

Abstract
Large-scale genome engineering in yeast is feasible primarily due to prodigious homology-directed DNA repair (HDR), a plethora of genetic tools, and simple conversion between haploid and diploid forms. However, a major challenge to rationally building multi-gene processes in yeast arises due to the combinatorics of combining all of the individual edits into the same strain. Here, we present an approach for scalable, precise, multi-site genome editing that combines all edits into a single strain without the need for selection markers by using CRISPR-Cas9 and gene drives. First, we show that engineered loci become resistant to the corresponding CRISPR reagent, allowing the enrichment of distinct genotypes. Next, we demonstrate a highly efficient gene drive that selectively eliminates specific loci by integrating CRISPR-Cas9 mediated Double-Strand Break (DSB) generation and homology-directed recombination with yeast sexual assortment. The method enables Marker-less Enrichment and Recombination of Genetically Engineered loci (MERGE) in yeast. We show that MERGE converts single heterologous yeast loci to homozygous loci at ∼100% efficiency, independent of chromosomal location. Furthermore, MERGE is equally efficient at converting and combining loci, thus identifying viable intermediate genotypes. Finally, we establish the feasibility of MERGE by engineering a fungal carotenoid biosynthesis pathway and most of the human α proteasome core into yeast. MERGE, therefore, lays the foundation for marker-less, highly efficient, and scalable combinatorial genome editing in yeast.
Introduction
Baker’s yeast has long served as a convenient chassis for bioengineering owing to its genetic tractability, versatile metabolism, and ease of culture in the lab. Decades of fundamental research, together with the development of high throughput toolkits and genome engineering capacities, have established yeast as an ideal model eukaryote for system genetics and synthetic biology1. The availability of many selectable genetic markers and simple conversion between haploid and diploid forms has provided avenues to easily combine pairs of genetically engineered loci to understand gene-gene interactions at a global scale2. For more extensive genetic alterations, yeast’s highly efficient Homologous Recombination (HR) pathway even enables the synthesis of entire chromosomes, although this approach requires iterative use of selection markers and tedious repetitive procedures3. Nonetheless, the ability to alter large contiguous segments of genomic loci has many applications, such as genome minimization4, multiplex genome editing5, and the total synthesis of the Mycoplasma and E. coli genomes using yeast HR6, 7.
In spite of the progress in whole genome engineering, editing intermediate numbers of independent genomic loci–greater than two and in discontiguous regions of the genome–still presents a significant challenge. While strategies exist for E. coli8 (e.g., MAGE), diploid organisms such as yeast present the additional editing challenge of multiple alleles for each (independently assorting) genomic locus. Moreover, while high throughput cloning strategies and the reduced cost of de novo DNA synthesis now allow swapping entire heterologous pathways or protein complexes into yeast, such efforts frequently require the deletion of corresponding yeast loci, such as the case for efforts to systematically humanize yeast genes9, 10, which may entail replacing genes at their corresponding genomic loci to retain native regulation. Beyond these aspects, the expression of each new gene often reveals incompatibilities associated with the engineered pathway. Thus, there is a need for rapid, multi-site, progressive genome editing strategies to address these issues.
The efficiency and speed of CRISPR-Cas9-based genome engineering allow straightforward editing of multiple yeast loci in a single strain, eliminating the need for markers11–15. However, the approach gets progressively more challenging for multi-gene systems when the fitness of the intermediate genotypes is unknown (Figure S1). Therefore, rationally building heterologous genetic modules in a yeast surrogate requires a highly scalable and combinatorial genome editing technology.
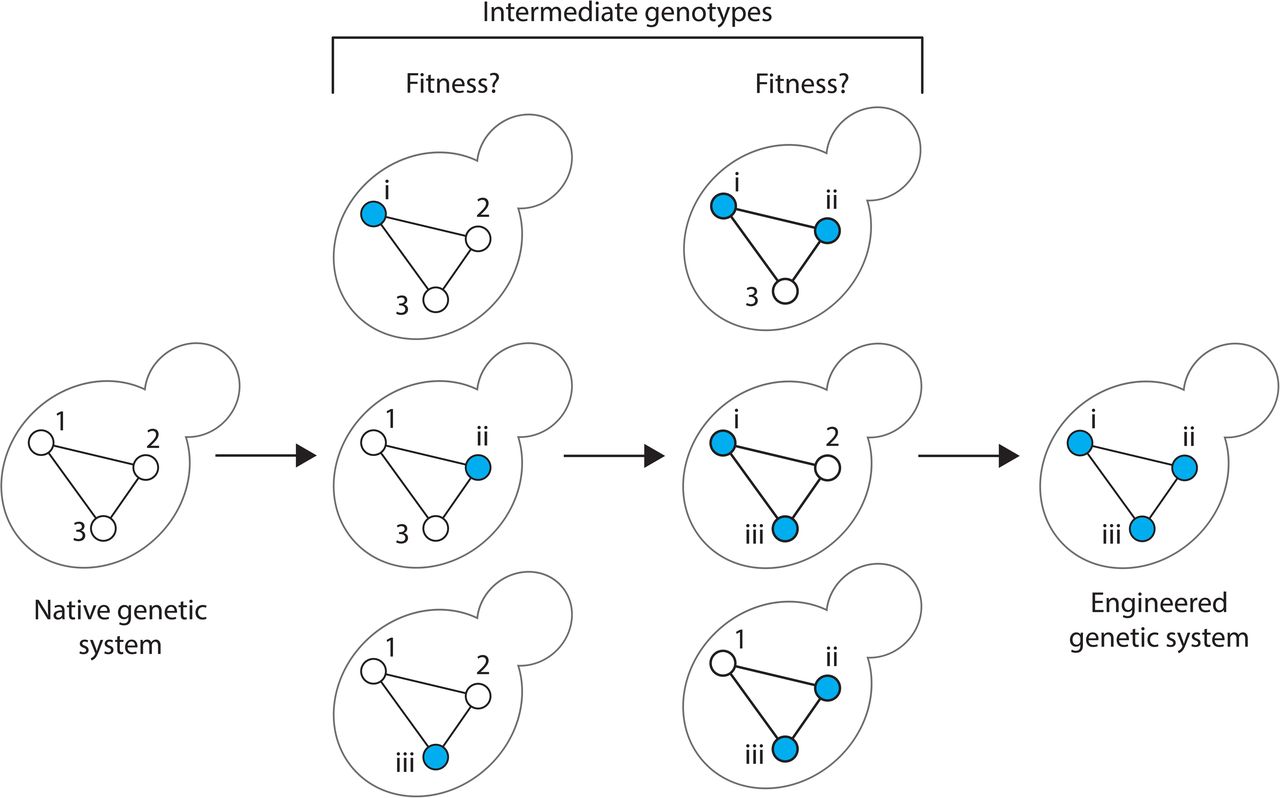
Let’s examine a simple 3-gene pathway. A sequential replacement strategy to fully engineer this module will navigate 6 possible intermediate genotypes. The following formula can calculate the number of genotypes: nCr = (n!/n − r!) (r!), where n represents a total number of genes to be combined (for example, depicted above, n = 3). And r represents the combined intermediates, like for a single-gene intermediates r = 1 and similarly for a 2-gene intermediates r = 2. Furthermore, while the strategy explores 6 intermediate genotypes, the distinct routes to complete pathway engineering are 9 as some genotypes may require gene 1 and then gene 2, or gene 2 and then gene 1. The higher the number of genes to be engineered, the more the number of intermediate genotypes to be explored. As a reference, for the humanization of the heptameric α proteasome core, a total of 126 intermediate hybrid yeast-human genotypes exist towards humanizing the entire yeast α core.
