

Abstract
The SARS-CoV-2 main protease (3CLpro) has an indispensable role in the viral life cycle and is a therapeutic target for the treatment of COVID-19. The potential of 3CLpro-inhibitors to select for drug-resistant variants needs to be established. Therefore SARS-CoV-2 was passaged in vitro in the presence of increasing concentrations of ALG-097161, a probe compound designed in the context of a 3CLpro drug discovery program. We identified a combination of amino acid substitutions in 3CLpro (L50F E166A L167F) that is associated with > 20x increase in EC50 values for ALG-097161, nirmatrelvir (PF-07321332) and PF-00835231. While two of the single substitutions (E166A and L167F) provide low-level resistance to the inhibitors in a biochemical assay, the triple mutant results in the highest levels of resistance (6- to 72-fold). All substitutions are associated with a significant loss of enzymatic 3CLpro activity, suggesting a reduction in viral fitness. Structural biology analysis indicates that the different substitutions reduce the number of inhibitor/enzyme interactions while the binding of the substrate is maintained. These observations will be important for the interpretation of resistance development to 3CLpro inhibitors in the clinical setting.
Introduction
There is an urgent need for potent and safe antiviral drugs for the treatment and prophylaxis of SARS-CoV-2 infections. Highly efficacious and safe viral protease inhibitors have contributed significantly to the effective treatment of infections with HIV and HCV. Coronaviruses have two proteases, the main protease 3CLpro (or Mpro) and the papain-like protease. 3CLpro is a cysteine protease that cleaves the two polyproteins (pp1a and pp1ab) of SARS-CoV-2 at eleven different sites, resulting in various non-structural proteins, which are key for viral replication [1]. The substrate of 3CLpro presents a distinct glutamine at the P1 site (Leu-Gln/Ser, Ala, Gly), while no known human proteases recognize this cleavage site [2, 3]. 3CLpro can thus be considered a highly attractive drug target for the development of SARS-CoV-2 antivirals [4]. The potential of 3CLpro inhibitors has become apparent with the development of nirmatrelvir (PF-07321332), a peptidomimetic reversible covalent inhibitor that is co-formulated with the pharmacokinetic enhancer ritonavir (the resulting combination being marketed as Paxlovid) [5]. When treatment is initiated during the first days after symptom onset, it results in roughly 90% protection against severe COVID-19 and hospitalization [6]. We recently showed that nirmatrelvir is equipotent in vitro against the current SARS-CoV-2 variants of concern (VoC). Nirmatrelvir protects Syrian Golden hamsters from intranasal infection with different VoCs and prevents transmission to untreated co-housed sentinels [7, 8]. Other clinical candidate 3CLpro inhibitors include ensitrelvir (S-217622), a non-peptidic, non-covalent SARS-CoV-2 3CLpro inhibitor [9, 10], and PBI-0451 [11]. The clinical development of lufotrelvir, an intravenous pro-drug of PF-00835321 has been discontinued [12, 13].
Treatment with antivirals can result in the selection of resistant viral variants and subsequent therapeutic failure. This has been described extensively in the treatment of (chronic/persistent or acute) viral infections caused by HIV, HBV, HCV, herpesviruses or influenza [14, 15]. Importantly, transmission of resistant viruses has been reported for HIV and influenza [16, 17]. For SARS-CoV-2, selection of resistant isolates has only been described for remdesivir, a polymerase inhibitor. In vitro selection with remdesivir results in the emergence of resistance-associated mutations. Yet, in the clinical setting, treatment with remdesivir so far only led to the selection of mutations that are associated with low level resistance [18, 19]. A causal effect between SARS-CoV-2 resistance to any replicase inhibitors and therapy failure has not yet been demonstrated, most likely because these inhibitors are not yet widely used in the clinic and/or resistant variants might have a fitness disadvantage.
Here we report on a pathway by which SARS-CoV-2 achieves significant resistance and cross-resistance to 3CLpro inhibitors during serial passage in cell culture in the presence of a first generation 3CLpro inhibitor, ALG-097161. This molecule was prepared as a tool compound in the context of a drug discovery program.
Results
SARS-CoV-2 acquires resistance to 3CLpro Inhibitors during passage with ALG-097161
ALG-097161 inhibits the replication of SARS-CoV-2-GHB, a prototypic Wuhan isolate (Belgium/GHB-03021/2020; EPI_ISL_407976; 2020-02-03 [20]), in VeroE6 cells by 50% at a concentration of 0.59 μM, and has no effect on the viability of uninfected host cells at concentrations up to 10 μM (Table 1). Since VeroE6 cells have a high efflux of some chemotypes, all selection experiments, antiviral assays and toxicity assays were performed in the presence of the P-glycoprotein (Pgp) efflux inhibitor CP-100356 (0.5 μM) [21]. The antiviral activity of ALG-097161 can be ascribed to inhibition of 3CLpro (IC50 = 0.014 μM - Table 3) and there is no relevant inhibitory effect on human Cathepsin L (IC50 > 10 μM – data not shown). For comparative reasons the chemical structures of ALG-097161 and the clinical 3CLpro inhibitors nirmatrelvir and PF-00835231 are shown in Figure 1.
