

ABSTRACT
Engineering synthetic heterotrophy (i.e., growth on non-native substrates) is key to efficient bio-based valorization of various renewable (e.g., lignocellulosic biomass) and waste (e.g., plastics) substrates. Among these, engineering hemicellulosic pentose utilization has been well-explored in Saccharomyces cerevisiae (yeast) over several decades but genetic factors that constrain maximum growth rate remain elusive. Through a systematic analysis (flux balancing, directed evolution, functional genomics, and network modeling), we find that once global regulatory response is appropriately remodeled, wild-type-like growth profiles can be achieved with minimal metabolic engineering effort. This indicates that intrinsic yeast metabolism is highly adaptable to growth on non-native substrates. We identified that extrinsic factors – specifically, genes that direct flux of pentoses into central carbon metabolism – are rate limiting. We also find that deletion of endogenous genes to promote growth demonstrate inconsistent outcomes that are genetic-context- and condition-dependent. For the most part, these knockouts also lead to deleterious pleiotropic effects that decrease the robustness of strains against inhibitors commonly found in lignocellulosic hydrolysate. Thus, perturbation of intrinsic factors (e.g., metabolic, regulatory genes) provides incremental and inconsistent benefits at best and at worst, is detrimental. Not only do the findings and approach described here expedite the design-build-test cycle but also simplifies the engineering process. Overall, this work provides insight into the limitations and pitfalls to realizing efficient synthetic heterotrophy and provides a novel paradigm to engineer the same.
INTRODUCTION
Engineering metabolism for growth on non-native substrates (i.e., synthetic heterotrophy) has been an outstanding challenge for several decades in various microbial species (1–4). The traditional approach is to take a purely metabolic perspective and constitutively overexpression catabolic genes that input substrate to central carbon metabolism with the expectation that the native cellular systems can direct subsequent steps required for growth. In case initial designs do not lead to rapid growth, flux balancing (5), adaptive laboratory evolution (ALE) (6, 7), functional genomics (8, 9), and directed evolution (6, 10) approaches are used. More recently, systems-level analysis of regulatory structures in engineered strains have been used to identify dysregulated pathways and corrective interventions have provided avenues to improve engineering outcomes (11–13). Unifying to these approaches is that they are reactive interventions that try to rectify inefficiencies introduced due to strain engineering but not proactive in circumventing undesirable outcomes by integrating heterologous activity with native microbial physiological functions. Prototypical of this approach is engineering the yeast Saccharomyces cerevisiae for complete and rapid utilization of lignocellulosic pentoses (14–17). In prior work, we demonstrated the benefits of integrating global cellular regulation to enable rapid growth and complete xylose catabolism (18). Here, we engineer strains of yeast for rapid growth and pentose (xylose, arabinose) utilization and assess intrinsic and extrinsic constraints and pitfalls that control desirable phenotypic outcomes. We find that limitations are largely extrinsic, and that intrinsic yeast metabolism is highly adaptable to rapid growth on these non-native substrates.
To achieve this, we first modified our semi-synthetic regulon system – based on synthetically activating the galactose (GAL) system by the non-native substrate – for efficient utilization of arabinose through the isomerase pathway (19). We demonstrate that while initial outcomes – like with xylose, previously – are superior to constitutively overexpressing the same genes, growth rates were lower than that on the native substrate, galactose. To identify factors that constrain growth on either pentose, we performed pathway balancing, directed evolution, and systems biology-driven functional genomics (Figure 1). To our surprise, we found that when cells can coordinate global growth responses with substrate use, growth is largely extrinsically controlled – i.e., limited by the upstream/heterologous pathway that controls non-native substrate uptake and flux to central carbon metabolism. We also found through parallel investigations that genetic interventions that prune cellular regulatory networks to improve strain performance often led to pleiotropic defects with mid-to-severe fitness costs. Thus, our findings suggest that a repurposed native substrate utilization regulon when synergized with an optimized upstream heterologous metabolic module may be sufficient to attain optimal performance and additional genetic modifications are, at best, unnecessary, and at worst, deleterious.

The goal of the work is to identify factors that control growth adaptability of yeast on non-native substrates like pentoses, xylose and arabinose.