Synthetic Genetic Array (SGA) analysis permits the combination of loci in a fitness-driven manner, requiring markers linked to the modified loci and haploid-specific selections16. Using an SGA-like strategy to build a heterologous multi-gene system would need several unique markers linked to each gene. However, the lack of adequate selection markers limits its application. The Green Monster (GM) method bypasses the marker dependency by using Green Fluorescent Protein (GFP) expression as a readout to combine engineered loci17. However, implementing the GM strategy can be challenging for heterologous pathway engineering with an added burden of expressing fluorescent cassettes18. Thus, accomplishing large-scale combinatorial genome editing in yeast necessitates new genetic tools that circumvent these requirements.
In this work, we describe a novel, CRISPR-Cas9-based method to readily combine genetically engineered loci without the need for markers and exogenous repair templates. The approach involves generating CRISPR-Cas9-mediated Double-Strand Breaks (DSBs) and highly efficient Homology Directed Repair (HDR) along with yeast mating and sporulation to randomly assort edited sites performing multi-site and marker-less combinations of engineered loci. This selective and successive elimination of specific yeast loci mimics a gene drive19–22 (Figure S2) and facilitates Marker-less Enrichment and Recombination of Genetically Engineered yeast loci (MERGE).

Every locus in yeast follows a normal (1:1) inheritance pattern of segregation. However, CRISPR reagent targeted to a haploid or a diploid locus leads to lethality. Similarly, a heterozygous yeast locus follows 1 (1, wild-type) : 1 (i, engineered) inheritance pattern. CRISPR reagent targeted to one of the heterozygous alleles initiates homologous recombination using the CRISPR-resistance homologous chromosome as a repair template, thus enabling efficient conversion to homozygous loci, mimicking a gene drive. The subsequent segregation pattern of 4 : 0 allows propagation of only desired loci.
We show that CRISPR-mediated selection operates similarly to classical selection markers, enabling MERGE to efficiently explore many combinations of genetically engineered loci, revealing a fitness-driven path to engineering any heterologous system in yeast. We further demonstrate that MERGE enables rapid assembly of an entire carotenoid biosynthesis pathway by performing a multi-site and marker-less combination of distinct engineered loci. Finally, using a multiplexed version of MERGE, we humanize a near-complete α proteasome core (6 of 7 subunits) in yeast while revealing a fitness-driven path to the humanization of complex processes.
Results & Discussion
CRISPR-Cas9 allows marker-less selection and enrichment of unique genotypes in yeast (CELECT)
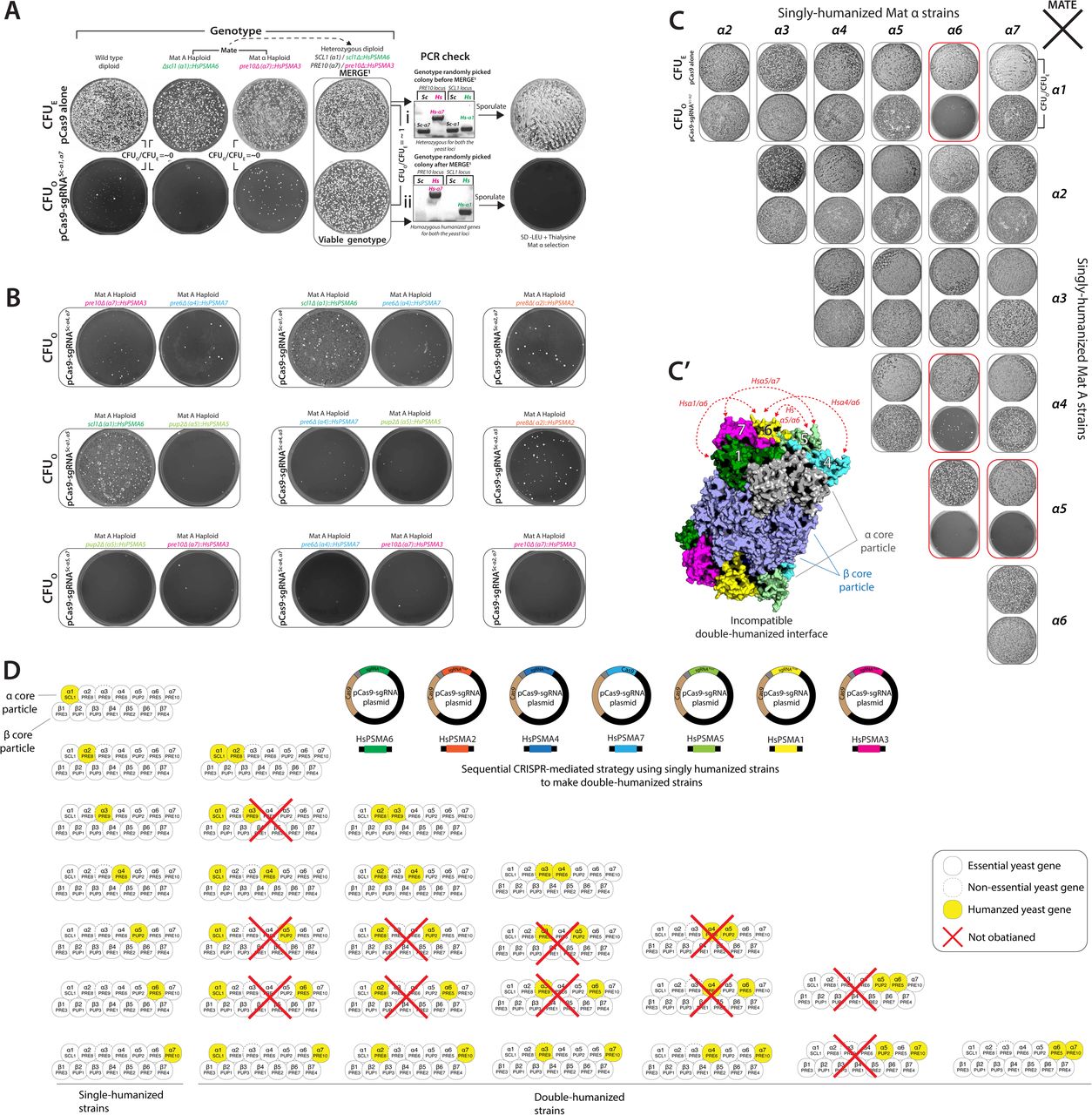
CRISPR-Cas9 enables the precise and marker-less editing of both essential and non-essential yeast loci owing to significantly lower error-prone Non-Homologous End Joining (NHEJ) in yeast relative to HDR23. Therefore, Cas9-sgRNA-induced lethality serves as a rapid test for a functional CRISPR reagent in yeast (Colony Forming Units Observed or CFUO = ∼0) compared to the Cas9 alone (Colony Forming Units Expected or CFUE). To verify the ON-target activity of CRISPR reagents, the transformation of a plasmid carrying expression cassettes for both Cas9 and a sgRNA targeting a specific locus (pCas9-sgRNAlocus) in a yeast strain that harbors a corresponding engineered locus resistant to further targeting allows the survival of colonies similar to the pCas9 alone (CFUO/CFUE = ∼1) (Figure 1A).