Phenotypic resistance associated with the L50F E166A L167F mutation profile as determined in VeroE6 cells
Phenotypic resistance associated with the L50F E166A L167F mutation profile as determined in a cell-based 3CLpro reporter assay
Chemical structures of ALG-097161, Nirmatrelvir and PF-00835231
To identify resistance development to 3CLpro inhibitors, we passaged SARS-CoV-2-GHB in VeroE6 cells in the presence of increasing concentrations of ALG-097161. The selection started at a concentration of 0.4 μM and for each next passage the virus was cultured at the same concentration, a 3× higher concentration and a 3× lower concentration (Figure 2). The culture demonstrating virus breakthrough, as observed by a significant cytopathic effect, at the highest concentration was then used for the next passage. In this way, the concentration of ALG-097161 could be increased gradually to 5 μM at passage 8 (p8, day 28) and maintained for one passage at 15 μM but had to be decreased again to 5 μM for the subsequent passages in order to allow viral replication until p12 (day 39) (Figure 2). Whole genome sequencing was performed on RNA purified from harvested supernatants at passages 5, 8 and 12. At p5 no dominant mutations (>50 %) were identified in the 3CLpro gene but at p8 and p12 dominant mutations in 3CLpro were detected that result in amino acid substitutions L50F and E166A by p8 and L50F, E166A and L167F by p12. As a control, SARS-CoV-2 was also passaged, at the same frequency, in the absence of compound and no mutations were identified in the 3CLpro gene of the resulting samples at any passage (data not shown).

Passaging SARS-CoV-2-GHB (Wuhan) in VeroE6 cells in the presence of increasing concentrations of ALG-097161 (and the efflux inhibitor CP-100356). Selection was initiated at 0.4 μM. At every passage several new cultures were started with the same concentration as well as a lower and a higher concentration. The passage with the highest compound concentration that could be maintained was selected for the following passage. At passage 5, 8 and 12, vRNA in the cell culture medium was sequenced. Substitutions in the 3CLpro that were found at these passages are indicated.
For phenotypic analysis, a new virus stock (p13) was grown in the presence of 4 μM ALG-097161, and the presence of L50F E166A L167F was confirmed by genotypic analysis. Antiviral testing with this stock clearly indicates strong resistance to ALG-097161 and cross-resistance to both nirmatrelvir and PF-00835231. The EC50 values of all these 3CLpro inhibitors is increased > 10x (Table 1). As expected, the sensitivity for the polymerase inhibitor GS-441524 remains unchanged.
Resistance to 3CLpro inhibitors in a cell-based reporter assay
In a second series of experiments, we aimed to confirm the observed resistance profile in an independent cell-based assay. To this end, we engineered, using a reverse genetics approach [22], a virus stock that carries the three mutations. While the correct sequence was confirmed, the replication capacity of this recombinant mutant virus was too low to allow antiviral experiments.
For this reason, we set out to confirm the resistance profile in another type of cell-based assay. We adapted a previously described cell-based reporter assay of SARS-CoV-2 3CLpro enzymatic function [23]. In this gain-of-function assay, inhibition of 3CLpro results in an increased eGFP signal. Introducing the three amino acid changes L50F E166A L167F into the construct confirmed the resistance profile. Indeed, a 23- and 28-fold loss of potency for ALG-097161 and nirmatrelvir respectively, was observed (Table 2). The resistance level of the triple mutant against PF-00835231 was 6.9-fold.
