 inhibitor suppressed NEPC progression in vivo
To determine whether QC6352 could suppress NEPC growth in vivo, we treated mice bearing PSTR tumors and 144-13 tumors subcutaneously in immune-deficient mice. We found that QC6352 treatment dramatically suppressed the growth of PSTR cells (Figure 6A-B) and 144-13 cells (Figure 6C-D), as shown by reduced tumor sizes and tumor weights. To ensure QC6352 has similar effects on tumor progression in immune-competent host, we treated PSTR GEMM with QC6352. Similarly, QC6352 treatment in PSTR GEMM model led to a significant reduction in tumor burden as shown by tumor weights (Figure 6E). We then examined the effects of QC6352 treatment on tumor cell proliferation. We found that QC6352 treatment led to a significantly reduced proliferation in PSTR GEMM as shown by Ki67 IHC staining (Figure 6F). We did not observe noticeable toxicity as shown by measurement of peripheral blood count and measurement of weight of major organs (Suppl. Figure 5A-B), consistent with the findings in the original study.34 Together, our data suggest that KDM4 inhibitor QC6352 can significantly delay NEPC progression in vivo.

(A-B) QC6352 treatment (50 mg/kg daily) suppressed the growth of subQ implanted PSTR cells as shown by weekly measurement of tumor volumes (A) and measurement of tumor weights at endpoint (B). (C-D) QC6352 treatment suppressed the growth of subQ implanted 144-13 cells as shown by weekly measurement of tumor volumes (C) and measurement of tumor volume and tumor weights at end point (D). (E) QC6352 treatment suppressed the growth of primary tumors in PSTR GEMM as shown by tumor weights at end point. (F) QC6352 treatment reduced NEPC proliferation in vivo as shown by IHC staining of Ki66 in tumors from PSTR mice.
DISCUSSIONS
Neuroendocrine prostate cancer (NEPC) has been increasingly observed in clinic, and most of these NEPC occurs in patients who develop therapeutic resistance to potent androgen pathway inhibitors. Despite a high response rate to platinum-based chemotherapy that is of transient nature, there are no life-prolonging therapeutic options for NEPC patients. In recent years, epigenetic dysregulation has emerged as a hallmark for NEPC and targeting epigenetic regulators has become a promising approach. Here we identified KDM4A as an important regulator of NEPC progression and demonstrated that targeting KDM4A can be a promising therapeutic approach.
KDM4A is an epigenetic modifier and its dysregulation may alter the expression of many genes. Our findings that KDM4A regulates MYC expression in NEPC provides a novel mechanism for how KDM4A exerts its effects on prostate cancer progression. MYC has previously been shown to be a key player of prostate cancer progression and prostate specific expression of MYC was sufficient to induce prostate cancer and promote tumor progression in mouse models.40-42 Our study reveals a mechanism by which Myc is activated and Myc plays a role in NEPC, providing a novel insight into how epigenetic modifier regulates oncogene expression. This novel finding also promoted us to examine whether such regulation occurs in other cancer types. We analyzed a publicly available RNA-seq datasets43 and found Myc mRNA was downregulated in mouse squamous cell carcinoma cell lines upon Kdm4a KO compared to control cells (Suppl. Figure 6A). Also, QC632 treatment led to reduced expression of MYC mRNA in triple negative breast cancer (TNBC)35 (Suppl. Figure 6A). Furthermore, we also examined a publicly available ChIP-seq dataset (KDM4A) in TNBC35 and found that KDM4A binds to MYC promoter/gene body in TNBC (Suppl. Figure 6B). These findings suggest that the regulation of MYC expression by KDM4A may also contribute to tumor progression in other cancer types with KDM4A overexpression.
Using genetically engineered mouse models (GEMMs), we demonstrated that Kdm4a KO significantly reduced the incidence of NEPC that are present in the TRAMP mice and extended the overall survival. Since the expression of Cre recombinase is driven by Probasin promoter that starts at puberty (∼ 4-6 weeks), Cre-dependent Kdm4a KO occurred during the tumor initiation phase. Thus, we could not rule out that the KDM4A is also necessary for tumor initiation and that in part contributes to the effects of Kdm4a KO on tumor progression and survival observed in KT mice. Future experiments using transgenic mouse expressing inducible Cre controlled by tamoxifen or doxycycline to inactivate Kdm4a at various stages of prostate cancer progression are needed to address the roles of KDM4A in tumor initiation and progression.