(A) pCas9-sgRNA1 targeted to any yeast locus (locus 1) leads to lethality (CFUO) compared to the vector without sgRNA (pCas9 alone; CFUE). However, the modification of locus 1 to i prevents further targeting by the corresponding CRISPR reagent (CFUO). A simple readout of CFUO/CFUE enables the identification of modified genotypes. (B) Each CRISPR reagent targeted to several wild-type yeast loci shows near 100% lethality (Gray bars or CRISPR-sensitive loci). However, after editing the loci (by generating single-humanized yeast α proteasome genes, ade2Δ::kanMX and inserting carotenoid genes at landing pad loci), the strains show resistance to the corresponding CRISPR reagent (Blue bars or CRISPR-resistant loci, CFUO/CFUE = ∼1). (C) Each unique genetically modified strain can be enriched from the mixture using the corresponding CRISPR reagent (CELECT). The transformation of pCas9 alone serves as a control (i). Haploid ade2Δ::kanMX strain quantifies the efficiency of the CRISPR selection (pCas9-sgRNAADE2), enriching only the ade2Δ homozygous genotypes (red-colony phenotype) (ii). The transformation of pCas9-sgRNARPT5 causes lethality as all the strains in the mix harbor a wild-type copy of the RPT5 gene (iii). Similarly, each CRISPR reagent specific to individual yeast α proteasome genes also selected the corresponding humanized strains as demonstrated by PCR-based genotyping of randomly picked colonies (iv).
We tested this strategy by humanizing α proteasome core genes in yeast that are functionally replaceable by their human counterparts24. Using CRISPR-Cas9, we replaced each yeast α proteasome gene with its human ortholog at the native loci (Figure S3). Additionally, we modified non-essential loci to test if the CRISPR-mediated selection is broadly applicable and found that all engineered strains are resistant to the corresponding CRISPR-Cas9-sgRNA mediated lethality (CFUO/CFUE = ∼1, Figure 1B, Figure S4A & S4B). Henceforth, we refer to the CRISPR-Cas9-mediated selection of genotypes as CELECT.
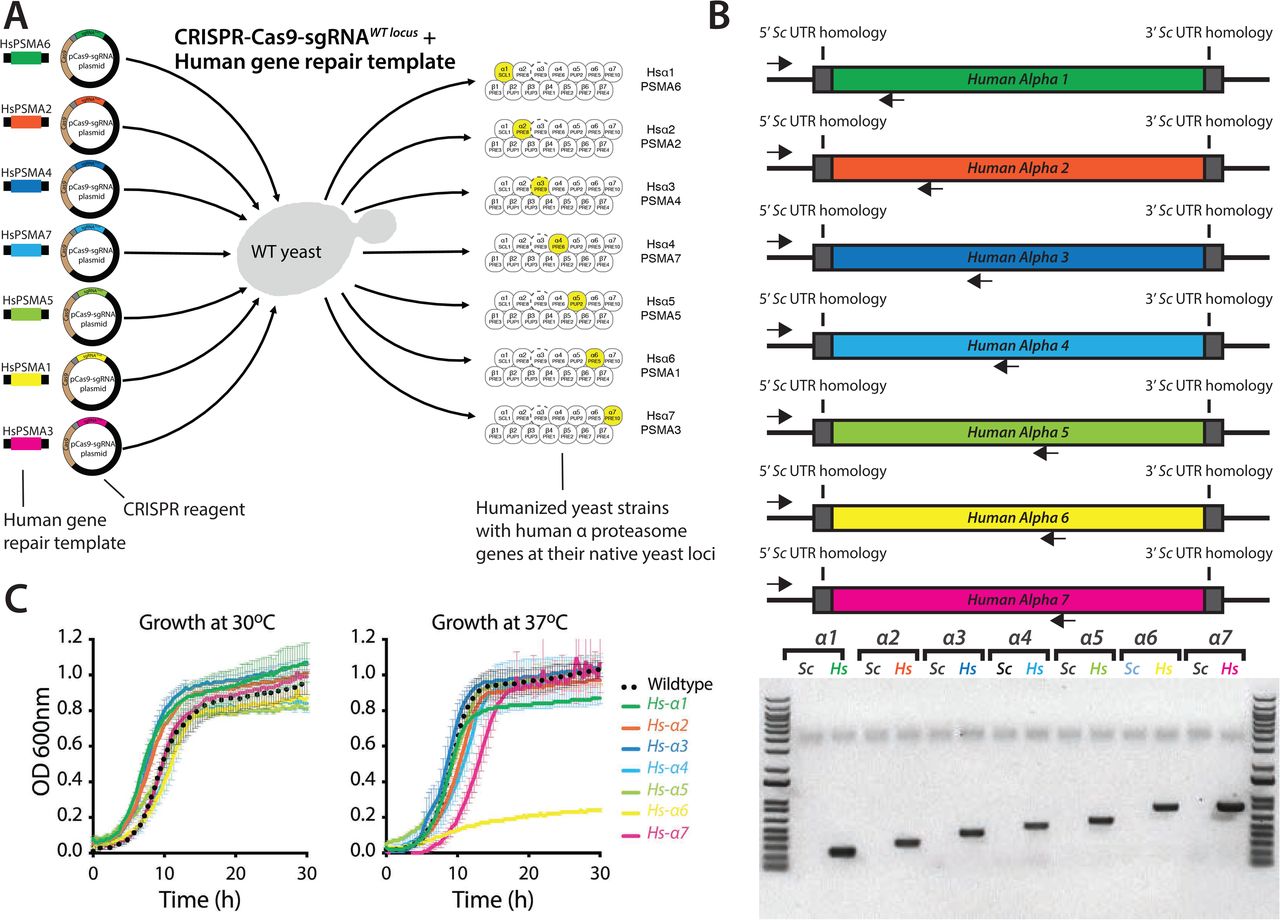