Effect of mutations on 3CLpro enzymatic activity and dimerization
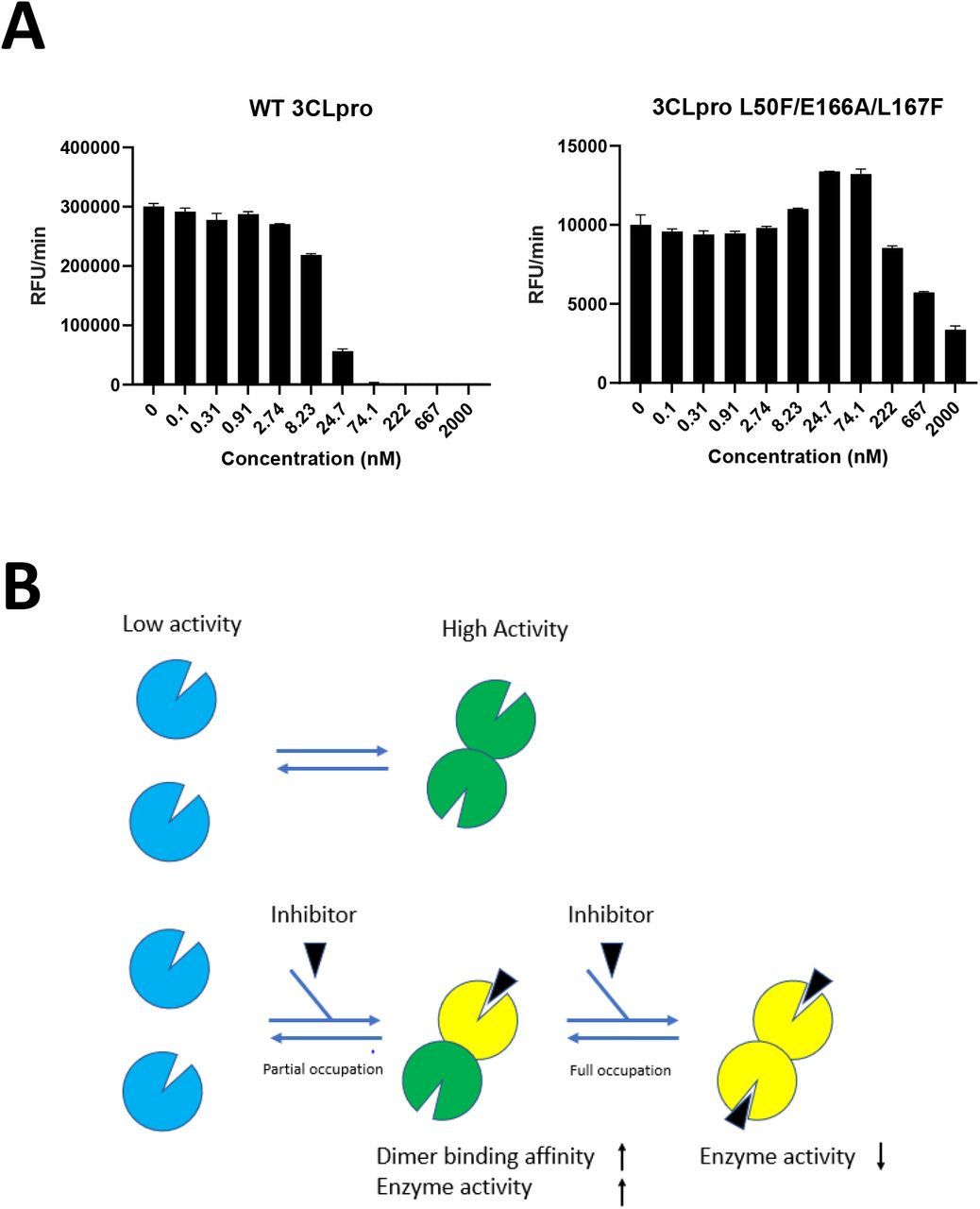
Recombinant 3CLpro proteins were produced with the L50F, E166A and L167F substitutions alone or combined, and their enzymatic activity was tested in a FRET assay using a peptide substrate featuring the canonical glutamine cleavage site [24]. In this assay, wild-type (WT) 3CLpro shows linear product conversion in the enzyme concentration range of 0.5-12.5 nM (Figure 3A). Compared with the WT protein, all tested mutants display reduced enzymatic activity (Figure 3B). The E166A and L167F, as single mutants, reduce the activity to about 15% of wild-type, whereas the L50F substitution reduces the activity to as low as 0.5%. The enzyme containing the three substitutions L50F, E166A and L167F, is less compromised than L50F alone, with 5.3% activity as compared with WT. The loss in enzymatic activity for each mutant can be attributed to a reduced binding affinity for the substrate and a reduced formation of active dimers (Figure 4). The enzymes with E166A and L167F are less impaired than the enzyme with L50F. In comparison, the triple mutant exhibits an intermediate phenotype for both parameters (Figure 4).

(A) The enzymatic activity of WT 3CLpro was measured in a FRET assay. (B) Comparison of enzymatic activity between WT 3CLpro and mutated enzymes. Each data point represents a single measurement.

(A) Initial velocities of enzyme titration of WT and mutant 3CLpros were fitted as described (see methods) to calculate the monomer dimer equilibrium dissociation constant. (B) 3CLpro dimerization binding affinity for each enzyme. Each data point represents a single measurement.