We showed that KDM4 inhibitor QC6352 not only suppresses cell proliferation, but also induces apoptosis in NEPC cells. However, we did not observe any noticeable increase in apoptosis in KDM4A KD/KO NEPC cells as shown by western blot for cleaved caspase 3 and cleaved PARP1 (Suppl. Figure 6C). Since QC6352 also inhibit other KDM4 family members, the apoptosis observed in QC6352-treated cells may be attributed to simultaneous inhibition of multiple KDM4 family members in NEPC cells. This notion is supported by the previous reports that KDM4 family members, such as KDM4B and KDM4D, play a role in DNA damage response.44,45 Also, KDM4A plays a role in replication stress.43 Interestingly, GSEA analysis identified apoptosis signature in Kdm4a-KD PTR cells compared to control cells (Suppl. Figure 7D), although it does not rank as the top pathways. This finding suggests that inhibition of KDM4A may also contribute to apoptosis induced by QC6352. However, future studies are needed to delineate the functions of individual KDM4 family members in QC6352-induced apoptosis NEPC.
In summary, our study established KDM4A as an important epigenetic regulator that drives NEPC progression and can serve as an effective therapeutic target.
MATERIALS and METHODS
Mice strains
Pb-Cre4; Pten loxP/loxP (Pten pc-/-), Pb-Cre4; Pten loxP/loxP; Smad4 loxP/loxP (aka Ptenpc-/- Smad4pc-/-), and Pb-Cre4; Pten loxP/loxP; Trp53 loxP/loxP (aka Ptenpc-/- Trp53pc-/-) models were developed previously46. Rb1loxP/loxP strain (FVB;129-Rb1tm2Brn/Nci) was obtained from NCI Mouse Repository. Kdm4aloxP/loxP (Strain #:029424) and TRAMP mice (stock # 003135) were obtained from Jackson Laboratory. Ptenpc-/- Smad4pc-/- and Ptenpc-/- Trp53pc-/- mice were crossed with Rb1loxP/loxP mice to generate Pb-Cre4; Pten loxP/loxP; Smad4 loxP/loxP Trp53 loxP/loxP Rb1loxP/loxP (Ptenpc-/- Smad4pc-/- Trp53pc-/- Rb1pc-/-, PSTR) and Pb-Cre4; Pten loxP/loxP; Trp53 loxP/loxP; Rb1loxP/loxP mice (Ptenpc-/- Trp53pc-/- Rb1pc-/-, PTR). The genotyping of the Pb-Cre4 transgene and all the conditional alleles were performed using conventional PCR as described previously.46-49 The copy number of TRAMP transgene will be determined by quantitative PCR50 and only mice that are homozygous for the transgene were used in this study. Mice were maintained in pathogen-free conditions at M.D. Anderson Cancer Center. All manipulations were approved under MD Anderson Cancer Center (MDACC) Institutional Animal Care and Use Committee (IACUC) under protocol number 00001713-RN01.
Human prostate tumor tissues and patient-derived xenografts (PDX)
FFPE Primary castration resistant prostate tumor tissues were obtained from Dr. Patricia Troncoso and Dr. Miao Zhang MD Anderson Tissue Bank. FFPE and fresh tumor tissues from PDXs were obtained from Dr. Nora Navone (GU PDX core). The Movember PDX TMA has been described previously.23
Cell lines and cell cultures
PTR and PSTR cell lines were isolated from Ptenpc-/-Trp53pc-/ Rb1pc-/- and isolated from Ptenpc-/- Smad4c-/-Trp53pc-/- Rb1pc-/- mice. All cell lines tested for mycoplasma were negative within 6 months of performing the experiments. Cell line authentication was not performed. 293T, NCI-H660, LASCPC-01, and MYC-CaP cells were obtained from ATCC. 144-13 was generated from PDX MDA PCa 144-13.26 293FT cells were obtained from Thermo Fisher Scientific Inc. 144-13, LASCPC-01 and NCI-H660 cells were cultured in HITES medium supplemented with 5% fetal bovine serum using ATCC-formulated RPMI-1640 Medium (Catalog No.30-2001) with the following components to the base medium: 0.005 mg/ml Insulin, 0.01 mg/ml Transferrin, 30nM Sodium selenite (final conc.), 10 nM Hydrocortisone (final conc.), 10 nM beta-estradiol (final conc.), extra 2mM L-glutamine (for final conc. of 4 mM).