(A) For each gene, a ‘knockout’ (KO) plasmid (CRISPR reagent) is constructed that expresses the Cas9 endonuclease and a sgRNA with a targeting sequence to the gene of interest. To replace the gene of interest, the CRISPR reagent is co-transformed into yeast with a linear repair DNA, consisting of the replacing gene sequence flanked by long (60-200bp) homology sequences to the flanking region of the genome. Cas9 and the sgRNA are co-expressed and target the gene of interest for cutting, allowing repair by homologous recombination (HR) with the provided repair DNA. (B) After obtaining transformants, sequencing across the junction reveals successful replacement (as shown for the human α proteasome subunits replaced at the native yeast loci). PCR for the yeast locus (Sc) is negative, whereas for a human gene (Hs) is positive. (C) The growth of singly-humanized α-subunits is not significantly impaired. Each curve represents an averaging of 4 replicates for each indicated strain, grown in YPD for 24 hours. No strains exhibit significantly impaired growth, though strains with human subunits α4, 5, and 6 show some extension of lag phase at 30°C and marginally slowed doubling times at 30°C. The behavior is similar at 37°C except in Hs-α6, which exhibited a significant growth defect.
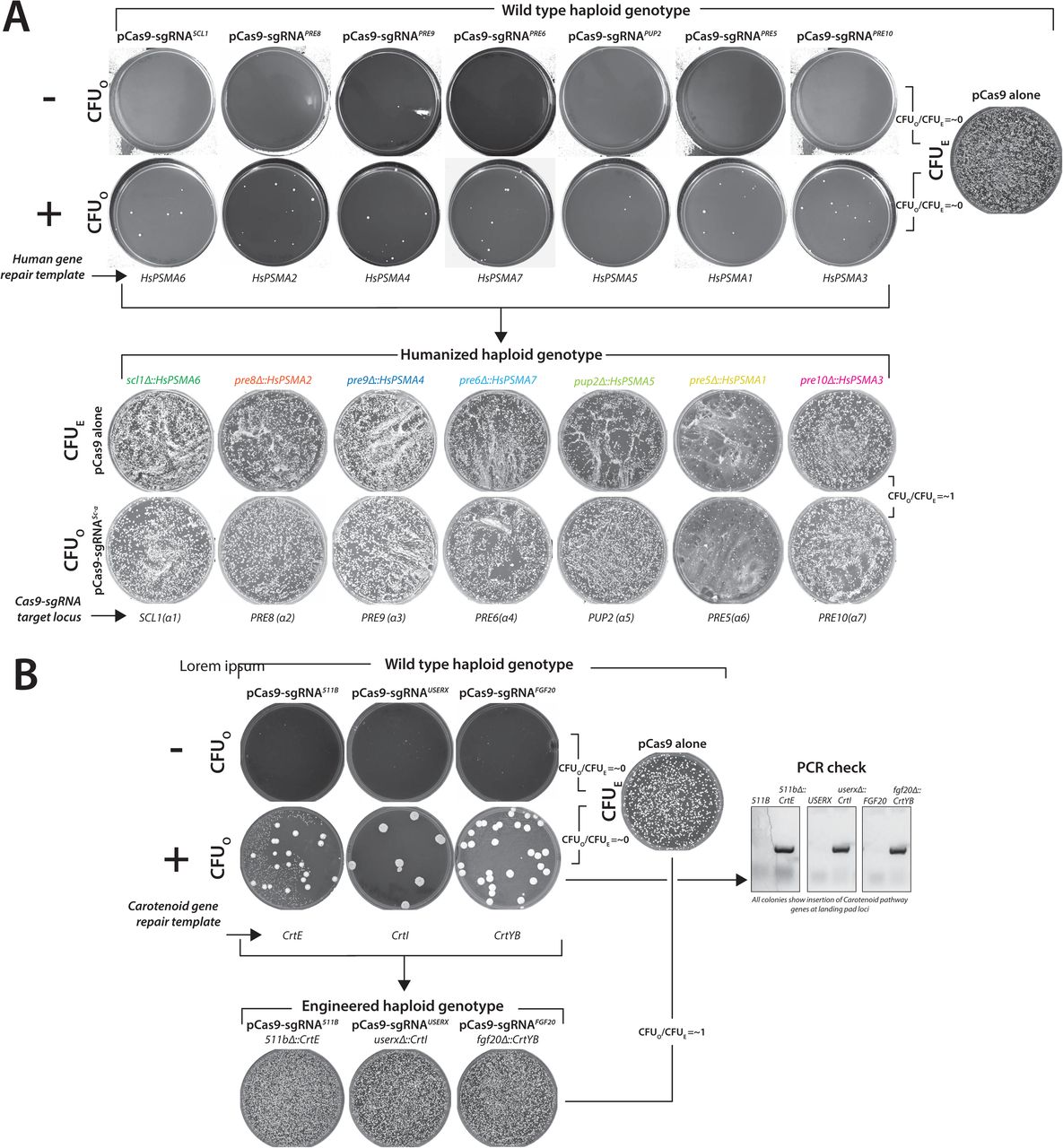
(A) CRISPR reagent (pCas9-sgRNAlocus) targeted to any yeast locus often leads to lethality with few surviving colonies (CFUO). The corresponding transformation of a control plasmid (pCas9 alone) estimates the transformation efficiency (CFUE). CRISPR reagents targeting 7 yeast α proteasome genes show lethality (CFUO/CFUE = ∼0) with or without the repair template. After humanizing the corresponding yeast loci, each single-humanized haploid strain shows resistance to further targeting by the corresponding CRISPR reagent, respectively (CFUO/CFUE = ∼1). (B) CRISPR reagents targeted to landing pad loci (511B, USERX and FGF20) enabled editing by introducing carotenoid gene transcription units CrtE, CrtI and CrtYB (provided as PCR fragment repair templates). The engineered strains show resistance to further cutting by the corresponding CRISPR reagents (CFUO/CFUE = ∼1). PCR-based genotyping confirmed the integration of each carotenoid transcription unit for every colony tested.
