

Abstract
Intracellular pathogens are auxotrophic for many metabolites and must rely on the host. While this reliance is well established, how pathogens manipulate host metabolism to their benefit is not understood. For intracellular pathogens, distinguishing the origin of the metabolite as host- or pathogen-derived is challenging. The obligate intracellular parasite Toxoplasma gondii alters the host cell by a pre-invasion process known as “kiss and spit”, where the contents of the parasite rhoptry organelles are secreted into the host cytoplasm before invasion occurs. This separation of microbe from the host offers a rare opportunity to demonstrate pathogen manipulation of the host. Using mass spectrometry-based metabolomics, we determined that kiss and spit changed host metabolites in nucleotide synthesis, the pentose phosphate pathway, glycolysis, and amino acid synthesis. An increase in 2,3-bisphosphoglycerate (2,3-BPG) abundance led us to hypothesize that high levels of host 2,3-BPG contribute to the activation of host cytosolic nucleosidase II (cN-II) to alter purine availability. Treatment with the cN-II inhibitor fludarabine and a cell line with a cN-II genetic knockout reduced T. gondii growth. Our results demonstrate that T. gondii rhoptry contents discharged during kiss and spit remodel host metabolism. They also suggest that T. gondii manipulates the host cN-II enzyme to acquire its necessary purine metabolites.
Introduction
Intracellular pathogens need a suitable host cell to replicate and provide a nutrient supply. Most of these host cells are not highly metabolically active, so these microorganisms need to reprogram their host cell to support microbial replication. While this is an area of high interest to the microbiology field, to our knowledge, there are no studies on how secreted microbial proteins change host metabolism in the absence of microbial replication. If we could determine how pathogens manipulate the host metabolism, we could define novel drug targets called Host-Directed Therapies (HDTs). HDTs will be especially effective against intracellular pathogens that are reliant on host metabolic supplementation. For example, the obligate intracellular parasite Toxoplasma gondii relies on the host cell to provide arginine, tyrosine, tryptophan, purines, cholesterol, or sphingolipids and relies on the host cell to synthesize these essential metabolites and import them (1–8). These auxotrophies present an opportunity for HDTs that inhibit pathways that T. gondii requires but a healthy uninfected host cell does not. Current therapeutics for T. gondii treatment are limited, must be given in combinations, and all target parasite metabolism (9). To develop a new generation of HDTs that limit the growth of T. gondii, we must characterize how the parasite changes host metabolism.
Direct quantification of the host metabolome during infection is complicated by the technical challenge of separating T. gondii and host cell metabolites. Instead, studies have used genetically manipulated host cells to identify essential host pathways for T. gondii growth, including arginine synthesis and cholesterol scavenging (1, 10). Other work has used gene expression analysis and proteomics to show that T. gondii changes the transcription and translation of enzymes in multiple host pathways including the pentose phosphate pathway, glycolysis, and nucleotide synthesis (11–13).
T. gondii is incapable of synthesizing purines and must import them from their host (14, 15). Purines must be fully dephosphorylated before they can be taken up and used by the parasite (7). Purine nucleotide levels are regulated by host nucleotidases that hydrolyze these nucleotides into nucleosides (16) which are then transported to the interior of the parasite (17). There are purine transporters inside the parasite membrane that transport purine nucleobases or nucleosides into the parasite cytosol (22). 5’-nucleotidases dephosphorylate non-cyclic nucleoside monophosphates to nucleosides and inorganic phosphate. At least seven human 5’-nucleotidases with different subcellular localization have been identified: ecto-5’-nucleotidase, cytosolic 5’-nucleotidase IA, cytosolic 5’-nucleotidase IB, cytosolic 5’-nucleotidase II (cN-II), cytosolic 5’-nucleotidase III, cytosolic deoxynucleotidase, and mitochondrial deoxynucleotidase (18). Among them, cN-II catalyzes both the hydrolysis of several nucleoside monophosphates and the phosphate transfer from a nucleoside monophosphate donor to the 5’ position of a nucleoside acceptor (19–21). cN-II acts on substrates such as IMP, GMP, and their corresponding deoxy-derivates (21). cN-II has been reported in many human and vertebrate tissues. Its activity is high in cells with elevated DNA synthesis as well as in cancerous tissue compared to its normal parental tissue, making it a target for cancer chemotherapeutics (21). cN-II activity is modulated by effector molecules such as 2,3-bisphosphoglycerate (2,3-BPG), ATP, and GTP in an allosteric site (21, 22).