RESULTS
Integration of catabolism and global regulation enables rapid growth on pentoses
Previously, we demonstrated that coupling activation of galactose (GAL)-responsive regulon to catabolism and growth on non-native substrate, xylose, enabled faster growth and more complete utilization when compared to the constitutive overexpression of the same genes. Here, we wanted to assess whether the benefit was unique to xylose or if growth on other substrates, like arabinose, could also benefit from activation of the GAL regulon. Since the Gal3p-Syn4.1 was engineered to activate the GAL regulon in a xylose-dependent manner, we wondered if it could be activated by arabinose since the two sugars are structurally similar. We assessed activation of GAL1p-EGFP by wild-type Gal3p or engineered Gal3p-Syn4.1 in the presence of native Gal2p and/or engineered Gal2p-2.1 permease (18). We observed that a Gal3p-Syn4.1 and Gal2p-2.1 co-expressing strain showed activation on arabinose with low background and high dynamic range (Figure 2A). Interestingly, arabinose showed higher activation of GAL1p-EGFP than xylose, even though both Gal3p-Syn4.1 and Gal2p-2.1 were engineered for activity on xylose (Figure 2B). Given robust activation, we constructed a semi-integrant strain by integrating accessory genes including, sensor (GAL3-Syn4.1), transporter (GAL2-2.1), and transaldolase (TAL1) – expressed under GAL promoters – to generate a “REG” (regulon) strain background (Figure 2C). Similarly, as control, and to compare the with the traditional engineering approach, we integrated GAL2-2.1 and TAL1 under strong constitutive promoters to generate the “CONS” parental strain. We then assessed the growth of both strains transformed with araBAD or XYLA*3-XKS1 arabinose and xylose, respectively. In ARA-REG and XYL-REG all genes were expressed under GAL-responsive promoters whereas in ARA-CONS and XYL-CONS all genes were under constitutive promoters (Figure 2C). On both carbon sources, the strains that coordinated GAL regulon activation with substrate assimilation demonstrated higher growth rate (μmax; ARA-REG = 0.14 ± 0.04 h-1, XYL-REG = 0.17 ± 0.01 h-1) compared to those that constitutively overexpressed the same genes (μmax; ARA-CONS = 0.06 ± 0.01 h-1, XYL-CONS = 0.11 ± 0.01 h-1) (Figure 2D-E).

A) Dose-dependent GAL1p-EGFP activation by arabinose of strain expressing Gal3p (WT) or Gal3p-Syn4.1 in and Gal2p (WT) or Gal2p-2.1. B) Comparison of GAL1p activation by 2 % of native sugar, galactose, and non-native sugars – xylose and arabinose. C) Genetic structure of semi-integrant REG and CONS strain backgrounds. Growth curve of REG and CONS strains on D) arabinose and E) xylose when transformed with respective arabinose and xylose isomerase pathway genes, respectively. REG and CONS strains use GAL-responsive and constitutive promoters, respectively, for all genes indicated (Table S1 and S2).
Perturbation of intrinsic factors result in modest improvements
In traditional metabolic engineering strategies, adaptive laboratory evolution (ALE) or rational data-driven approaches are often used to improve growth rates of initial strain designs, since growth rates and biomass yields are most often suboptimal. We decided to use a (data-driven) systems biology approach to identify “intrinsic” genetic targets to modify so that may lead to improved growth rates of our REG strains on xylose and arabinose (i.e., pentoses). To identify these, we identified differentially expressed genes between pentoses (non-native substrates) and galactose (a native substrate) – since growth rate on the latter has evolved to function harmoniously as part of the GAL regulon. Identifying the differences between two and closing the gap between them may aid in better synchronizing GAL regulation with pentose metabolism. We chose not to investigate previously identified genetic targets due to the vast difference in the transcriptional phenotype of REG strains relative to traditional CONS-type designs (18).
We performed RNA-seq on strains growing on arabinose (ARA-REG), xylose (XYL-REG), and galactose (WT). We observed that while the overall profiles of all three substrates were different, the two pentoses clustered closer together than with the hexose, galactose (Figure 3A). Differential gene expression (DGE) analysis also revealed that a total of 865 genes were differentially regulated between ARA-REG and GAL-REG strains and 1455 genes between XYL-REG and GAL-REG strains (Figure S1) (p-value < 0.05 after Benjamini–Hochberg correction). We reasoned genes that are either directly regulated by GAL regulon (e.g., GAL2, GAL4, etc.) make a small fraction of the transcriptome and would not be differentially expressed in pentoses and hexoses. Rather, differences would arise due to metabolic differences between pentoses and hexoses (e.g., energetic content, cofactor balance, etc.) that impose an alternate and complex, albeit indirect, regulatory control. This would explain why the transcriptome profile of the two pentoses were more alike compared to galactose. Therefore, we also hypothesized that if we performed DGE analysis on pentose (combined xylose + arabinose) vs. galactose, we may be able to identify “intrinsic” factors that sense and distinguish growth on pentoses vs. a hexose. We found 480 genes that were upregulated and 358 genes that were downregulated genes in pentoses vs. galactose (Figure 3B).