siRNA/shRNA knockdown and CRISPR/Cas9 genome editing
siRNAs were ordered from Dharmacon Inc (human KDM4A: GUAUGAUCUUCCAGACUUA; GUGCGGAGUCUACCAAUUU; mouse Kdm4a: GAACAUCCUACGACGAUAU; GUUCGUGAGUUCCGCAAGA; human MYC: AACGUUAGCUUCACCAACA; AACGUUAGCUUCACCAACA; mouse Myc: GGACACACAACGUCUUYGGA; UCGAAACUCUGGUGCAUAA). Lentiviral shRNA plasmids were obtained from Sigma Aldrich (Human KDM4A: shKDM4A#1 TRCN0000013493, shKDM4A#2 TRCN0000013494, shKDM4A#3 TRCN0000013495; Mouse Kdm4a: shKdm4a#2 TRCN0000103526). Mouse shKdm4a#4 plasmid was obtained from VectorBuilder of which sequence of siKdm4a#4 (GUUCGUGAGUUCCGCAAGA) was cloned into the lentiviral vector (VB210619-1017gjm). Lentiviruses were packaged in 293FT cells using second generation packaging vectors, psPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259). Synthetic guided RNA (sgRNA) was ordered from Synthego Inc. Recombinant cas9 was obtained from Thermo Fisher Scientific Inc. sgRNAs and recombinant cas9 were transfected into cells according to the protocol from the manufacturer. Single clones were selected, and Kdm4a KO clones were confirmed by western blot analysis.
Chemicals and inhibitors
QC6352 (HY-104048), NCGC00247743 (HY-112308), Enzalutamide (HY-70002), GSK126 (HY-13470), MYCi975 (MYCi975) were ordered from MedChemExpress Inc. Incucyte Caspase 3/7 Dye for Apoptosis (Cat#4440) was ordered from Sartorius.
Western Blot Analysis
Cells were lysed on ice using RIPA buffer (Boston BioProducts) supplemented with Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Proteins with loading buffer (20 µg) were subjected to SDS-PAGE and transferred onto a nitrocellulose membrane. Membrane was blocked in 2% non-fat dry milk for an hour before being incubated with primary antibodies prepared in TBST consisting of 0.5% BSA overnight at 4°C. Next, the membrane was washed 3 times with Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST) and was incubated with HRP-conjugated secondary antibody for 2 h in room temperature. After washing 3 times with TBST, the membrane was exposed to Clarity Western ECL Substrate (Bio-Rad) according to the protocol and imaged with Azure Biosystems c600. The following antibodies were used in this study: KDM4A (ab191433, Abcam), beta-Actin (MA5-15739, Invitrogen), cleaved-PARP (Mouse specific #9544; human specific #9546, Cell Signaling Technology) cleaved-caspase 3 (#9662, Cell Signaling Technology), cMyc (ab32072, Abcam), c-Anti-Rabbit IgG HRP-linked antibody (#7074, Cell Signaling Technology), Anti-Mouse IgG HRP-linked antibody (#7076, Cell Signaling Technology).
Immunohistochemistry analyses
Tissues were fixed in 10% formalin overnight and embedded in paraffin. Immunohistochemical (IHC) analysis was performed as described earlier (Aguirre et al., 2003). Primary antibodies used for immunohistochemistry are as follows: KDM4A (ab191433, Abcam), KDM4A (3393S, CST), Ki-67 (GTX16667, GeneTex), Synaptophysin (M7315, Dako), AR (ab133273, Abcam), Chromogranin A (20086, ImmunoStar). Secondary antibodies used are as follows: anti-rabbit(RMR622L, Biocare), anti-mouse(8125S, CST). The immunohistochemistry signals were developed with DAB Quanto(TA-125-QHDX, Epredia).
Cell proliferation, Foci-formation, soft-agar colony forming, and apoptosis assays
For cell proliferation assay, 5×102-2×103 cells per well were seeded in 96-well plate with or without corresponding treatment. After 5 days, absorbance was measured using Cell Counting Kit-8 (B34034, Bimake) according to the manufacture’s manual. For foci-formation assay, 1×103 cells were seeded in 6-well plates and cultured for 5 to 7 days before they were fixed and stained with crystal violet as described.51 For soft-agar colony forming assay, 2.5×103 cells were seeded in 6-well. Cells were cultured for 3 weeks. Colony sizes and number were measured as described.52 Apoptosis was detected by flow cytometry analysis of cells stained with Annexin V and PI staining or by Cytation 5 using Caspase3/7 dye for apoptosis (Cat#4440, Sartorius).
Xenograft Studies
PSTR (1×105 cells/100ul per mouse) and 144-13 (2×106/100ul per mouse) cells were prepared in 1:1 PBS and Matrigel (BD Biosciences) and injected into flanks of 6-week-old nude mice (n = 5-10 per group). Mice were treated with 50 mg/kg QC6352 (once a day) when tumors reached 100mm3. Tumor growth was monitored by measuring tumor sizes twice a week. Tumor Volume was calculated according to the formula: Volume = length × width2/2. All xenograft experiments were approved by the MD Anderson IACUC under protocol number #00001713-RN01.