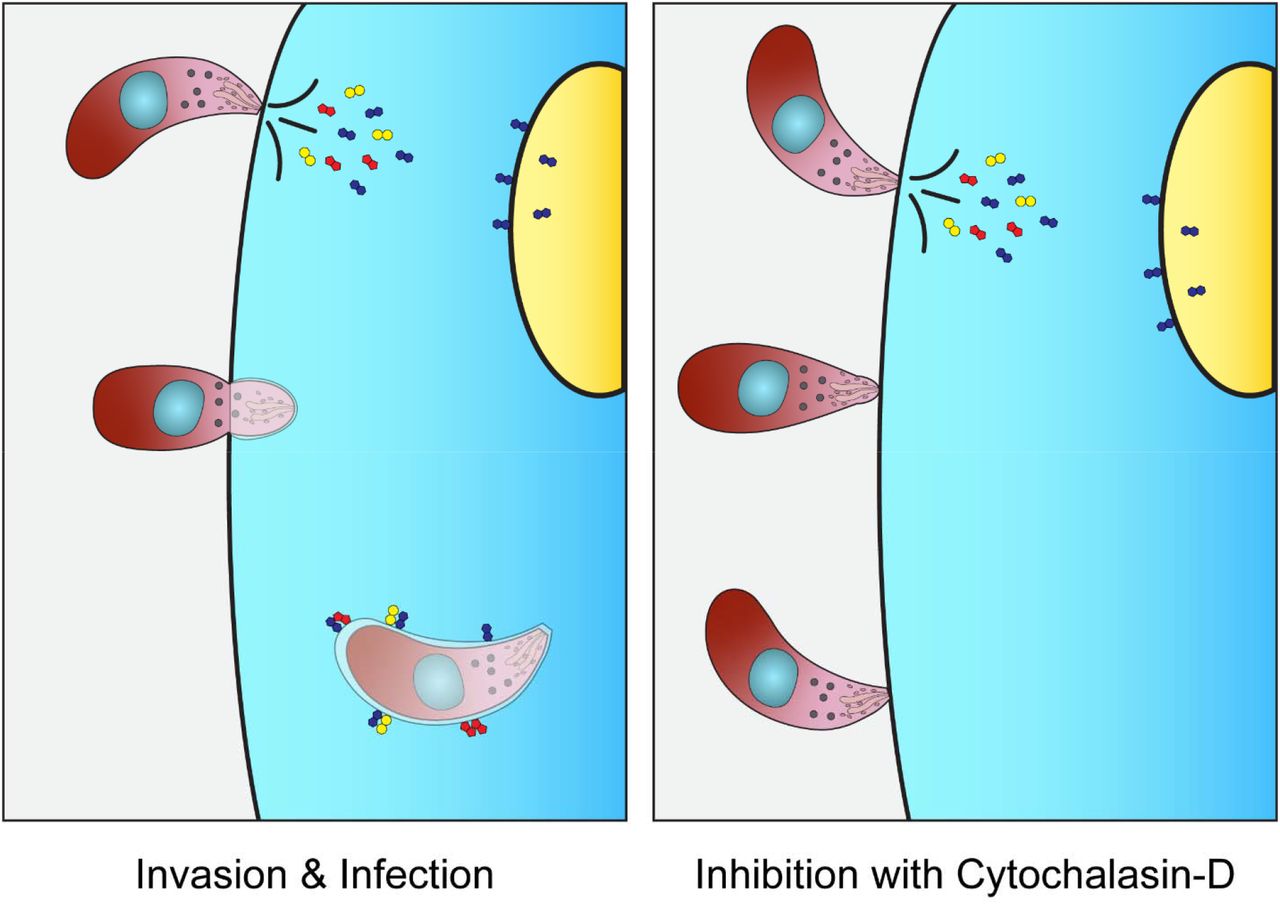
A concentrated secretion of parasite factors occurs in a pre-invasion process known as kiss and spit (Figure 1) when the contents of the T. gondii rhoptry organelles are secreted into the host cytoplasm (23, 24). The rhoptries contain an estimated fifty proteins and lipids, most of which are functionally uncharacterized, and the impact of kiss and spit on host metabolism is unknown (24–29). Cytochalasin D (30, 31) or Mycalolide B (32–35) function as actin polymerization inhibitors that prevent invasion, allowing the host changes associated with kiss and spit to be studied independently of parasite invasion and replication. In this study, we determined how T. gondii rhoptry contents discharged during kiss and spit remodel the host metabolism using mass spectrometry-based metabolomics. In our prior metabolomics studies of T. gondii infected host cells, we saw an increased abundance of 2,3-BPG (36). This result was surprising because 2,3-BPG is an intermediate in a glycolytic shunt that decreases glycolysis ATP synthesis by 50%, and we had assumed that T. gondii infection would increase host energy synthesis. In the present research study, we have found that T. gondii kiss and spit generates high levels of 2,3-BPG, similar to full infection. We hypothesize that high levels of 2,3-BPG act as an allosteric regulator of cN-II enzyme to upregulate its activity, which in turn generates purine nucleosides for T. gondii. We also examine the FDA-approved cN-II inhibitor, fludarabine, to block T. gondii replication and purine abundance.

Kiss and spit is the pre-invasion process when the contents of the parasite rhoptry organelles are secreted into the host cytoplasm (Left panel). It was studied using the actin inhibitor Cytochalasin D (right panel), which allows the parasite to attach but not invade the host cell. Some rhoptry proteins are directed to the nucleus of the host cell, others form the parasitophorous vacuoles (PV), and others are believed to be secreted into the host cytoplasm.
Results
T. gondii kiss and spit selectively remodels host cell metabolism
We performed a time course mass spectrometry-based analysis of the impact of T. gondii on host cell metabolism using a kiss and spit tissue culture model. By pretreating T. gondii with the actin polymerization inhibitor cytochalasin D, we allowed T. gondii to secrete the contents of the rhoptries into host cells while preventing infection (37). This metabolic system is simplified compared to our full infection model, with only host metabolism and a discrete pool of parasite rhoptry content. Our analysis found that the parasite rhoptry proteins changed the host metabolism in nucleotide synthesis, the pentose phosphate pathway, glycolysis, amino acid synthesis, and the abundance of the signaling molecules myo-inositol and cyclic-AMP (Figure 2).

Heatmap shows metabolite abundance in human cells over 12 hours after T. gondii kiss and spit. Triplicate kiss and spit treated and untreated dishes of HFFs were metabolically quenched, and metabolites were extracted at 5 times points over time (1.5,3, 6, 9, and 12 HPK&S). Metabolomes were quantified using HPLC-MS and metabolites were identified with known standards. Kiss and spit treated sample abundances were averaged and normalized to the average control abundance then log base 2 transformed (Log2 (Kiss and Spit Abundance/Control Abundance)) with blue being less abundant and yellow more abundant. Metabolites were chosen for inclusion based on whether they could be confidently identified, and their abundance changed during infection. Values represented in this heat map can be found in Table S1.
Several nucleotide metabolites increased in abundance after kiss and spit, including multiple phosphorylated forms of purines and pyrimidines. CDP, UDP, GDP, GTP, and CTP were all moderately more abundant between 6- and 12-hours post kiss and spit (HPK&S), as was the precursor metabolite hypoxanthine. While these changes were not large in amplitude there was a consistent increase in abundance across a range of nucleotide metabolites.