To further verify if the resistance of humanized strains is exclusive to its corresponding CRISPR reagent, all single-humanized α proteasome strains were mixed in culture (Figure 1C). To quantify the enrichment of unique genotypes, we inoculated a ade2Δ::kanMX haploid strain in the mixture. The pCas9-sgRNAADE2 exclusively enriched for resistant ade2Δ genotype, whereas all other genotypes in the mix harboring a wild type ADE2 locus are inviable (Figure 1C-ii). Conversely, the transformation of pCas9-sgRNARPT5 targeting RPT5 (a base subunit of the proteasome complex), for which all the strains in the mix harbor a wild-type copy, shows no survivors (Figure 1C-iii). We demonstrate that each CRISPR reagent targeting yeast α proteasome genes selected a corresponding humanized genotype from a mix, respectively (Figure 1C-iv). Thus, CRISPR-Cas9-mediated resistance functions similarly to conventional antibiotic or auxotrophic markers in yeast.
MERGE0 is nearly 100% efficient at converting loci irrespective of the yeast gene location on the chromosome
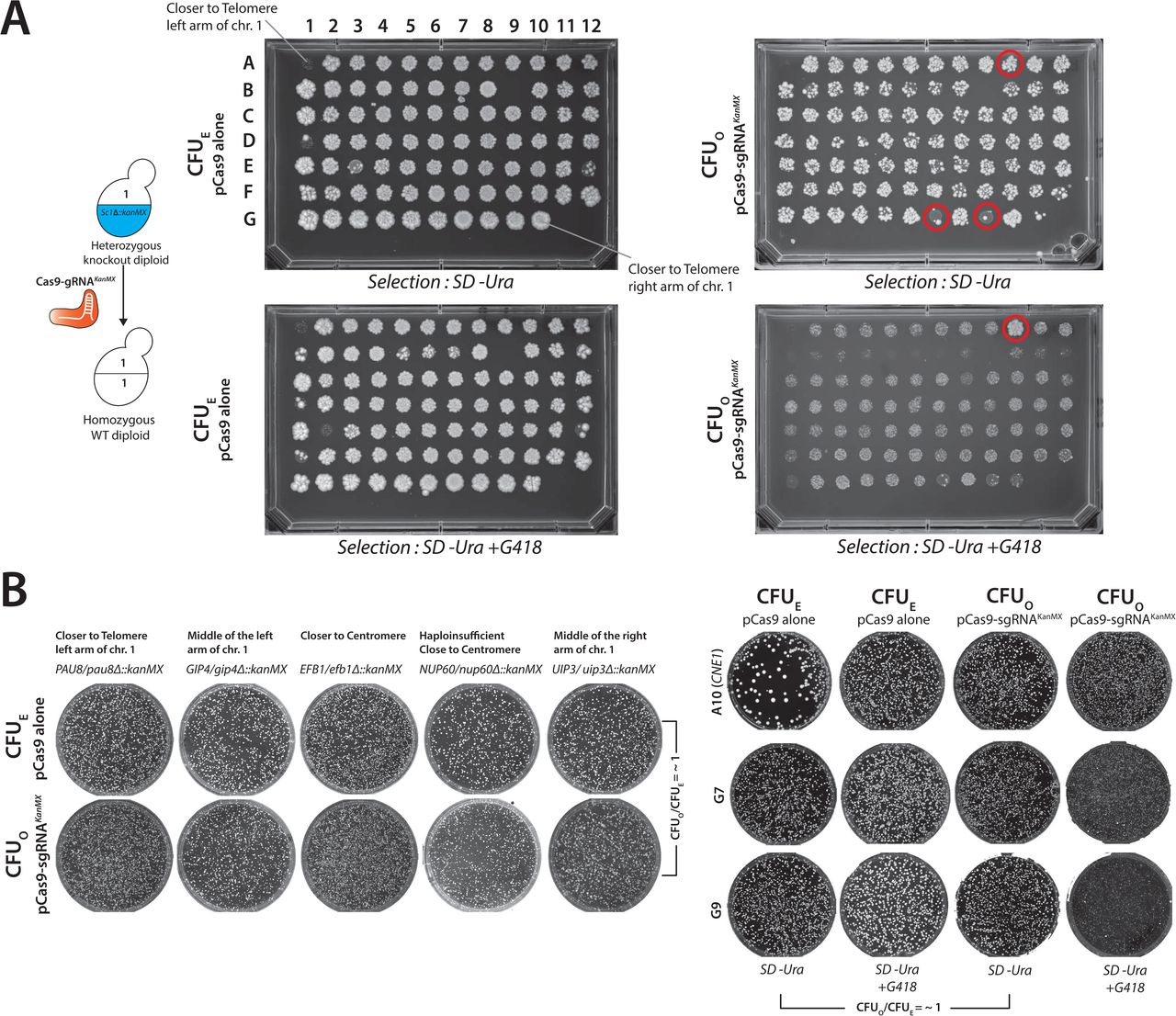
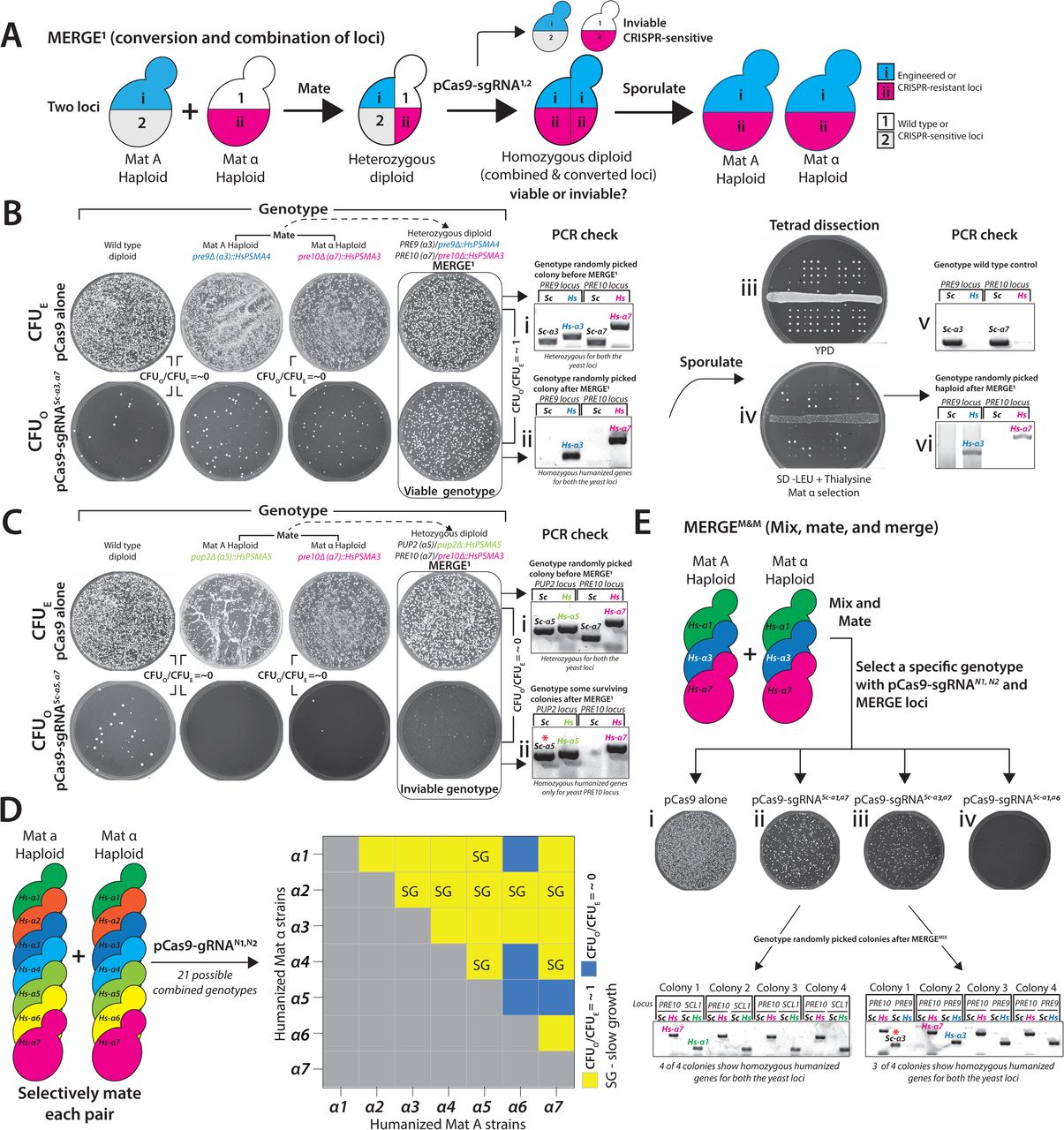
Mating of engineered haploid strains with wild-type generates a heterozygous genotype. CRISPR-Cas9 reagent targeted to either allele should enable the conversion to homozygous diploid at high efficiency while also enriching the desired genotype (MERGE level 0 or MERGE0) (Figure 2A). We used ADE2/ade2Δ::kanMX heterozygous knockout diploid (hetKO) strain to quantify the efficiency of MERGE0. The wild type ADE2 allele is susceptible to pCas9-sgRNAADE2 mediated DSB. In contrast, the ade2Δ::kanMX allele is resistant, proving a readout of conversion as the loss-of-function of ADE2 results in a red color colony phenotype.

(A) Mating haploid yeast strains, each harboring a different allele at a single locus, enables the combination as a heterozygote. CRISPR reagent targeted to one of the alleles (CRISPR-sensitive locus) initiates recombination using the homologous chromosome with a CRISPR-resistant locus as a repair template. Sporulation of the resulting homozygous diploid yields the desired genotype in both mating types. (B) CRISPR reagent (pCas9-sgRNAADE2) targeted to the wild type ADE2 locus quantifies the efficiency of MERGE0. The transformation of pCas9-sgRNAADE2 in the wild type haploid or diploid strains is lethal, with few surviving red colonies suggesting efficient ON-target activity (CFUO/CFUE = ∼0). However, the transformation of pCas9-sgRNAADE2 in heterozygous diploid ADE2/ade2Δ::kanMX strain shows no lethality (CFUO/CFUE = ∼1). Instead, all surviving red colonies suggest efficient