A) Differential expression volcano plot for transcriptome profile of galactose vs. xylose and arabinose combined. B) PCA plot of the three conditions AR#; ARA-REG grown on arabinose, XR#; XYL-REG grown on xylose, G#; WT grown on galactose; C) Network topology obtained of mutual information network obtained by mapping differentially expressed genes onto the yeast gene regulatory network. D) 24 highly connected genes identified from the mutual information network (with p < 0.05) that are targets for deletion to improve growth on pentoses.
Given the large number of differences between the two conditions (pentose vs. galactose), we wanted to identify core, highly-connected, regulatory elements that are the major contributors of observed differential phenotypes, which requires analysis of inferred gene regulatory networks (GRN). We mined our data using three different GRNs – EGRIN (20), YEASTRACT (21), and CLR (22) – to identify potential factors that are responsible for divergent transcriptional phenotypes between pentoses and galactose. We then associated the significant genes (log2 > 1.5, p-value < 0.05) within our DGE data with the corresponding node within the three networks. Using “Betweenness Centrality” (BC) value for each of the remaining nodes, we collected the identities of the top nodes to compare against the list of potential targets generated by examining the DGE data alone. Exploring this network, we found that the enriched nodes broadly related to six gene ontology (GO) terms (Figure 3C, Figure S2). We determined a list of 24 targets for knock-out to understand their effect on growth and phenotype (Figure 3D). We generated barcoded deletion (KO) libraries of these “intrinsic” factors in wild-type and REG strain backgrounds and performed enrichment on glucose, galactose, xylose, and arabinose (Figure 4A). Comparing population shifts using barcodes, we identified 8 genes that displayed positive fitness on both xylose and arabinose but not on galactose – indicating a role in controlling growth primarily on pentose. We then tested their growth individually on galactose, xylose, and arabinose (Figure 4B-D). Of these genes, only ΔGLN3 was either neutral or beneficial in pentose – all other deletions were detrimental for growth on xylose. Further, the growth rate of this mutant (and all others) was still lower than that of the parental strain on galactose, indicating additional limitations. Overall, the results of this approach were disappointing. And while we can employ several strategies to further improve growth (e.g., combine deletions, ALE, etc.), we decided to focus on the upstream metabolic module.

A) Fitness map of knock-out library of 25 genes identified from the mutual information network. The scale represents fitness ranging from blue (negative) to orange (positive). Growth rates of KOs with positive fitness on B) galactose, C) xylose (GAL1p-XYLA*3 GAL10p-XKS1), D) arabinose (GAL1p-araB GAL10p-araA GAL7p-araD).
Pentose metabolism is largely extrinsically controlled
Our studies have so far focused on identifying “intrinsic” factors that may be controlling/bottlenecking growth on pentoses and given the inconsistent benefits, we wondered whether the limitations were, in fact, “extrinsic”. The former implies that the native metabolic and/or regulatory capacity of this yeast is inherently limited for effective pentose metabolism and improving growth rate requires vast restructuring of associated (intrinsic) networks. The latter implies that there are no inherent limitations in this yeast and that it is already poised for rapid growth on pentoses, but the observed low growth rates are due to suboptimal design of the upstream metabolic module that includes the heterologous (extrinsic) genes. To assess whether the “extrinsic limitation” paradigm has any merits, we needed to optimize the design of the upstream metabolic module responsible for substrate uptake and flux into central carbon metabolism (i.e., glycolysis).