The pentose phosphate pathway (PPP), which is primarily known for generating the biosynthetic cofactor NADPH and the nucleotide precursor ribose 5-phosphate, was also impacted by kiss and spit. The first step in the PPP generates gluconolactone-6-phosphate via glucose-6-phosphate dehydrogenase (38). This step commits carbon to the pentose phosphate pathway and is believed to be the rate-limiting step of the human PPP (38). Gluconolactone-6-phosphate was depleted in host cells at all time points HPK&S with a 2.2-fold lower abundance at 12 HPK&S (Table S1 and Figure 2). In contrast, the next metabolite in the pathway, 6-phosphogluconate, was 1.8 and 2.6-fold more abundant at 9 and 12 HPK&S, respectively. Ribose-5-phosphate was similarly more abundant at the last two time points, indicating that kiss and spit may increase the flow of carbon through the PPP. Further down the PPP in the non-oxidative portion of the pathway sedoheptulose-7-phosphate (S7P) was 4.2 and 7.2-fold more abundant at 9 and 12 HPK&S, while the related metabolite sedoheptulose-1,7-bisphosphate (SBP) was 1.4 and 3.4-fold more abundant at the same time points. Octulose-1,8-bisphosphate follows a similar pattern to SBP, with 3.6- and 5.8-fold increases in abundance at 9 and 12 HPK&S, and although octulose-8-phosphate was not consistently above the limit of detection it also appeared to increase in abundance. The increase in abundance of SBP, S7P, OBP, and O8P shares a similar pattern to the T. gondii infection metabolome, although the host does not possess the sedoheptulose biphosphatase enzyme, present in T. gondii, that converts SBP to S7P (36). It is possible that the increase in SBP and OBP abundance is not connected to the PPP but is instead a byproduct of increased fructose bisphosphate aldolase activity in glycolysis.
Multiple glycolytic intermediates were more abundant after kiss and spit at 9 and12 HPK&S, including fructose-6-phosphate which was 1.6 and 2-fold more abundant, fructose-1,6-bisphosphate which was 1.2 and 4.2-fold more abundant, and 3-phosphoglycerate which was 1.4 and 1.6-fold more abundant. Phosphoenolpyruvate and 2,3-bisphosphoglycerate abundance followed a similar pattern, increasing 2.4-fold at 6 and 9 HPK&S and 3.8-fold at 12 HPK&S.
Two amino acid precursors, 3-phosphoserine and argininosuccinate, were increased in abundance by kiss and spit. 3-phosphoserine is the final metabolite in the serine biosynthetic pathway and is 2.2-fold more abundant at 9 and 12 HPK&S (39). Similarly, argininosuccinate is the final intermediate in the arginine synthesis pathway and it is 3 and 2.4-fold more abundant at 9 and 12 HPK&S, respectively (40).
Kiss and spit also altered the abundance of two metabolites that act as signaling molecules. Myo-inositol was depleted throughout the time course, at 2.4-fold less abundant at 12 HPK&S. In contrast cyclic AMP (cAMP) was more abundant after kiss and spit, with 4-fold increases observed at 6 and 9 HPK&S.
Control of kiss and spit
To ensure that our findings were due to kiss and spit, we examined host metabolism after treatment with heat-killed parasites or T. gondii infection conditioned media. Host cells were treated with heat-killed parasites or a heated media negative control for 12 hours and then had their metabolites extracted and analyzed. No changes to host metabolism were observed, indicating that the presence of parasites alone was not causing a metabolic shift in host cells. Conditioned media was taken from heavily infected cells and swapped onto uninfected dishes for 12 hours before metabolites were extracted and analyzed, using media from uninfected cells as a negative control. Again, we found no difference between the two conditions (Figure S1), indicating that factors secreted into the media during infection were not responsible for the changes observed after kiss and spit. To ensure that the changes we observed in host metabolism were not attributable to the T. gondii remaining in the dish we performed a control to measure the metabolic contribution of the parasites. 2 X 106 parasites were incubated in media with cytochalasin D for 12 hours before pelleting the parasites, washing them to remove the media, and then extracting metabolites. A blank control (media with cytochalasin D but no parasites) was treated identically. No metabolites were detectable in either case, indicating that the contribution of the parasites in the dish to our metabolic data is negligible, and the changes we observed were occurring within the host metabolism.

Heatmap shows metabolite abundance in human cells over 12 hours after Conditioned control treatment. Triplicates of treated and control dishes of HFFs were metabolically quenched, and metabolites were extracted at 12 HPT. Metabolomes were quantified using HPLC-MS and metabolites were identified with known standards. Abundances were averaged and normalized to the average control abundance then log base 2 transformed (Log2 (Abundance/Control Abundance) with blue being less abundant and yellow more abundant.
Conserved shifts in nucleotide metabolism for full infection and kiss and spit
In Figure 3A, we compared our previously published metabolomic analysis of full infection (36) and our current kiss and spit metabolomic data set. We found conserved changes in nine metabolites: SBP, S7P, OBP, 2,3-BPG, inosine, guanosine, GDP, GTP, and 6-PG. The metabolites sedoheptulose 7-phosphate (S7P) and sedoheptulose 1,7-bisphosphate (SBP) increased in abundance during full infection and kiss and spit, but not in uninfected host cells. While S7P is an intermediate in the pentose phosphate pathway that generates pentoses and ribose 5-phosphate for nucleotide synthesis, SBP cannot be easily used by the host because it does not contain the required bisphosphatase to dephosphorylate SBP (SBPase). SBPase is part of the photosynthetic pathway, thus T. gondii but not mammalian cells contain it (36). Thus, SBPase represents a novel step in T. gondii central carbon metabolism that allows T. gondii to energetically-drive ribose synthesis, via the energetically favorable dephosphorylation of SBP, without using NADP+. T. gondii SBPase drives carbon into S7P where it can flow into either ribose-5-phosphate and nucleotide metabolism or down the non-oxidative PPP and back into glycolysis.