conversion to the knockout locus. (C) Similarly, CRISPR reagents targeted to several wild-type yeast loci show near 100% lethality with few surviving colonies while simultaneously providing exogenous repair templates (oligo for ADE2 locus and PCR fragments for the remaining loci) (Red bars). However, each heterozygous diploid displays resistance to CRISPR reagents respectively (CFUO/CFUE = ∼1, Yellow bars). PCR confirmation verified the conversion to the CRISPR-resistant locus. (D) CRISPR reagent targeting KanMX cassette (pCas9-sgRNAKanMX) shows no OFF-target activity in a strain lacking the cassette (CFUO/CFUE = ∼1). However, haploid or diploid strains harboring KanMX cassettes as the only allele show lethality (CFUO/CFUE = ∼0), suggesting ON-target activity. However, diploid strain heterozygous for KanMX allele (SWH1/swh1Δ::kanMX) is viable (CFUO/CFUE = ∼1), suggesting conversion to the wild type allele as demonstrated by the loss of G418 resistance after performing MERGE0. Using heterozygous diploid knockout strains (hetKO) arrayed across the entire yeast chromosome I, MERGE0 similarly converted every knockout allele to the wild type locus (Yellow = CFUO/CFUE = ∼1), except in the case of CNE1 that showed resistance to the CRISPR reagent but did not lose the KanMX cassette. (E) Alternatively, using hetKO strains and targeting CRISPR reagent to the wild-type yeast locus enables a single-step gene essentiality assay in yeast. The remarkably high efficiency of MERGE0 converts every heterozygous yeast locus to homozygous null, therefore, allowing viability only if the gene is non-essential (as in the case of yeast proteasome α3 gene). All other essential loci show lethality after MERGE0.
The transformation of the wild-type haploid or diploid yeast with pCas9-sgRNAADE2 showed a lethal phenotype (∼0-20 CFUO on average) (Figure 2B). However, pCas9-sgRNAADE2 transformation in ADE2/ade2Δ::kanMX hetKO strain shows resistance while also converting the locus to Δade2::KanMX at ∼100% efficiency (Figures 2B & S5A). MERGE0 performs with a comparable proficiency at single-humanized α proteasome and landing pad loci (Figures 2C, S5B & S5C). To further test if MERGE0 can convert any heterozygous yeast locus independent of the position on a chromosome, we explored the strategy across many yeast genes located on chromosome 1. We designed a CRISPR reagent to target KanMX (pCAS9-sgRNAKanMX) in hetKO diploid strains harboring the KanMX cassette instead of a yeast gene. MERGE0 converted all heterozygous loci to homozygous wild-type alleles, respectively, with simultaneous loss of KanMX cassette (except for CNE1) (Figure 2D, S6A & S6B).