Ligand-induced protease activation with low concentration of active site inhibitor has previously been described for MERS 3CLpro [25]. This effect can be explained by the inhibitor stabilizing dimer formation by occupying only one of the two binding sites, while full occupancy of the two dimer active sites results in complete enzyme inhibition (Figure 5B). We obtained comparable results and found that the impaired enzymatic activity and dimer formation of the mutants can be partially rescued with low concentrations (around 0.05 μM) of ALG-097161, while higher concentrations inhibit product formation (Figure 5A).

(A) Comparison of ALG-097161 dose-response for WT and L50F E166A L167F 3CLpro. Three independent experiments were performed. The figure shows the results of one representative experiment. (B) Model for ligand-induced dimerization and enzymatic activation at low concentration of inhibitor.
Resistance to 3CLpro inhibitors in the biochemical assay
Based on the characterization of the different enzymes (see above), we decided to use all enzymes at a concentration of 50 nM to quantify the activity of the inhibitors. ALG-097161, nirmatrelvir and PF-00835231 all inhibit WT 3CLpro with IC50 values ranging from 13-23 nM (Table 3). For ALG-097161 the IC50 increases 5-6-fold with single substitutions E166A and L167F, and reaches 35-fold vs. the enzyme with L50F E166A L167F. In comparison, the potency of nirmatrelvir is more affected by mutation E166A vs. L167F (10-fold vs. 4.4-fold) and reaches 72-fold change on the enzyme with the 3 substitutions. For PF-00835231 the shift in IC50 is in general lower with a 6.0-fold change vs. the enzyme with L50F E166A L167F. The effect of the L50F substitution could not be determined under these conditions due to its low intrinsic enzymatic activity.
Structural Biology and Computational Chemistry
Experimental data with the 3CLpro system and computational chemistry investigations were used to rationalize the phenotype observed for ALG-097161 in vitro.
A series of high-resolution X-ray structures of proprietary ligands in complex with WT 3CLpro were solved, including a data set refined to 1.3 Å for an inhibitor with substantial similarity to ALG-097161, in particular identical P2 and P3 substitutions. These data afforded detailed understanding of the conformation of the 3CLpro binding site and provided an excellent starting point for in silico investigations of ALG-097161.
The conformational space of ALG-097161 was explored extensively and a large set of representative ligand structures was evaluated through de novo docking calculations. These computations yielded a well-converged and thoroughly validated predicted binding mode of ALG-097161 covalently bound to the catalytic site of WT 3CLpro (Figure 6).

Predicted binding mode of ALG-097161 covalently bound to the catalytic site of WT 3CLpro. Left panel: ALG-097161 (carbon: cyan, nitrogen: navy; oxygen: red) covalently docked to WT 3CLpro (gray cartoon and surface; key side chains shown with carbon: green, nitrogen: navy; oxygen: red). Right panel: ALG-097161 binds covalently to the catalytic C145 and forms seven hydrogen bonds with 3CLpro: warhead to C145 in the oxyanion hole; P1 lactam to H163, E166 and F140; peptide backbone to H164 and E166; P3 indole to E166. The fused bicyclic P2 substitution maximizes Van der Waals interactions in the S2 sub-pocket.
In line with the crystal structures obtained with similar compounds, ALG-097161 is predicted to form a covalent bond from the activated carbon atom of its hydroxy-methyl-bearing warhead to the side chain of the catalytic C145 residue, anchoring the inhibitor into the binding cleft of the 3CLpro enzyme. In addition, ALG-097161 forms seven direct hydrogen bonds with the binding site. The oxygen atom of the inhibitor’s warhead forms a hydrogen bond to the backbone NH of C145 in the oxyanion hole. The carbonyl oxygen of the P1 lactam hydrogen-bonds to the side chain of H163, while the NH of the P1 lactam forms a hydrogen-bonding network via a bifurcated interaction to the side chain of E166 and the backbone carbonyl of F140. The peptide backbone of ALG-097161 anchors itself with hydrogen bonds to the backbone carbonyl of H164 and the backbone HN of E166. The NH function of the P3 indole hydrogen-bonds the inhibitor to the backbone carbonyl of E166. The fused bicyclic P2 substitution of ALG-097161 maximizes Van der Waals interactions via extensive productive contacts to the protein side chains that form the S2 sub-pocket of WT 3CLpro.
The detailed understanding of ALG-097161 interactions with WT 3CLpro from our in silico analysis, combined with broader structural biology observations, provides a logical rationale for the mutations observed during in vitro passaging of the compound in cell culture (Figure 7).

ALG-097161 (carbon: cyan, nitrogen: navy; oxygen: red) modeled in the L50F E166A L167F 3CLpro (Post-MD pose). The overall binding mode of ALG-097161 is conserved, but one hydrogen bond from the lactam moiety is lost due to E166A mutation. Hydrogen bonds to the backbone of A164 are conserved, despite the distortion required to accommodate the L167F substitution.