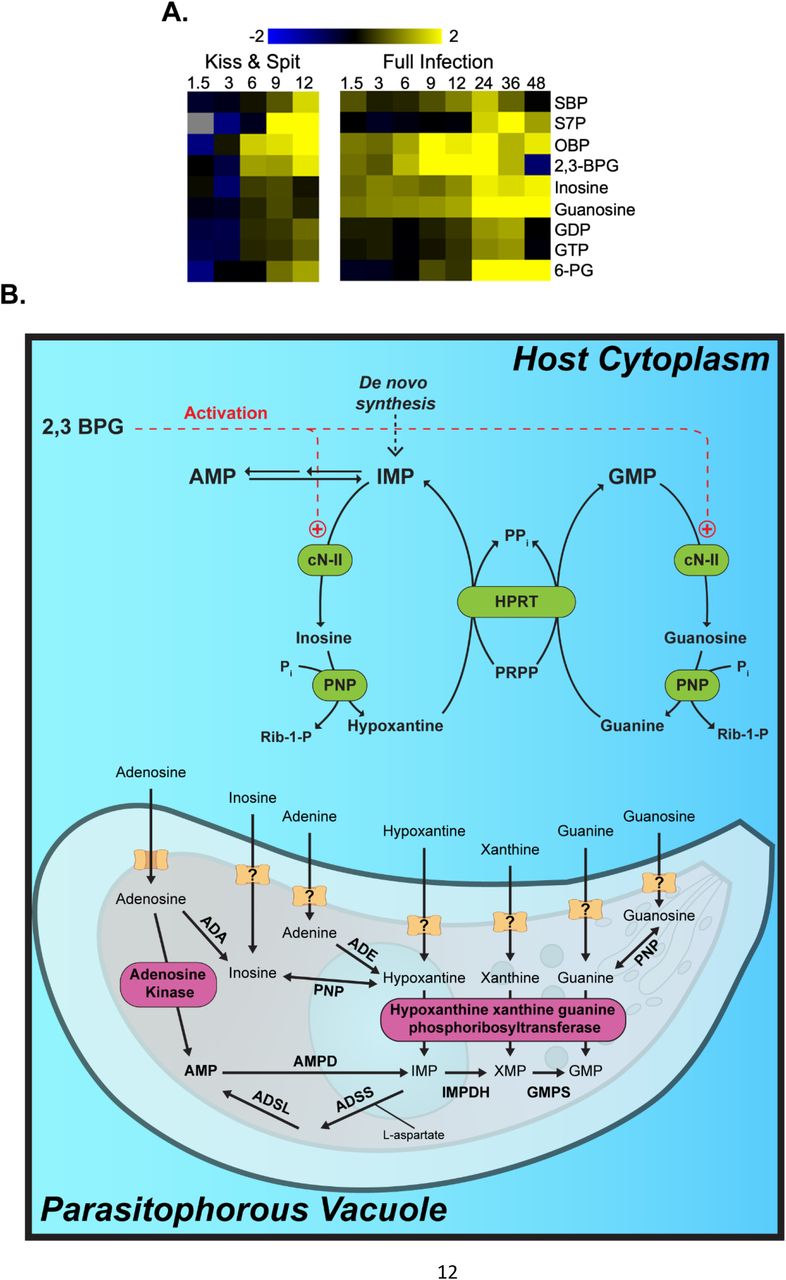
(A) Heat map of metabolites that are significantly up (yellow) or down (blue) regulated in response to kiss and spit (left panel) or full infection (right panel). Log 2 scale bright yellow is a 4-fold change. Gray indicates peaks are at or below the limit of detection. Hours post-infection are across the top of the heat map, 1.5, 3, 6, 9, 12, 24, 36, or 48. 6-phosphoglucanate (6-PG) is an intermediate in the pentose phosphate pathway. (B). The hypothesis of how T. gondii infection stimulates the activity of host Cytosolic nucleotidase II enzyme (cN-II). T. gondii kiss and spit and full infection produce abundant host 2,3BPG. 2, 3 BPG acts as an activator of the host cN-II enzyme. The activation of cN-II by ATP is shown (+). cN-II hydrolyzes nucleotide monophosphates such as GMP and IMP to produce guanosine and inosine, respectively. These nucleosides are then transported to the interior of parasite cytosol to produce RNA molecules. Adenosine kinase (AK) and HXGPRT represent the major pathways for salvage and incorporation into the Toxoplasma nucleotide pool. Figures modified from Pesi, et al. (62) and Fox & Bzik (47). cN-II: cytosolic 5′-nucleotidase II; PNP: purine nucleoside phosphorylase; HPRT: hypoxanthine guanine phosphoribosyl transferase; XO: xanthine oxidase; ADA, Adenosine deaminase; ADE, adenine deaminase; ADSL, adenylosucccinate lyase; ADSS, adenylosuccinate synthetase; AMPD, AMP deaminase; GMPS, GMP synthetase; HXGPRT, hypoxanthine_xanthine_guanine phosphoribosyltransferase; IMPDH, inosine 50-monophosphate dehydrogenase; PNP, purine nucleoside phosphorylase; PRPP, 5-phosphoribosyl-1-pyrophosphate.
Kiss and spit increase 2,3-BPG abundance and connection to purine metabolism
2,3-BPG abundance increased 2.6-fold at 6, 2.4-fold at 9, and 3.8-fold at 12 HPK&S (Table S1 and Figure 3B). 2,3-BPG is not part of the normal glycolytic pathway and is a known allosteric regulator. 2,3-BPG is synthesized in the Rapoport-Luebering shunt, which is a two-step pathway around the phosphoglycerate kinase step in glycolysis (41) Because it has been shown that 2,3-BPG is an allosteric activator of cN-II (22, 42), we hypothesized that high levels of 2,3-BPG upregulate cN-II activity, which in turn generates purine nucleosides for uptake by T. gondii (22, 42) (Figure 3B).
cN-II catalyzes both the hydrolysis of several nucleoside monophosphates and the phosphate transfer from a nucleoside monophosphate donor to the 5’ position of a nucleoside acceptor (19–21) (Figure 3B). cN-II acts on substrates such as IMP, GMP, and their corresponding deoxy-derivates, producing inosine and guanosine respectively (19–21). Inosine and guanosine abundance increased both during T. gondii infection but also after kiss and spit (Figure 3A). The gene expression analysis of cN-II and bisphosphoglycerate mutase (BPGM), the enzyme responsible for most of the 2,3-BPG synthesis, shows that both enzymes are approximately 2-fold more expressed during early full infection (36) (Figure S2).

(A) Fold change (Infected/ uninfected) of host BPGM gene expression during T. gondii full infection. (B) Fold change (Infected/ uninfected) of host cN-II gene expression during T. gondii full infection. Data were generated in our previous publication (36).