(A) A schematic of possible genotypes after the ADE2 locus in a heterozygous diploid ADE2/ade2Δ::kanMX strain is subjected to a CRISPR-Cas9-mediated DSB followed by sporulation. The repair via NHEJ could mutate the locus (ade2*) or repair via HDR converts the locus to ade2Δ::kanMX, either scenario leads to a red colony phenotype. However, post sporulation, the prior scenario will show only 50% G418 resistant colonies compared to the latter outcome, which will exhibit 100% G418 resistance. Tetrad dissection of pooled cells from heterozygous diploid strains before MERGE0 (pCas9 alone) or after MERGE0 (pCas9-sgRNAADE2) shows that before MERGE0 cells appear as 2 : 2 (red : white) and 2 : 2 (G418- resistant : G418-sensitive) phenotype. However, after MERGE0, cells show 4 : 0 (red : white) phenotype. Furthermore, all 4 spores in the post MERGE0 strain show 4 : 0 G418 resistance. (B) Schematic shows CRISPR reagent targeted to one of the heterozygous alleles (wild type yeast gene) in a diploid strain permits efficient conversion to homozygous humanized loci. MERGE0 efficiently converts wild-type yeast loci α proteasome genes to humanized loci (CFUO/CFUE= ∼0). PCR-based genotyping verified the conversion to the CRISPR-resistant humanized loci for every colony tested. (C) Schematic shows how CRISPR reagent targeted to the landing pad loci in a heterozygous diploid enables conversion to engineered carotenoid loci. MERGE0 converts heterozygous landing pad loci (511B, USERX, and FGF20) to engineered carotenoid loci (CFUO/CFUE = ∼1). PCR-based genotyping shows heterozygous status of the loci before MERGE0 and conversion to engineered carotenoid loci after MERGE0. Sporulation yields haploids of both mating types with only engineered loci.
Furthermore, using CRISPR-Cas9 to target a wild-type yeast locus instead of the KanMX cassette in a hetKO diploid strains, MERGE0 enables single-step gene essentiality screening in yeast, significantly increasing efficiency compared to the classical methods such as Tetrad dissection or SGA analysis25, 26. Except for a non-essential α3 gene, all six essential α proteasome genes are inviable post-transformation of the corresponding CRISPR reagents (Figure 2E).
MERGE1 permits a fitness-driven combination of the engineered loci
Given the high efficiency of MERGE0 at converting one yeast locus, we next tested if the method works similarly to convert and combine two distinct loci (MERGE level 1 or MERGE1). Mating yeast strains, each with one modified locus, facilitate the combination as heterozygotes at two separate loci. CRISPR-Cas9 with two sgRNAs that target each unique locus allows simultaneous conversion to paired CRISPR-resistant loci, respectively (Figure 3A). To test the pipeline across all humanized α proteasome strains, we first used MERGE0 to move all singly humanized α proteasome loci from BY4741 background to SGA strain background in both mating-types (Figure S4B). To verify if MERGE1 can simultaneously convert two distinct loci, we tested the efficiency of combining paired-humanized α3/α7 and α1/α7 genotypes. We show that MERGE1 is highly efficient as each heterozygous genotype is resistant to the double-sgRNA CRISPR-Cas9 reagent while converting and combining two humanized loci (Figure 3B, S7A). The CRISPR reagent is lethal in wild-type and singly humanized strains (Figure 3B & S7B). The sporulation of viable diploids yields haploid double-humanized strains in both mating-types. Genotyping randomly picked colonies confirms the conversion to humanized loci (Figure 3B). Additionally, the double-humanized α7/α1 genotype, while viable, shows a sporulation defect (a phenotype associated with Hsα7) (Figure S7A). Thus, MERGE1 allows the survival and enrichment of only viable homozygous double-humanized loci without the requirement of any diploid-specific selection. However, a double-humanized α5/α7 is inviable as a combined genotype, as evidenced by double-sgRNA CRISPR-Cas9-mediated lethality (CFUO/CFUE = ∼0). PCR-based genotyping of a surviving colony shows homozygosity of only the Hsα7 locus (Figure 3C). In contrast, while a single-humanized Hsα6 strain is temperature-sensitive (TS) at 37°C, combination with the neighboring humanized α7 gene rescues the phenotype (Figure S7A′). However, the TS phenotype is associated with one of the variants of Hsα6 (Variant 1, 37>Glycine) (Figure S8A), whereas another common variant (Variant 2, 37>Valine) shows no growth defect at 37°C (Figure S8B). Thus, MERGE1 revealed the fitness of paired-humanized genotypes similar to synthetic genetic interactions without the need for linked markers or haploid-specific selections16.

(A) Schematic shows a CRISPR reagent (pCas9-sgRNAKanMX) targeted to the KanMX allele in a hetKO strain converts the locus to homozygous wild-type. The hetKO strains were arrayed as genes close to the left-arm of a telomere through the centromere to the right arm of a telomere. The strains were transformed with pCas9 alone (CFUE) or with pCas9-sgRNAKanMX (CFUO) and spotted on either SD-Ura or SD-Ura with G418 selection. Every genotype shows spots with a similar density of colonies showing no lethality due to the CRISPR reagent (CFUO/CFUE = ∼1). The spots on SD-Ura with G418 selection show G418 resistance in the case of pCas9 alone transformants. However, all but one (A10 - CNE1) strain transformed with pCas9-sgRNAKanMX lost the KanMX cassette resulting in G418 sensitivity. (B) MERGE0 mediated conversion of loci was tested on several unique hetKO genotypes on Petri plates, demonstrating agreement with 96-well spot assays. All tested strains show no lethality due to the CRISPR reagent (CFUO/CFUE = ∼1). Ambiguous spots on a 96-well plate (A10, G7 and G9) were further analyzed on Petri plates. G7 and G9 spots show no lethality due to CRISPR reagent (CFUO/CFUE = ∼1) and loss of KanMX cassette. While the A10 (CNE1) strain is resistant to the CRISPR reagent, the cells retain G418 resistance. It is unclear why this behavior occurs.