First, we looked at the effect of plasmid copy number. For arabinose, there was no difference in growth rate when araA-araB-araD genes were expressed on high- or low-copy plasmids, whereas for xylose, we only observed growth when the XYLA*3-XKS1 gene dose was high (Figure S3). In either case, changing plasmids backbone did not improve growth rate. Next, we hypothesized that balancing expression of these heterologous genes may be required to enhance growth rate. For arabinose, we created all six combinations of gene-promoter pairings and assessed their performance. Surprisingly, we found marked improvement in growth rate – from 0.14 ± 0.04 h-1 in the original design to 0.27 ± 0.03 h-1 in the best re-design (Figure 5A). To understand the cause of this behavioral change, we compared the relative expression levels of the araA, araB, and araD using quantitative reverse transcription PCR (qPCR) (Figure 5B). We observed that in the poor performing combinations, expression of araB (ribulokinase) was the highest, whereas, in high performing combinations, expression level followed this pattern araA > araB > araD. In addition, we found a strong positive correlation between growth rate and relative expression of araA:araB and araA:araD, respectively (Figure 5C-E). The success of this approach encouraged us to attempt the same on xylose and found similar improvements – from 0.17 ± 0.01 h-1 to 0.24 ± 0.01 h-1 (Figure S4). These are comparable to the growth rate of yeast on galactose when GAL1-7-10 are expressed from a plasmid (0.24 ± 0.03 h-1) (Figure S5). Next, we used directed evolution to improve the growth rate on arabinose further. We randomly mutagenized the six arabinose pathway combinations, adding barcodes to track the lineages and enriched this library size of 108 variants in minimal (SC+Ara) and complex (YPAA) arabinose media. The growth rate over subcultures increased from 0.12 h-1 to 0.22 h-1 and 0.18 h-1 to 0.26 h-1 in SC+Ara and YPAA, respectively (Figure 5G, S6). Using barcodes, we tracked the performance of promoter-gene combinations. We observed that initially all the six plasmids start at similar abundance, but the araA-B-D (under GAL1-10-7p, respectively) was the most abundant at the end of enrichment in SC+Ara. We picked single colonies from each condition and calculated their growth rates and identified 4 variants that showed the highest growth rates (0.35 ± 0.04 h-1) from the SC+Ara enriched culture (Figure S7). We sequenced the barcodes to identify the lineage and the whole cassette to identify the mutations and found that three of the six initial designs were represented in the four best variants (named N-3, N-6, N-12, and N-16) (Table S3). Re-transformation into parent background strain indicated that the four variants attained the same