Chemical inhibition of cN-II reduces T. gondii replication
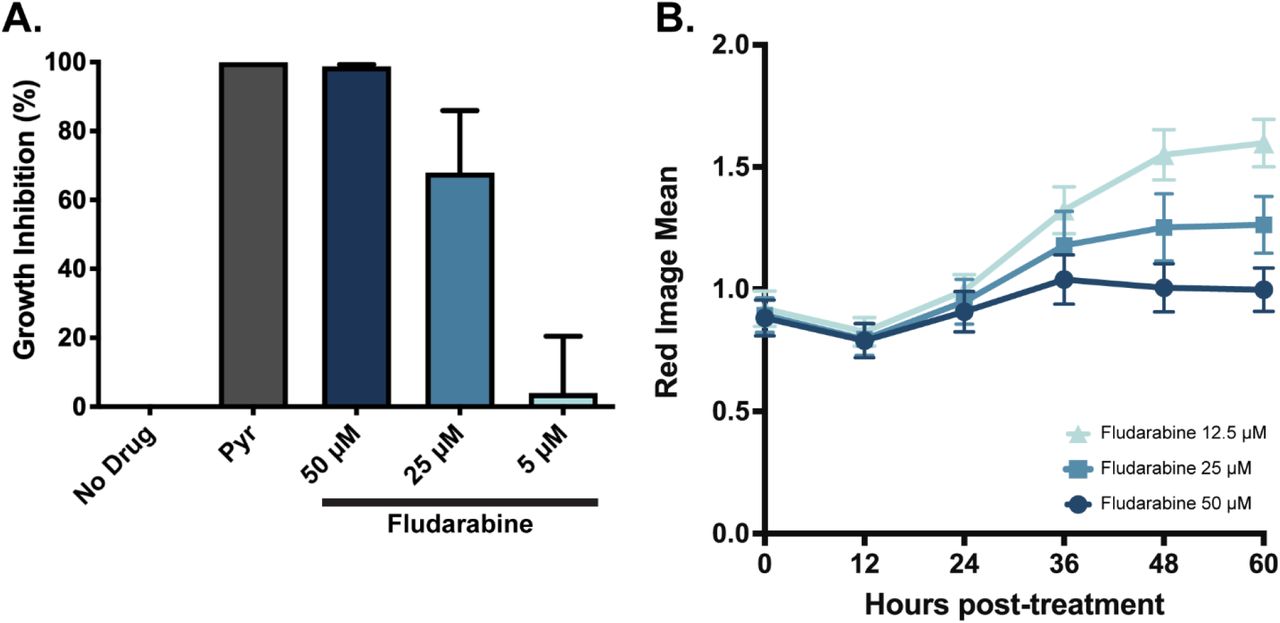
To test the importance of the host cN-II activity to T. gondii growth, we performed a [3H]uracil uptake assay in the presence of 50, 25, or 5 μM of the cN-II inhibitor fludarabine. As a negative control for growth inhibition, T. gondii was grown untreated, and as a positive control, parasites were treated with 1μM pyrimethamine, which completely inhibits growth. Uninfected host cells were also assayed to determine background host uptake of [3H]uracil. We found that fludarabine inhibits T. gondii growth in a dose dependent manner, with 50μM completely halting replication (Figure 4A). We also measured the replication of mCherry-expressing T. gondii in HFF cells in presence of fludarabine for 60 hours (Figure 4B). We confirmed that fludarabine inhibits T. gondii growth in a dose dependent manner, and this inhibition is maintained at least for 60 hours.

(A) Rate of [3H] uracil uptake by replicating T. gondii under five conditions: no treatment negative control for growth inhibition, 1μM pyrimethamine positive control for total growth inhibition, and either 50, 25, or 5μM fludarabine treatment. Uninfected cells were also assayed for a baseline level of [3H] uracil uptake by host cells. Uracil uptake was measured, host incorporation was subtracted, and eachcondition was normalized to the uninhibited DMSO control condition to determine percent growth inhibition. (B) Replication of cherry ME49 tachyzoites during 60 HPK&S with three concentrations of fludarabine. Red Image Mean was obtained at different time points with an Incucyte machine. Error bars represent the standard deviation of three replicates by condition.
Genetic knockout of cN-II affects T. gondii replication
We took advantage of the existence of a cN-II-knockout in a breast cancer cell line denominated MDA-MB231 to determine the enzyme’s importance to parasite replication (43, 44). We measured the growth of T. gondii in the absence of cN-II by quantitative polymerase chain reaction (qPCR). T. gondii replicated significantly less in host cells without cN-II at 12 HPI but not 24 HPI (Figure 5A). One explanation for this phenomenon is that the absence of the host cN-II enzyme reduces the availability of the purine pool for the replication of the parasite at early time points; but by 24 HPI, there is a compensatory effect that allows the parasite to recover and grow faster due to the interconversion of purines, especially in this breast cancer cell line. The growth reduction was significant but less evident under starvation media, likely because this growth environment was already poor for nucleotides (Figure 5B). Cancer cells have faster nucleotide metabolism to satisfy their high replication rate.

Replication of T. gondii in cN-II knock-out MDA-MB231 host cells by relative quantification of T. gondii in MDA-MB231 cN-II KO host cells. qPCR was performed using T.gondii SAG1 primers normalized to host housekeeping gene PPIA2. 1/ CT ratio was calculated. (A) normal media with 10% FBS and (B) starvation media with 1% FBS at 12 and 24 HPI. Each bar represents the mean of nine replicates and SD. Statistical analyses were performed one-way Anova by Turkey multiple comparisons at Alpha =0.05 using Prism software.
Genetic knockout of HXPGRT enzyme affects T. gondii purine acquisition
To understand the mechanism of growth inhibition, we performed metabolomics on the fludarabine treated parasites. Unfortunately, due to the rapid turnover of purine metabolites in replicating T. gondii, we were not able to see consistent and significant differences in purine metabolites in wild-type parasites (Figure S3). To reduce the effect of purine interconversion in the parasite, we used Pru parasites with a genetic deletion of the hypoxanthine-guanine phosphoribosyl transferase (HXGPRT) enzyme (PruΔHXGPRT). HXGPRT rephosphorylates imported purines, but it is not essential for T. gondii due to the activity of adenosine kinase (AK) and the conversion of AMP to IMP, XMP, or GMP (45). We performed metabolomics comparing the parental Pru WT and PruΔHXGPRT strains in the presence and absence of fludarabine (Figure 6). Fludarabine affected the abundance of purines in T. gondii infected-HFF cells at 24 HPI (Figure 6B). IMP and GMP, the preferred substrates of the cN-II enzyme, accumulated when treated with fludarabine, as we were expecting (Figure 6B). The nucleobase products of the cN-II reaction, inosine, guanosine, and xanthine were less abundant with fludarabine treatment, although not always reaching statical significance (Figure 6B). AMP, which is the preferred substrate for cytosolic 5’nucleotidase I (46) is significantly less abundant in PruΔHXGPRT parasites treated with fludarabine, highlighting that parasites maybe be using more adenine to compensate for the reductions in inosine and guanosine.