Residues 166 and 167 are located within 5Å of the bound inhibitors (ALG-097161 as described above; PF-00835231 and nirmatrelvir from PDBs 6xhm and 7rfw, respectively) and consequently the observed substitutions have a direct impact on compound / target interaction. E166 plays a key role in compound binding, with no less than three hydrogen bonds formed with the inhibitors. The E166A substitution eliminates one of the hydrogen bonds made within the P1 sub-pocket. Interactions with this sub-pocket are known to be important for substrate recognition by coronaviral 3CLpro enzymes, and its occupancy, usually by a lactam moiety, is a key driver for inhibitor potency. It is therefore unsurprising that all three inhibitors are adversely affected by the E166A change. Meanwhile, L167F results in the presence of a bulkier side chain on the neighboring position. While this change does not directly conflict with compound binding, L167 is in close contact with other residues, including Phe185. Molecular Dynamics (MD) simulations on the L50F E166A L167F enzyme suggest that some distortion of the sub-pocket might be necessary to accommodate the bulkier Phe residue. In particular, the loop containing L167F moves away from F185 and the distance between the Cαs of F185 and L167F / P168 consistently increases, resulting in a more open binding site and a sub-optimal fit of the inhibitor, even though interactions with the A166 backbone are conserved (Figure 8). A comparable change was observed with PF-00835231 and nirmatrelvir. Interestingly, all three inhibitors (ALG-097161, PF-00835231 or nirmatrelvir) were affected by the L167F change.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed effect of the L167F substitution. Accommodation of the bulkier F167 residue results in some distortion of the binding site and in a possibly sub-optimal fit. Top Left: Binding mode of ALG-097161 in WT 3CLpro (minimized MD frame at 5 ns simulated time). Protein represented as surface (colored per atom type) and ligand as green sticks. Top right: surface view for ALG-097161 in L50F E166A L167F triple mutant (minimized MD frame at 5 ns simulated time). Ligand represented as cyan sticks. Note the more open surface around the indole moiety. Bottom: Overlay of the two MD frames. WT protein displayed as pale green cartoon and L50F E166A L167F mutant displayed as pale blue cartoon. Distances between Cα of F185 and L167/F167 (red dots) and between Cα of F185 and P168 (yellow dots) are indicated.
L50F falls outside of direct Van der Waals or hydrogen-bond contact range to ALG-097161. However, the L50 side chain is located within 2.7 Å of Q189 and L50F may precipitate an adjustment in the placement of the Q189 side chain. High resolution X-ray data on close analogues of ALG-097161 (not shown) indicate that Q189 can hydrogen-bond to the peptide backbone of this inhibitor class via an extensive network of ordered water molecules. While it is possible that the L50F mutation affects the rotamer distribution of Q189 and water-mediated interactions, a rigorous assessment of how those changes impact enzyme activity and/or inhibitor binding would require more elaborated techniques (e.g., free energy perturbation) that are beyond the purpose of this work.
Finally, it is anticipated that the natural SARS CoV-2 polyprotein substrates, that are processed by the 3CLpro, are less affected by the L50F, E166A and/or L167F substitutions, due to their larger size and approximately two-fold higher number of polar interactions with the protease (as observed in 2q6g.pdb for SARS CoV-1 3CLpro in complex with bound substrate).
Discussion
Nirmatrelvir is the first oral SARS-CoV-2 (3CLpro) protease inhibitor developed for the treatment of COVID-19. Highly potent protease inhibitors have also been developed against HIV and HCV [26]. To avoid resistance development, HIV and HCV protease inhibitors are being used, with great success, in fixed dose combinations with other directly acting antiviral drugs that have a different mechanism of action. As a single agent several of these inhibitors select (rapidly) for drug-resistant variants. SARS-CoV-2 infections are typically acute in nature; hence treatment can be of short duration which may largely reduce the likelihood that resistant variants develop. Yet also for acute infections, the emergence of drug-resistant variants is a serious concern, certainly in case that such variants can be efficiently transmit via the airborne route resulting in a loss of therapeutic options for patients. For example, the first-generation influenza drugs, amantadanes, are no longer recommended as resistance to amantadine and rimantadine is widespread [17].
To monitor clinical drug-resistance development it is crucial to identify the mutations involved. To that end, we used the 3CLpro inhibitor ALG-097161, a probe compound in the context of a drug discovery program, to select for resistant variants in vitro by passaging SARS-CoV-2 in presence of increasing drug concentrations. While similar experiments have been performed with SARS-CoV-2 neutralizing antibodies and polymerase inhibitors [19, 27], this is, to our knowledge, the first description for a 3CLpro inhibitor.