maximum growth rate as that on galactose (0.29 ± 0.01 h-1). Since the mutations were distributed throughout the cassettes, we quantified the expression levels of araA, araB, and araD in the four strains and we found a strong positive correlation between growth rate and relative expression of araA:araB and negative correlation between araB:araD, indicating that the directed evolution campaign likely altered both activity and expression in each strain differently (Figure S8). These results highlight a key insight about yeast: it’s ability to utilize pentoses is largely limited only by “extrinsic” factors (upstream pathway, especially heterologous enzyme activity) and minimally by any “intrinsic” factor (i.e., native regulation or metabolic pathway).

A) Comparison of growth performance of different promoter-gene combination. B) Gene expression analysis of the six strains by qPCR. Pearson correlation coefficient between growth rate and gene expression levels of C) araA:araB, D) araB:araD, and E) araA:araD, respectively. G) Enrichment of mutagenic araBAD library in SC+Ara. H) Fitness heatmap of strain dynamics during enrichment on arabinose, reveled by sequencing barcodes. I) Growth curves and J) qPCR expression profile of araBAD genes encoded in plasmids isolated from top performing strains isolated after directed evolution.
Engineering intrinsic factors lead to pleiotropic fitness trade-offs
We next assessed whether the global regulatory elements identified through our network analysis would benefit our best designs (araA-B-D under GAL1-10-7p and XYLA*3-XKS1 under GAL1-7p). Deleting each of the 8 genes in this strain did not result in any improvements over the parental strains (Figure 6A). Here too, we could explore a combinatorial deletion of ALE strategy to enhance growth rate. However, given that each strain with had already attained aerobic growth rate equivalent to the maximum described for this yeast on glucose and galactose (0.22 h-1 – 0.25 h-1), with high biomass yields and short lag, we expect that improvements would be insignificant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
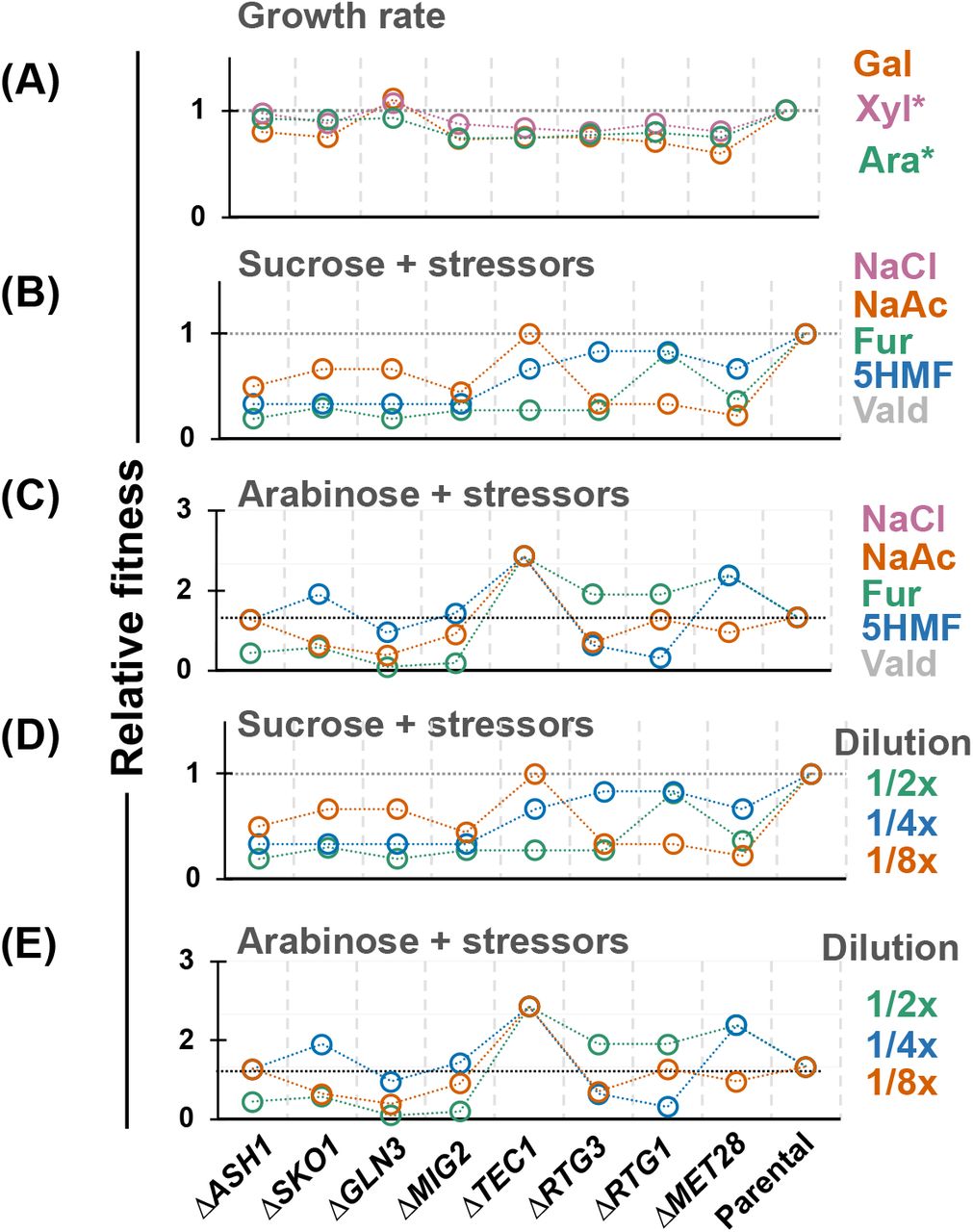
A) Growth rate of the optimized plasmids in KO-strains. Fitness of KOs in presence of individual stressors when grown on B) sucrose and C) arabinose or in the mixture of stressors on D) sucrose and E) arabinose. The relative fitness is expressed as growth of deletion strains over the fitness of parental strain under the same conditions. NaCl = sodium chloride; NaAc = sodium acetate; Fur = furfural; 5HMF = 5-hydroxymethylfurfural; Vald = veratraldehyde. 1/2x, 1/4x, 1/8x = serial dilutions of the five stressors combined. Xyl* = GAL10p-XYLA*3 GAL7p-XKS1; Ara* = GAL1p-araA GAL10p-araB GAL7p-araD in respective REG backgrounds.