(A) Experiment # 1. (B) Experiment #2 (C) Experiment # 3. (D) Experiment # 4. The abundance of purine metabolites at 24 HPI. Each bar represents the mean abundance of three replicates normalized to the mean abundance of the uninfected control. Statistical analyses were performed with two-way ANOVA with Fisher LSD test at Alpha =0.05 using Prism software.

Two individual experiments of Abundance of purine metabolites in HFF cell line infected with Pru WT or PruΔHXGPRT T. gondii and treated with fludarabine at 24 HPI. Each bar represents the abundance mean of three replicates normalized to the abundance mean of the uninfected control. Statistical analyses were performed with two-way ANOVA with Fisher LSD test at Alpha =0.05 using Prism software.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A). Metabolomic methodology (B) Abundance of purine metabolites in HFF cell line infected with Pru WT or PruΔHXGPRT T. gondii and treated with fludarabine at 24 HPI. Each bar represents the abundance mean of three independent experiments normalized to the abundance mean of the uninfected control. Statistical analyses were performed with two-way ANOVA with Fisher LSD test at Alpha =0.05 using Prism software.
Discussion
Kiss and spit (Figure 1) is a process where the T. gondii parasite secretes rhoptry proteins into the host cell (24). After kiss and spit occurs, the parasite uses actin polymerization to enter the host cell (24). Treating parasites with the actin polymerization inhibitor cytochalasin D allows kiss and spit to occur without invasion and replication (34, 35). We discovered that kiss and spit changes host metabolite abundance in multiple pathways including glycolysis, the pentose phosphate pathway, amino acid synthesis, and nucleotide synthesis (Figure 2). Our kiss and spit/metabolomic analysis is the first study to determine how the parasite secreted contents remodel the host cell metabolism.
T. gondii is an obligate intracellular parasite that cannot replicate outside of its host cell. They may exist in extracellular forms, but only for a short time without any replication (47). T. gondii needs to scavenge many metabolites from their host to replicate. Nucleotides are essential to different cellular functions that involve DNA and RNA synthesis, chemical energy (ATP), nucleotide-based enzyme cofactors (NAD, FAD), and secondary messengers (cAMP) (47, 48). Most apicomplexans can produce pyrimidines de novo but none of them can produce purines (49). Thus, they rely on the importation of purines from their host to supply their requirement for purine nucleotides (50, 51). All purines must be fully dephosphorylated before they can be taken up and used by the parasite. The cell membranes are a barrier to nucleotides transport, but nucleosides can pass in either direction(52). In T. gondii, three purine transporters have been characterized so far, TgAT1, TgAT2, and TgNT1 (47). The identification of host enzymes used by the parasite in purine salvage is essential for designing better drugs against parasites.
Our previous metabolomic profile of full infection (36) and the current kiss and spit metabolomic profile show that the parasite changes the abundance of many host metabolites related to nucleotide synthesis such as SBP, S7P, OBP, 6-PG, 2,3-BPG, and the purines inosine, guanosine, GDP and GTP (Figure 3). A surprising finding from the kiss and spit metabolome was the quadrupling of host 2,3-BPG abundance at 12 HPK&S. Host Bisphosphoglycerate Mutase (BPGM), the main source of 2,3-BPG, is transcriptionally upregulated in infected host cells during the first nine hours of infection (36) (Figure S2). T. gondii BPGM has yet to be identified, which makes it difficult to estimate the parasite’s contribution to 2,3-BPG synthesis. In this study, we tested the hypothesis that kiss and spit increases 2,3-BPG synthesis and that the metabolite then acts as an allosteric activator of the cN-II enzyme which in turn generates purine nucleosides for T. gondii (Figure 3B).
Gene expression analysis of T. gondii full infection in HFF cells showed a high abundance of the host’s cN-II which could correlate to cN-II protein level (Figure S2). We also found a high abundance of the metabolite 2,3 BPG (Figure 3A). Previous research has demonstrated that cN-II activity is modulated by substrate binding and by effector molecules such as 2,3 BPG, ATP, and GTP in an allosteric site (21, 53). cN-II dephosphorylates the purines IMP, GMP (and sometimes XMP) to inosine, guanosine, and xanthine respectively (19–21). cN-II enzymes are present not only in human cells but are also present in other eukaryote and prokaryote organisms. PfISN1 is the first 5’ nucleotidase reported in Plasmodium falciparum, which is an IMP-specific nucleotidase that is allosterically activated by ATP(16). Also, a human ecto-nucleotidase has been reported to produce adenosine from AMP in the T. gondii infection context (54, 55).
To understand the role of host cN-II in T. gondii infection, we grew T. gondii in the presence of fludarabine, a cN-II inhibitor and a purine analog (54). Fludarabine inhibited parasite replication in a dose dependent manner (Figure 4). We evaluated the effect of the genetic knock-out of cN-II in T. gondii infection environment. T. gondii replicated less in host cells with cN-II KO at 12 HPI (Figure 5). To understand the fludarabine inhibition mechanism of T. gondii replication, we performed a metabolomic analysis. Metabolomic analysis of fludarabine treatment of PruΔHXGPRT parasites showed increased the abundance of the cN-II substrates GMP and IMP and reduction of the products guanosine and inosine (Figure 6B).