The selection process indicated that there is a significant barrier to resistance. The first changes were observed only between passage 5 and 8 (20-30 days). These were the double substitution L50F E166A in 3CLpro, which then evolved to the triple substitution L50F E166A L167F. Phenotyping this triple mutant virus revealed a 63-fold increase in the EC50 for ALG-097161. Moreover, significant cross-resistance (>10-fold increase EC50) was observed with nirmatrelvir, the active component of Paxlovid, and PF-00835231, an earlier 3CLpro clinical candidate. Next, mutant viruses were engineered using a reverse genetics approach, but the titer of these virus stocks was insufficient to allow further phenotypic characterizations. This is a first indication of the impaired replication capacity of the resistant variants. To confirm the phenotype of the L50F E166A L167F mutant, two alternative approaches were used. First, a cell-based assay was used with heterologous expression of wild-type and mutated 3CLpro and a substrate linked to a reporter protein. The EC50 values for ALG-097161 and nirmatrelvir in this assay increased 23- and 28-fold respectively as compared with wild-type. In a second approach we studied wild-type and mutant enzymes. Enzymes carrying the single substitution L50F, E166A or L167F were found to have markedly lower enzymatic activity (<20% as compared to wild-type). In particular the L50F mutant enzyme had only 0.5% activity compared to wild-type which could be restored to 5.3% when also E166A and L167F were introduced. This is a second indication that the mutations are associated with an impaired replication capacity. Each of the enzymes was also less efficiently inhibited by the 3CLpro inhibitors. The largest increase in IC50 was observed when all three substitutions L50F E166A L167F were combined. For this triple-mutant enzyme the IC50 for ALG-097161 and nirmatrelvir increase by 35- and 72-fold respectively.
A structural biology analysis shows that the substitutions decrease interactions between the enzyme and the inhibitor. E166A causes a direct loss of a hydrogen bound with the lactam-moiety that is found in all the inhibitors tested here, L167F increases the size of the binding pocket causing a decrease of the Van der Waals forces between enzyme and inhibitor, and L50F is thought to change the rotamer confirmation of Q189 which, for the wild-type, has many interactions with the inhibitors through a network of water molecules.
The loss of interactions, resulting from amino acid substitutions associated with resistance, might have a lower effect on substrates than on inhibitors, as the former rely on a significantly higher number of interactions for binding. Consequently, these resistance-associated mutations seem to allow a better discrimination between substrate and inhibitor at the expense of intrinsic enzymatic activity.
At this moment peer-reviewed information on SARS-CoV-2 3CLpro resistance is not readily available. The Paxlovid label indicates that E166V is more common in nirmatrelvir/ritonavir-treated subjects relative to placebo-treated subjects, and that E166A is associated with a 33-fold increase in Ki for nirmatrelvir [28]. Both these observations are in accordance with the results described above.
Our report emphasizes the need for additional research to elucidate potential resistance pathways of SARS-CoV-2 inhibitors in vitro. It is also a starting point for surveillance for nirmatrelvir resistance and indicates the potential need for combination therapies with different classes of SARS-CoV-2 inhibitors.
Methods
Compounds, cells, viruses, proteins and peptides
ALG-097161, Nirmatrelvir and PF-00835231 were synthesized by Aligos Therapeutics and purified to >95% purity. GS-441524 was obtained from MedChem Express (cat no HY-103586). The African green monkey kidney Vero E6 cell line was purchased from ATCC (catalog no. CRL-1586™) and maintained in DMEM (gibco cat no 41965-039) supplemented with 10% v/v heat-inactivated FCS. The SARS-CoV-2 BetaCov/Belgium/GHB-03021/2020 (EPI ISL407976|2020-02-03) isolate was obtained from a Belgian patient returning from Wuhan in February 2020. The isolate was passaged 7 times on VeroE6 cells which introduced two series of amino acid deletions in the spike protein [20].
SARS-CoV-2 3CLpro wild-type and mutant enzymes were produced as previously described [29]. Peptide substrate (Dabcyl-KTSAVLQSGFRKM-E(Edans)-NH2 for FRET was sourced from Biopeptide (San Diego, CA) at >95% purity.
Genotypic analysis
RNA was extracted using the NucleoSpin RNA virus kit (Macherey-Nagel) according to the manufacturer’s instructions. Reverse transcription was carried out via SuperScript IV and cDNA was posteriorly amplified using Q5® High-Fidelity DNA Polymerase (NEB) with the ARTIC nCov-2019 primers, following the recommendations in the sequencing protocol of the ARTIC Network (https://artic.network/ncov-2019). Samples were multiplexed following the manufacturer’s recommendations using the Oxford Nanopore Native Barcoding Expansion kits NBD104 (1-12) and NBD114 (13-24), in conjunction with Ligation Sequencing Kit 109 (Oxford Nanopore). Sequencing was carried out on a MinION sequencer using R9.4.1 flow cells and MinKNOW 2.0 software.