One consideration not yet accounted for is the suitability these strains for bioprocessing and their resilience to growth inhibitors found in lignocellulosic hydrolysates. Given that all deletion targets are highly-connected genes that control key cellular processes, we were concerned that dysregulation major networks may lead to undesirable pleiotropic effects. Since resilience against stress is a complex phenotype, often requiring concerted response from gene networks, we tested the fitness of all single KO strains on bioprocessing relevant stressors – individually, and in combination (Figure 6B-E). Using parental strain as reference, we observed that performance of strains under stress was highly context dependent. For example, in sucrose medium, all deletions lost fitness in single or mixed inhibitor cultures (Figure 6C, E). Conversely, on arabinose (with GAL regulon activated), certain KO strains had improved tolerance to stressors (e.g., ΔTEC1, ΔMET28) (Figure 6D, F). ΔGLN3 has previously been shown to improve fitness under isobutanol stress (23); however, in our study, it was less fit than the parental strain under all stress conditions. Interestingly, it did demonstrate improved growth rate in a sub-optimal upstream metabolic design in arabinose (Figure 4E) but lost that benefit in a more optimized design (Figure 6C, E). Collectively, these results highlight that deleting genes to remodel expression profiles to enhance a single phenotype (e.g., growth rate on a non-native substrate) can lead to some improvements, but they are often accompanied by negative pleiotropic effects that make the strain less suitable for eventual bioprocessing applications.
DISCUSSION
Despite decades of effort engineering synthetic heterotrophy with S. cerevisiae, there is not formalized understanding of what limits metabolic adaptability of this yeast for growth on pentoses. It is important to differentiate between substrate utilization/uptake from growth since the former can be readily achieved by diverting flux to unwanted or dead-end byproducts (e.g., Crabtree metabolites, organic acids, pentitols) often with native substrate supplementation to support biomass generation. However, valorization to high-value products requires efficient catabolism to central metabolic products whose concentrations are regulated and associated with growth. Initial studies in literature focused on upstream (extrinsic) elements (e.g., heterologous gene expression/activity, transporter engineering, etc. (24–28)) whereas recent focus has been more intrinsic, focusing on functional genomics, adaptively laboratory evolution (ALE), and often network/systems analysis, to identify inherent limitations (5–13). Successes have been abundantly reported with improvements in growth along with a series of deletion and overexpression targets (PHO13, ALD6, ASK10, YPR1, SNF6, RGT1, CAT8, MSN4, GPD1, CCH1, ADH6, BUD21, ALP1, ISC1, RPL20B, COX4, ISU1, SSK2, YLR042c, CYC8, PHD1, TEC1, ARR1, etc. (13, 29–35)). Interestingly, many targets are unique to individual studies, suggesting the context dependence of targets on initial strain designs. Prototypical is a recent report of using a combination of all the aforementioned approaches and extensive analysis to engineer a strain of yeast that can grow aerobically with high growth rate (0.26 h-1) and short lag phase (9-15 h) on xylose (13). Through our work, we posit that requirement for extensive engineering to identify intrinsic limitations is only required when the upstream/extrinsic metabolic module is sub-optimal. Indeed, constitutive overexpression of upstream metabolic genes results in significant stress (18) and low growth rates that must be compensated for through extensive strain engineering focused on preventing cellular detection of stress rather than activating growth supporting modifications. We demonstrated that with an optimal upstream module, strains can attain near-wild-type growth profiles (fast specific growth rate, short lag phase, high biomass yield) if a growth-associated regulon – the GAL regulon – is activated. Importantly, our final strain designs have only one gene deleted (other than the Leloir pathway genes) – GRE3 – to minimize oxidation of substrate pentoses to pentitols. Many genes that have previously been identified as important inactivation targets to improve growth on pentoses are intact. We also overexpress only a minimal upstream metabolic module (TAL1 and GAL2-2.1) along with the specific pentose metabolic genes (araBAD or XYLA*3-XKS1). Our work strongly argues that pentose metabolism is largely extrinsically controlled and there are no major intrinsic regulatory or metabolic limitations for growth and the insights present a paradigm shift in engineering synthetic heterotrophy.
An additional advantage of this approach is the preservation of native regulatory systems that are otherwise dysregulated in traditional engineering approaches. We found that while deletion of endogenous genes could improve growth in strains with sub-optimal upstream modules under certain conditions, they are associated with pleiotropic defects and are less robust in the presence of common lignocellulosic inhibitors. This is not surprising since most genetic interventions aim to suppress cellular responses to stress rather than promote growth. Thus, strains engineered for synthetic heterotrophy through extensive gene inactivation are less robust for eventual bioprocess applications. Overall, our approach is minimalistic and holistic – our strains maintain native regulatory systems and even exploited them toward the engineering goal (i.e., the GAL regulon). This approach significantly simplifies and expedites the design-build-test cycle for synthetic heterotrophy and demonstrates that intrinsic adaptability of yeast toward growth on non-native substrates. We expect that this insight will expand the utility of this yeast for valorizing current and emerging waste and/abundant substrates.
DATA AVAILABILITY
RNA-seq sequencing data has been submitted to NCBI SRA and is available under accession PRJNA837644. R script for mutual information network is available at https://github.com/nair-lab/yeast-MINet.