Quantitative RT-PCR and RNA-sequencing
Cell mRNA was isolated by Direct-zol RNA MiniPrep (50ug) (#11-331, ZymoResearch). iScript cDNA Synthesis Kit (Bio-Rad) was used to generate cDNA for quantitative PCR (qPCR) analysis using PerfeCTa reg SYBR reg Green SuperMix Reaction Mixes (QuantaBio). The primers used for qPCR analysis. Cell mRNA was isolated by RNeasy Kit (Qiagen) and reverse transcribed using Superscript III cDNA Synthesis Kit (Life Technology). Quantitative PCR was performed using SYBR-GreenER Kit (Life Technology). The primers used were listed in Suppl. Table 4. RNA-seq of PTR cells with shRNA control and shRNA Kdm4a#2 were performed on total RNA (3 replicates) using NEBNext® Ultra II Directional RNA Library Prep Kit for Illumina NextSeq at the Cancer Genomics Center, The University of Texas Health Science Center at Houston. The raw data will be submitted to the NCBI Gene Expression Omnibus (GEO). Processed human RNA-seq datasets from previous studies (Grasso et al.,53 Abida et al.,25 and Beltran et al.20) were downloaded from cBioportal.54,55 Publicly available RNA-seq datasets (GSE158467, GSE90891, GSE95293, GSE137953, and GSE95293) were downloaded from NCBI GEO. DeSeq2 was used to identified differentially expressed genes. DEGs were used for gene set enrichment analysis (GSEA) as described.47
Chromatin-immunoprecipitation (ChIP), ChIP-sequencing, and Cut and Run
ChIP-seq was performed in 144-13 cells as described51 using the following antibodies at the MD Anderson Epigenomics Profiling Core: anti-KDM4A antibody ((#5766, lot 021110, Schuele Laboratory), H3K27ac (ab4729, Abcam), H3K9me3 (C15410056, Diagenode), H3K36me3 (ab9050, Abcam). Libraries were prepared using NEBNext Ultra DNA Library Kit (E7370). Sequencing was performed using an Illumina HiSeq 2500 instrument. Reads were aligned to a reference genome Hg19 as described.56 The raw data will be submitted to the NCBI Gene Expression Omnibus (GEO). Publicly available ChIP-seq dataset GSE95190 was downloaded from NCBI GEO.
Statistical analysis
All the experiments were replicated at least twice in the laboratory except for microarray and ChIP. Data are presented as mean ± SD unless indicated otherwise. Student’s t test assuming two-tailed distributions was used to calculate statistical significance between groups. **** P < 0.0001, *** P < 0.001; ** P < 0.005; * P < 0.05.
Author Contributions
G.W. contributed to the study’s conception and design of this study. G.W., C.M., M.Z., X.L., F.W., A.G.H., X.S., D.L., J.P., M.Z., P.T., and J.Z. performed the experiments and acquired, analyzed, and interpreted the data (e.g., statistical analysis, biostatistics, computational analysis). P.S., N.N., E.M., and R.S. provided key reagents. A.K.J., M.G.L, P.C., C.J.L., A.A., provided scientific inputs for the development of the project. C.M., S.H.L and G.W. contributed to the writing and editing the manuscript.
Competing Interests statement
CJL reports receiving commercial research grants from Bayer, Sanofi, Janssen, Astellas Pharma, Pfizer; and honoraria from Bayer, Janssen, Sanofi, Astellas Pharma. No potential conflicts of interest were disclosed by the other authors.
Acknowledgements
We thank Sarah E. Townsend for editing. G.W. is supported by funding from MDACC (Moon Shot, IRG, and PCRP), UT STARs Award (The University of Texas System Board of Regents), NIH [R00 CA194289 (GW), P50 CA140388(C. Logothetis, S.-H. Lin)], DoD [DOD-PCRP-Idea W81XWH-21-1-0522(GW)]. S.H.L. is supported by grants from the NIH R01CA174798 (S.-H. Lin), NIH 5P50CA140388 (C. Logothetis, S.-H. Lin), and Cancer Prevention Research Institute of Texas grants RP150179 & RP190252 (S.-H. Lin). This study is supported by NIH P30CA016672 for the use of Research Animal Support Facility, Flow Cytometry and Cellular Imaging Core Facility, and Functional Genomics Core at MD Anderson Cancer Center. We thank the support from the Cancer Prevention and Research Institute of Texas (CPRIT RP180734) for the RNA-seq service provided at Cancer Genomics Center, The University of Texas Health Science Center at Houston. We thank the support from Epigenomics Profiling Core, Center for Cancer Epigenetics and the Department of Epigenetics and Molecular Carcinogenesis for the ChIP-seq services.
Footnotes
Conflict of interest: C. J. Logothetis reports receiving commercial research grants from Bayer, Sanofi, Janssen, Astellas Pharma, Pfizer; and honoraria from Bayer, Janssen, Sanofi, Astellas Pharma. No potential conflicts of interest were disclosed by the other authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}