Antiviral testing
The 50% effective concentration (EC50), the concentration of compound required for fifty percent antiviral activity, of different antivirals against SARS-CoV-2 wild-type (WT) or the L50F E166A L167F mutant virus was determined on VeroE6 cells as follows. On day −1, the test compounds were serially diluted in 100 μL assay medium (DMEM supplemented with 2% v/v FCS) in 96-well plates. In a next step, 50 μL of VeroE6 cells (25,000 cells/well) was added together with 2 μM of the MDR1-inhibitor CP-100356 (final concentration 0.5 μM). The plates were incubated (37°C, 5% CO2 and 95% relative humidity) overnight. On day 0, 50 μL of SARS-CoV-2 WT or the triple mutant strain at a multiplicity of infection (MOI) of 0.001 tissue culture infectious dose (TCID50) per cell was added and the plates were stored in a humidified incubator at 37°C and 5% CO2. In absence of antiviral activity, the VeroE6 cells undergo a cytopathic effect. Cell viability was determined 4 days p.i. using a viability staining with MTS [30]. The percentage of antiviral activity was calculated by subtracting background and normalizing to the untreated-uninfected control wells, and the EC50 was determined using logarithmic interpolation.
Generation and rescue of recombinant viruses
We used the in-yeast transformation-associated recombination (TAR) cloning method as described previously with some adaptations [22]. In sum, the whole SARS-CoV-2 genome was encoded in 12 overlapping DNA fragments. These so-called WU-Fragments and a TAR-vector are homologous recombined in yeast forming the yeast artificial chromosome (YAC). The SARS-CoV-2 3CLpro is encoded on WU-Fragment 5 and was replaced with newly generated and overlapping PCR products to introduce the three amino acid changes L50F E166A L167F. The overlapping PCR products were made via reverse transcription from RNA purified from vRNA of the virus stock obtained from the selection. In brief, cDNA was generated from RNA by LunaScript RT SuperMix (NEB). PCR reactions targeting the 3CLpro gene were performed using Q5® High-Fidelity DNA Polymerase (NEB) and the resulting PCR products were mixed and matched to recombine SARSCoV-2 3CLpro L50F, E166A, L167F, SARSCoV-2 3CLpro L50F and SARSCoV-2 3CLpro E166A, L167F. All PCR products were purified with the High Pure PCR Product Purification Kit (Roche) before being used for TAR cloning.
In vitro transcription was performed for EagI-cleaved YACs and PCR-amplified SARS-CoV-2 N gene using the T7 RiboMAX Large Scale RNA production system (Promega) as described previously [22]. Transcribed capped mRNA was electroporated into baby hamster kidney (BHK-21) cells expressing SARS-CoV N protein. Electroporated cells were co-cultured with susceptible Vero TMPRSS2 cells to produce passage 0 (P.0) of the recombinant viruses. Passage 1 virus stocks were produced in VeroE6 cells or Calu-3 cells, but only low-titer stocks could be obtained.
3CLpro FRET-based assay
The FRET-based assay was performed similarly to the SAMDI-MS assay. Assays were performed in 20 μL volume in 384-well Non-binding Low-volume plates (Greiner Bio-One; Monroe, NC) at ambient temperature. 3CLpro and its mutants were preincubated with inhibitors for 30 min. Reactions were initiated by the addition of a FRET-compatible peptide substrate Dabcyl-KTSAVLQSGFRKM-E(Edans)-NH2. Fluorescence was measured for 90 minutes at 2-min intervals using a 340/460 excitation/emission filters on a Envision plate reader (Perkin Elmer). The IC50 values were calculated by fitting the curves using a four-parameter equation in GraphPad Prism. To calculate the dimer dissociation constant (Kd), the velocities of enzyme titration of WT and mutant 3CLpros were fitted to the equations (1) and (2).


Equation (2) was described previously for the calculation of monomer dimer equilibrium dissociation constant (Kd) [25].
3CLpro cell-based reporter assay
293T cells were maintained at 37°C/5% CO2 in Dulbecco’s DMEM (Corning#10-013-CV) supplemented with 10% fetal bovine serum (Gibco #10091148) and 1% penicillin/streptomycin (Gibco #15140122). 293T cells were seeded in a 96-well plate at 5×105 cells/well and returned to incubation for 24h. The following day cells were treated with compound at a final DMSO concentration of 2% prior to transfection with 100 ng of the wild-type or triple mutant (L50F E166A L167F) plasmid with Lipofectime LTX (ThermoFisher#15338100) for 24h. Post 24h transfection GFP fluorescence was detected with a Victor (Perkin Elmer) using the Fluoroscein 485/535 setting. Inhibition was determined from the resulting fluorescent signal and graphed using a variable slope four parameter curve fitting with GraphPad Prism (v 9.2.0).