AUTHOR CONTRIBUTIONS
N.U.N., V.D.T., and D.C. conceived and designed the research project. V.D.T., S.F.S., D.C., and N.U.N. co-wrote the manuscript. V.D.T., S.F.S., V.E.G., and T.H. performed the experiments. V.D.T., S.F.S., and N.U.N. analyzed the data. All the authors have reviewed the manuscript and approved it for submission.
COMPETING INTERESTS
None.
MATERIALS AND METHODS
Strains and plasmids
The list of plasmids and strains used are listed in Supplementary Table S1 and S2. Strain W303-1a (MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15) and plasmids pIS374, pIS376, pIS385 were obtained from Euroscarf (Germany). The plasmids were constructed in the present using NEB-HiFi DNA assembly master mix from NEB (Beverly, MA).
Growth studies
Overnight inoculums were grown in the required dropout SC medium with sucrose (2 %). The culture was washed twice in the growth medium and resuspended at an initial OD600 of 0.1 with appropriate sugar (2 %) in 250 mL shake flasks containing 20 mL of media. OD600 measurement was checked at frequent time intervals (3-6 h) on SpectraMax M3 spectrophotometer (Molecular Devices). Growth rate was determined by plotting the values in GraphPad Prism following non-linear regression and using exponential growth equation, Y = Y0 exp(kX).
Directed evolution
Error prone PCR libraries of arabinose cassettes (pVDT14, pVDT38-42) were generated as described earlier (18). Six-barcoded primers (Table S4) were used to track the proportion of the lineage during the enrichment. The amplicons from error prone PCR were assembled into pRS413 plasmids using yeast gap-repair cloning. To attain high library size, the linear fragments were transformed into yeast by electroporation as described previously (36). We attained library size of ∼108 CFUs which was then subjected to enrichment on arabinose in 2YP as well SC media. The cell pellets from each passage were frozen for quantifying barcodes using amplicon sequencing (Genewiz, Cambridge, MA). At the end of enrichment, the library pool was plated on 2YP+Ara and SC+Ara and 18 colonies were randomly picked for growth rate determination. The plasmids from best variants were isolated from strain, transformed into E. coli. The corresponding plasmid isolated from E. coli was sequenced and re-transformed into REG-strain for growth rate determination.
Genomics integrations
Accessory cassettes (pVDT29, 30, 35, Table S1) were integrated into VEG16 (W303-1a, ΔGRE3, ΔGAL1, ΔGAL3, ΔGAL7, ΔGAL10) (18) strain via disintegrator plasmid(37). Counterselection was performed on SC medium with 1 g/L 5-FOA to remove the URA3 marker and the locus was amplified, and sequence confirmed.
Deletion library
To generate the deletion library, the cassettes for the required targets were amplified from the genomic DNA of the appropriate strains from the KO collection (38). Since the cassettes contained KANMX marker, the library was selected on YP+G418 (400 μg/mL). The pool was stored as a stock and used for studying the fitness by transforming it with plasmids for galactose, xylose, and arabinose utilizing cassettes.
qPCR expression analysis
Total yeast RNA isolation was performed on OD ∼ 1 of yeast cells collected from mid-log phase growth using the GeneJet RNA Purification Kit (Thermofisher Scientific Catalog #: K0731) according to the instructions for isolating yeast RNA. Two samples were collected for each growth condition. The concentration of total RNA isolated was obtained by measuring 10-fold dilutions from each sample on a spectrophotometer. Using these concentrations, 2.5 μg of total RNA was subjected to the ‘routine’ DNase treatment protocol using the Invitrogen Turbo DNA-free Kit (Thermofisher Scientific Catalog #: AM1907). Next, 20 % of the final reaction (10-50 μL, targeting 500 ng RNA) was used to synthesize cDNA for the sample using the Invitrogen SuperScript IV First-Strand Synthesis System (Thermofisher Scientific Catalog #: 18091050). The random hexamers supplied by the kit were used to prime the reverse transcriptase enzyme. cDNA samples were diluted 100-fold (2 μL into 198 μL dH2O), of which 2 μL was used as the template for a 15 μL qPCR reaction using the Applied Biosystems PowerUp SYBR Green Master Mix (Thermofisher Scientific Catalog #: A25741). Each sample was tested for the expression level of five genes: the three arabinose catabolic genes (araA, araB, and araD) as well as two housekeeping genes (TFC1 and UBC6) using the primers listed in Table S5. Reactions were run on an Applied Biosystems Quantstudio 5 Real-Time PCR Instrument. The relative expression level of each catabolic genes was calculated by subtracting the geometric mean of the cycle thresholds (Ct) of the two housekeeping genes from the Ct of that catabolic gene, followed by conversion of the log-base two Ct into a non-log value that can be compared across genes.