Using chemical or genetic inhibition of the cN-II enzyme in T. gondii infected cells, we expected to observe: (1) accumulation of cN-II substrates such as the nucleosides AMP, GMP, IMP and XMP (2) reduction of the abundance of the nucleobase products of this reaction such as adenosine, guanosine, inosine, and xanthine, respectively, and (3) reduction of the abundance of the nucleobase subproducts of this reaction such as guanine. T. gondii uses different routes to obtain purines, including salvaging host materials and interconverting purines inside the parasite (48, 52). T. gondii can transport and salvage the host nucleosides adenosine, inosine, and guanosine, as well as the host nucleobases adenine, hypoxanthine, xanthine, and guanine. At least nine enzymes have been reported in T. gondii, involved in both interconversion and salvage of host purine (6). Thus, we want to focus on the AK and HXGPRT enzymes used by the parasite to incorporate host purines into the parasite nucleotide pool.
T. gondii adenosine kinase (TgAK) uses host adenosine to form the nucleoside AMP in the interior of the parasite and is the main route of purine incorporation in the parasite (6). Parasites lacking TgAK can survive, due to the presence of the HXGPRT enzyme. T. gondii salvages hypoxanthine, xanthine, and guanine through HXGPRT by converting them into their respective nucleosides (6). TgHXGPRT has two isoforms encoded by a single gene and with two different intracellular localizations (56). AK and HXPGRT cannot be genetically disrupted simultaneously, suggesting that these routes are essential for the parasite’s purine metabolism and survival (6). Thus, T. gondii has a redundant and compensatory purine salvage pathway using AK and HXGPRT enzymes. When we used a PruΔHXGPRT parasite, we saw fludarabine inhibition of the host cN-II enzyme, observed by the accumulation of cN-II substrates and reduction of cN-II products (Figure 6).
The study of purine metabolism in apicomplexan parasites is not clear due to the challenge of completely separating the parasite from the host cell. The capture, transport, and salvage pathways necessary for purine acquisition have long been viewed as a significant Achilles heel of the parasite that may be targeted for chemotherapy (47). Current treatment strategies in human infections caused by T. gondii or P. falciparum are based on targeting parasite enzymes and blocking the accumulation of nucleotides. This validated approach to chemotherapy highlights the significance of further research to dissect the details of nucleotide metabolism in the Apicomplexa (47). T. gondii may be hijacking the host cN-II enzyme to promote replication, indicating it could be an important target for the development of new anti-parasitic drugs. Here we also propose that fludarabine, an FDA-approved medicine could be used for the treatment of toxoplasmosis. Fludarabine inhibits the cN-II enzyme and DNA polymerase, which might both be routes for killing a fast replicating, purine needy cell, like T. gondii.
In conclusion, we have established that T. gondii selectively remodels the metabolism of a putative host cell prior to invasion. We found conserved shifts in nucleotide metabolism, first observed during full infection and replicated after kiss and spit. Both kiss and spit and full infection both generated high levels of 2,3-BPG, which is an allosteric regulator of host cN-II. We have tested the hypothesis that high levels of 2,3-BPG upregulate cN-II activity, which in turn generates purine nucleosides for T. gondii. Chemical and genetic inhibition of host cN-II affects T. gondii replication and changes purine metabolism during infection. Identification of host enzymes, like cN-II, which play a key role in parasite replication is critical to developing the next generation of anti– Toxoplasma host directed therapies.
Methods
T. gondii Strains and Cell Culture
Low passage type II ME49 T. gondii was used in all kiss and spit experiments. Pru and PruΔHXGPRT, a gift from D. Soldati (57), were used for fludarabine metabolomics. Human Foreskin Fibroblasts (HFFs), MDA-MB231 parental or cN-II-KO MDA-MB231 cells (donation from Lars Petter Jordheim lab) were grown in DMEM with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, and 1% penicillin-streptomycin (Sigma-Aldrich). Once HFFs or MDA-MB231 cells were in deep quiescence, defined as 10 days post confluency, DMEM media was changed to metabolomic media, RPMI1640 supplemented with 2 mM L-glutamine, 1% FBS dialyzed against PBS (MW cutoff of 10 kD), 10mM HEPES, and 1% penicillin-streptomycin. After 35 hours, the media was again changed with metabolic media, 1 hour before treatment with T. gondii.
Kiss and Spit Time Course Metabolomics
HFF dishes in triplicate were treated with 2 x 106 tachyzoites that had been incubated with cytochalasin D (Sigma-Aldrich) or an equal addition of media by volume. An additional negative control of media only treated with cytochalasin D was added to a separate set of dishes. The final concentrationof cytochalasin D was 1.5 μM. At time points 1.5, 3-, 6-, 9-, and 12-HPK&S, dishes were washed three times with ice cold PBS, then quenched with 80:20 HPLC grade Methanol: Water (Sigma-Aldrich). Dishes were incubated on dry ice at -80°C for 15 minutes. Plates were scraped, the solution removed, and spun at 2500 x g for 5 minutes at 4°C. The supernatant was removed and stored on ice, then the pellet was washed again in quenching solution and re-spun. Supernatants were combined, dried down under N2, and stored at -80°C.
Samples were resuspended in 100 µL HPLC grade water (Fisher Optima) for analysis on a Thermo-Fisher Vanquish Horizon UHPLC coupled to an electrospray ionization source (HESI) part of a hybrid quadrupole-Orbitrap high resolution mass spectrometer (Q Exactive Orbitrap; Thermo Scientific). Chromatography was performed using a 100 mm x 2.1 mm x 1.7 µm BEH C18 column (Acquity) at 30°C. 20 µL of the sample was injected via an autosampler at 4°C and flow rate was 200 µL/min. Solvent A was 97:3 water/methanol with 10 mM tributylamine (TBA) (Sigma-Aldrich) adjusted to a pH of 8.2 using approximately 9 mM Acetate (final concentration, Sigma-Aldrich). Solvent B was 100% methanol with no TBA (Sigma-Aldrich). Products were eluted in 95% A / 5% B for 2.5 minutes, then a gradient of 95% A / 5% B to 5% A / 95% B over 14.5 minutes, then held for an additional 2.5 minutes at 5%A / 95%B. Finally, the gradient was returned to 95% A / 5% B over 0.5 minutes and held for 5 minutes to re-equilibrate the column. MS parameters included: scan in negative mode; scan range = 70 - 1000 m/z; Automatic Gain control (AGC) = 1e6, spray voltage = 3.0 kV, maximum ion collection time = 40 ms, and capillary temperature = 350C. Peaks were matched to known standards for identification. Data analysis was performed using the Metabolomics Analysis and Visualization Engine (MAVEN) software (58). Heat maps were generated using the Multi Experiment Viewer program.