Structural Biology and Computational Chemistry
Structural biology data were generated for a recombinant 3CLpro construct that was co-crystallized with multiple ligands of interest. Data sets were collected at synchrotron light sources, yielding final refinement resolutions in the 1.3 to 2.7 Å range. All calculations were conducted with the Schrodinger Software package (Schrodinger, LLC.).
Docking to WT 3CLpro
The conformational space of the ALG-097161 ligand was explored with Molecular Dynamics and Large Scale Low Mode sampling techniques as implemented in the Macrocycle Conformational Sampling tool (Force Field: OPLS3e; GB/SA electrostatics; 10 kcal/mol energy window for saving unique structures; 0.75 Å RMSD for defining conformer uniqueness; 5000 simulation cycles and LLMOD steps, each; eigenvector determination for each global minimum; extended torsional sampling). We identified 571 unique, low-energy conformers of ALG-097161. ~26% of the conformers were used as input ligand structures for docking calculations to ensure a robust and converged prediction of the ALG-097161 binding mode to WT 3CLpro.
De novo, covalent docking calculations were performed with Glide for each selected ALG-097161 input ligand structure. The receptor for covalent docking was based on a 1.3 Å X-ray structure of a close analogue of ALG-097161 that bears identical P2 and P3 moieties. The receptor structure was cleaned up and minimized with restraints ahead of docking using the Protein Preparation Wizard. C145 was specified as the reactive residue for covalent binding, and the centroid of the X-ray reference ligand was used to define the volume for placement of ALG-097161 in the binding site. A custom covalent reaction parameter file was created to define the interaction between the inhibitor warhead moiety and the catalytic C145. Core constraints for docking pose filtering were imposed, with RMSD tolerance of 3 Å with respect to selected heavy atoms common between ALG-097161 and the X-ray reference. High accuracy (Pose Prediction Thorough) docking methodology was implemented for pose prediction. Up to 20 predicted covalently-bound complexes of ALG-097161 and WT 3CLpro were collected for each starting input ligand conformer that was piped into docking, yielding a large ensemble of 2,237 computed ALG-097161 binding modes. The predicted binding modes were assessed using the same physics-based heavy atom RMSD order parameter utilized for pose filtering. Multiple binding modes presented RMSDs lower than 1 Å. The binding mode shown in Figure 6 has RMSD = 0.77 Å. In addition, the computed binding poses were evaluated for presence of hydrogen bonds and key Van der Waals interactions observed for close analogues in X-ray data.
Modeling of inhibitors in L50F E166A L167F mutant
PDB structures 6xhm (complex with PF-00835231) and 7rfw (nirmatrelvir) were used as starting point for modeling compounds in the L50F E166A L167F mutant 3CLpro. The receptor structure was cleaned up and minimized with restraints ahead of docking using the Protein Preparation Wizard.
Covalent docking was used for placement of ALG-097161. C145 was specified as the reactive residue for covalent binding, and the centroid of the X-ray reference ligand was used to define the investigated binding site. A custom covalent reaction parameter file was used to define the reaction between the warhead moiety and the catalytic C145. High accuracy (Pose Prediction Thorough) docking methodology was implemented for pose prediction. Initial docking poses within 4 kcal/mole (glide gscore) were collected for further processing. Residues within 6 Å of the bound compound were minimized after covalent bond formation. The top 5 binding mode suggestions were saved for visual inspection.
Initial structures for the L50F E166A L167F triple mutant were built by introducing the mutations directly on the WT complex. A first restrained minimization was done with the protein preparation wizard to remove any unacceptable clash. Next, a Desmond simulation system was built (see below) and a 100 ps energy minimization procedure was run with Desmond to reach the nearest energy minimum.
Molecular Dynamics (MD) simulations were performed using the Desmond-GPU package. A simulation system was first set up around the protein-inhibitor complex, including a 10 Å water box, Na+ ions necessary to neutralize the system and additional Na+ and Cl− ions to simulate a 0.15 M NaCl concentration. OPLS4 was selected as force field. The NPT ensemble (T = 300K, P = 1.01325 bar) was used. The system was relaxed prior to simulation. MD were run for a (simulated) period of 5 ns, with frames saved every 20 ps (total 250 frames). Final frames were energy-minimized (100 ps minimization protocol). Protein-ligand interactions were evaluated in the 1 ns - 5 ns period.
Footnotes
↵* Shared Last authorship
References