RNA-seq
Transcriptomics of strains WT, XYL-REG, ARA-REG were performed on the mid-log phase cultures grown on their respective carbon source (galactose, xylose, or arabinose). Cells pellets were washed twice in water and stored at −80 °C and outsourced to Genewiz Inc. for RNA extraction and sequencing. RNA-seq was performed on Illumina HiSeq. Raw FASTQ files were processed for differential expression analysis using GeneiousPrime. The possible adapter sequences and low-quality short-read (less than 50 bp) trimming were performed using BB-Trim package. The reads were aligned to the reference genome W303 obtained from Saccharomyces Genome Database (http://www.yeastgenome.org) using Bow-Tie2 package. The edgeR package was used to normalize the gene count based on library size and was converted to cpm (counts per million) using. DESEQ2 package was sued for differential gene expression analysis. Genes with p-values < 0.05 and fold change of ≥ 2 were considered as differentially expressed.
Amplicon sequencing analysis
The total DNA was extracted and quantified using a spectrophotometer and approximately 100 ng was used as template for PCRs. Unique, barcoded primers flanked by Illumina sequencing adaptors were used to generate the amplicons after 15-20 cycles of PCR for sequencing (Table S6). The barcoded samples were pooled and sent for amplicon sequencing (2×250 bp, Genewiz, New Jersey, USA). For each pooled sample, we received approximately 100,000 sequencing reads. These data were processed according to a previously described bioinformatic workflow using Geneious Prime® 2020.2.4 (39). Briefly, the reads were paired and merged using the BBMerge package and filtered for poor-quality reads using the BBDuk package. The reads were mapped using BowTie2 and gene expression was calculated and differential expression was determined using DESeq2 (40).
Network analysis
The S. cerevisiae genes that we consider as the ‘gold standard’ list of transcription factors is the unique set of genes identified in the file supplemental data File S1. Our analysis utilizes three yeast genetic regulatory networks (EGRIN, YEASTRACT, and CLR) (41) in the context of integrating regulatory and metabolic networks to enable more accurate prediction of yeast phenotypes in different growth conditions (42). The Environment and Gene Regulatory Influence Network (EGRIN) consists of 92 regulators and 2,588 interactions and was constructed using the cMonkey and Inferelator computational tools trained on expression data from 2,929 microarray experiments to identify gene clusters and potential regulatory genes. The YEASTRACT gene regulatory network was extracted directly from the YEASTRACT database2 and consists of 177 regulators and 31,075 regulatory associations based on both ‘direct evidence’ – chromatin immunoprecipitation (ChIP), ChIP-on-chip, electrophoretic mobility shift assay, or examination of the effect of TF binding site mutations on target gene expression – as well as ‘indirect evidence’ provided by gene expression changes in response to deletion, mutation, or overexpression of a given TF3.
The context likelihood of relatedness (CLR) algorithm quantifies the amount of mutual information shared between all possible gene pairs in each set of gene expression data. The regulatory network is then inferred by taking the distribution of calculated mutual information for all gene pairs and filtering out gene pairs whose mutual information content is below a chosen threshold with the remaining pairs considered to be connections in the network4. The R programming language was used for importing and manipulating data. R scripts can be found in https://github.com/nair-lab/yeast-MINet.
Stressor studies
We determined fitness of the strains in presence of various stressors encountered during biomanufacturing using plate assay. Briefly, we tested the growth of by performing spot dilution on 2YP supplemented with 2 % sucrose or arabinose and various concentration of the stressors (Table S7). The plates after incubation were imaged and the colonies were counted to determine the CFUs. The CFUs in the presence of the stressor was divided by the CFUs in absence of the stressor to determine the fitness of that strain. This value was normalized to the fitness of the parental strain and was expressed as the relative fitness.
ACKNOWLEDGEMENTS
The authors would like to thank current and former Nair lab members, Dr. Todd C. Chappell, and Dr. Karishma Mohan for helpful discussions. This work was supported by NIH grant #DP2HD91798, NSF grant #1935354, and Tufts Launchpad | Accelerator to N.U.N.
Footnotes
REFERENCES