Heat-Killed Parasite Control
Low pass ME49 parasites were lysed from host cells, counted, and heat-killed by incubation at 85°C for 30 minutes. As a negative control, empty media was also heated for the same amount of time. 2 x 106 heat-killed parasites, or an equivalent volume of empty media was then added to confluent and quiescent dishes of HFFs in triplicate. Dishes were incubated for 12 hours at 37°C before having their metabolites extracted and analyzed using the previously mentioned methodology.
Conditioned Media Control
Conditioned media was taken from heavily infected (MOI 0.75) cells prior to host cell lysis and the release of parasites into the media. Media from uninfected paired dishes of host cells served as the negative control. Confluent and quiescent dishes of HFFs were incubated in each media condition for 12 hours at 37°C before having their metabolites extracted and analyzed using the previously mentioned methodology.
Metabolomics with chemical inhibition of cN-II
HFF were seeded in 60 mm dishes in triplicate and allowed to reach confluency. 36h before the procedure, the DMEM media was changed to metabolomic media. Then, each dish was infected with 2 x 106 ME49 or Pru tachyzoites and treated with 50 μM fludarabine or solvent only control. At specific time points, dishes were washed three times with PBS, spun, and then metabolites were extracted and analyzed using the previously mentioned methodology (Figure 6A). 10 μM of AMP, GMP, IMP, Guanine, Guanosine, and Inosine -HPLC standard were analyzed simultaneously.
Fludarabine growth assay
Confluent HFFs were infected with low pass ME49 and were left to replicate for 4 hours along with uninfected controls. After 4 hours the media was changed on all cells, creating 5 different treatment populations: uninfected with DMSO, infected with DMSO, infected with 1μM pyrimethamine (Sigma-Aldrich), and either 50, 25, or 5μM fludarabine (sigma-Aldrich) treatment. Triplicate samples for each of the five conditions were grown for 48 hours, and later 1 μCi of [3H] uracil was added. After a 24-hour growth incubation, monolayers were fixed by adding ice cold 0.6 M Trichloroacetic acid (TCA) and incubating at 4°C for 1 hour to fix the cells. TCA was removed, the cells were washed with water for 4 hours, then 1 M NaOH was added to resolubilize the monolayer, and plates were shaken for one hour at room temperature. Each sample was diluted 1:10 in scintillation fluid and [3H] abundance was measured. The uninfected population measured baseline host [3H] uracil uptake, infected cells treated with DMSO were the control for normal growth, and infected cells treated with pyrimethamine were the control for growth inhibition. Background host [3H] incorporation was subtracted from all samples, then the following equation was used to calculate percent inhibition in each sample condition by normalizing to the negative control:
 Additionally, HFF cells were seeded in 24 well plates until confluency. Then, they were infected with 1 x106 mCherry ME49 tachyzoites and treated with 50, 25, or 12.5 μM fludarabine (Sigma-Aldrich) treatment. Time-point fluorescent readings were then taken with an IncuCyte S3 (Sartorius-Germany) plate reader at 565-605 excitation and 624-705 emission for 60 hours. Read Image Mean of three wells in each condition was calculated with the Incucyte software.
Additionally, HFF cells were seeded in 24 well plates until confluency. Then, they were infected with 1 x106 mCherry ME49 tachyzoites and treated with 50, 25, or 12.5 μM fludarabine (Sigma-Aldrich) treatment. Time-point fluorescent readings were then taken with an IncuCyte S3 (Sartorius-Germany) plate reader at 565-605 excitation and 624-705 emission for 60 hours. Read Image Mean of three wells in each condition was calculated with the Incucyte software.
Quantitative PCR
MDA-MB231 parental and cN-II-KO MDA-MB231 cells (donation from Lars Petter Jordheim lab) were seeded in 24-well plates. Genomic DNA from infected cells was isolated following the protocol developed in Boothroyd’s lab (59). Real-time qPCR was performed (Bio-Rad iTaq Universal SYBR Green Supermix) on an Applied Biosystems QuantStudio 7 Flex Real-Time PCR system. For quantification of T. gondii, primers for the SAG1 gene were used: SAG1 forward: 5’ –TGCCCAGCGGGTACTACAAG– 3’ and reverse: 5’ TGCCGTGTCGAGACTAGCAG– 3’(60). To normalize gene expression, a housekeeping host gene on MDA-MB231, the Human peptidylprolyl isomerase A (PPIA) gene, was used(61). Primers were designed using the NCBI primer design tool and snapGene program version 5.3.1. PPIA-forward: 5’-TGGTTATGGAGGCTTTGAGGTTT-3’ and reverse: 5’-TGCCAGCAAGCACTGTACATATAA-3’ Expression was calculated by normalizing the cycle threshold values (CT) to that of human PPIA for each sample (ΔCT). The ΔCT value was then used to calculate the 1/ ΔCT). Three biological replicates and three technical replicates were analyzed for each condition at specific time points in two independent experiments.
Supplementary Material
Supplemental Table 1: This table contains the exact values for the ratios represented in the Figure 2 heat map.
CONFLICT OF INTEREST
Competing interests
We confirm that none of the authors have any competing interests in accordance with eLife’ guidelines.
Funding
This work was funded by the National Institutes of Health (R01AI144016 LJK), and Morgridge Postdoctoral Fellowship supported by the Morgridge Institute for Research (GMGL). Funding bodies had no role in the design of the study and collection, analysis, and interpretation of data, and in writing the manuscript.
Authors’ contributions
Design of experiments: GMGL, WO & LJK. Conceptualization: WO, GMGL LJK; Funding acquisition: LJK; Investigation: GMGL & WO; Methodology: GMGL, WO; Project administration: LJK. Resources: LJK; Supervision: LJK. Visualization: AMTP, GMGL, WO; Writing: GMGL & WO; Writing – review & editing: AMTP & LJK. All authors read and approved the final manuscript.
Acknowledgments
We thank Bruno Martorelli Di Genova for their lab assistance with the growth inhibition assay. Also, we are grateful to the Jordheim lab in Claude Bernard Lyon University for providing us with the MDAMB231 cells.
REFERENCES