(A) Double-sgRNA CRISPR reagent (pCas9-sgRNAScα1,α7) targeted to two wild type yeast loci (proteasome α1 and α7 genes) quantifies the efficiency of MERGE1. The transformation of pCas9-sgRNAScα1,α7 in the wild type, and single-humanized Hsα1 or Hsα7 strains is lethal, suggesting ON-target activity (CFUO/CFUE = ∼0). However, the transformation of pCas9-sgRNAScα1,α7 in diploid heterozygous humanized strain shows no lethality (CFUO/CFUE = ∼1). PCR-based genotyping of surviving colonies after MERGE1 shows conversion of both yeast to the humanized loci compared to before MERGE1. (B) All tested single-humanized strains transformed with double-sgRNA CRISPR reagents show lethality, demonstrating that selection only allows survival of viable paired genotypes. (C) Mating each single-humanized strain of opposite mating-types (obtained via MERGE0) provided 21 heterozygous genotypes to test MERGE1. Heterozygous diploid humanized strains transformed with double-sgRNA CRISPR reagents specific to each yeast allele allow testing viability of all paired-humanized genotypes (CFUO). If the genotype is permitted, the strategy enables survival similar to the pCas9 alone transformation (CFUE). While the majority of combined double-humanized genotypes are viable (CFUO/CFUE = ∼1), specific genotypes (Hsα1α6, Hsα4α6, Hsα5α6 and Hsα5α7) were inviable, suggesting incompatible genotypes. Other combinations, such as those involving the genotypes with Hsα2, showed a slow-growth phenotype. (C’) Yeast proteasome structure (PDB-1RYP) with α and β cores show the humanized α subunits that are incompatible as paired genotypes. Except for α5 and α6, the subunits are missing neighboring interactions within the α core. (D) A sequential strategy using single-humanized strains acquired several pairwise combinations of human α subunits. However, several other genotypes could not be obtained, revealing the drawback of the method. MERGE1 is far more efficient in identifying incompatible paired genotypes.

(A) A growth assay performed on a single-humanized Hsα6 strain (Variant 1, amino acid residue 37>Glycine) shows a temperature-sensitive (TS) phenotype at 37°C (dotted red line) compared to growth at 30°C (solid red line). The combined genotype of Hsα6,α7 partially rescues the TS phenotype at 37°C (dotted blue line) compared to the growth at 30°C (solid blue line). (B) Spotted dilutions of single-humanized Hsα6 strains (Variant 1, amino acid residue 37>Glycine) and (Variant 2, amino acid residue 37>Valine) show Variant 2 with no growth defect at 30°C, 23°C and 37°C compared to Variant 1.

(A) The schematic shows the mating of haploid yeast strains, each harboring two different alleles, enabling the combination as a heterozygote for each locus. Double-sgRNA CRISPR reagent targeted to both the alleles (CRISPR-sensitive loci, 1 & 2) initiates recombination at both loci using the homologous chromosome with CRISPR-resistant loci (i & ii) as a repair template, thus, enabling the simultaneous combination of 2 loci. CFUO/CFUE = ∼1 suggests viable combined genotypes. Sporulation of the resulting homozygous diploid yields the desired combined genotype in both mating types. (B) Double-sgRNA CRISPR reagent (pCas9-sgRNAScα3,α7) targeted to two wild type yeast loci (proteasome α3 and α7 genes) quantifies the efficiency of MERGE1. The transformation of pCas9-sgRNAScα3,α7 in the wild type, and single-humanized haploid Hsα3 or Hsα7 strains is lethal, with few surviving colonies (CFUO/CFUE = ∼0). However, the transformation of pCas9-sgRNAScα3,α7 in a heterozygous diploid humanized strain shows no lethality (CFUO/CFUE = ∼1). PCR-based genotyping of surviving diploid strains after MERGE1 shows conversion of both yeast to the humanized loci (ii) compared to before MERGE1 (i). Sporulation generated the paired-humanized haploids in both mating-types (shown here as Tetrads spotted on YPD (iii) and Mat α selection-iv). PCR check of haploid strains confirms the combination of humanized loci (vi). PCR of wild-type yeast loci is shown as a reference (v). (C) Alternatively, double-sgRNA CRISPR reagent (pCas9-sgRNAScα5,α7) targeting two wild type yeast loci (proteasome α5 and α7 genes) in a corresponding heterozygous diploid humanized strain showed lethality, suggesting incompatible combination (CFUO/CFUE = ∼0). The transformation of pCas9-sgRNAScα5,α7 in the wild type, and single-humanized Hsα5 or Hsα7 strains is lethal, with few surviving colonies, suggesting ON-target activity (CFUO/CFUE = ∼0). PCR-based genotyping of surviving colonies after MERGE1 shows the conversion of only one (α5) yeast to the humanized locus (ii) compared to before MERGE1 showing heterozygous genotype at both loci (i). (D) MERGE0 generated 7 humanized α proteasome strains in each mating-type. Mating each single-humanized strain allows a systematic test for every viable double-humanized genotype (21 combined genotypes). Transformation of double-sgRNA CRISPR reagent uniquely targeting yeast genes in each paired heterozygote facilitates the combination of two humanized loci as homozygotes (CFUO/CFUE = ∼1, yellow) while also revealing genotypes that are not permitted (CFUO/CFUE = ∼0, blue). (E) Mixing and mating singly humanized genotypes (Hsα1, Hsα3 and Hsα7) allows a random combination of two humanized alleles after double-sgRNA CRISPR selection referred to as mix, mate, and MERGE (MERGEM&M). Transformation of pCas9 alone serves as a control (i). Double-sgRNA CRISPR reagents, pCas9-sgRNAScα1,α7 (ii) and pCas9-sgRNAScα3,α7 (iii), specifically enriched the corresponding double-humanized genotype while also converting yeast to humanized loci. PCR-based genotyping of randomly picked colonies confirms Hsα1α7 (4 of 4 colonies tested) and Hsα3α7 (3 of 4 colonies tested). Comparatively, the transformation of double-sgRNA CRISPR reagent, pCas9-sgRNAScα1,α6 (iv), shows no surviving colonies as the genotype does not exist in the mixture.
To systematically determine if there are specific pairwise restrictions to the humanization of α-subunits, we mated all haploid single-humanized strains obtaining diploid heterozygous genotypes (21 different genotypes). The corresponding CRISPR-Cas9-based selection and a simple readout of CFUO/CFUE identified the permitted double-humanized genotypes (17/21 genotypes). The data reveals that only specific double-humanized genotypes are viable, whereas some are not (Figure 3D & S7C). The incompatibility of paired genotypes comprising Hsα1/α6, Hsα4/α6, Hsα5/α6, and Hsα5/α7 may likely be due to the missing neighboring interactions within the α proteasome core, except in the case of Hsα5,α6 pair (Figure S7C′). A sequential editing strategy successfully engineered some paired-humanized genotypes to confirm that MERGE1 represents viable/fit paired genotypes. However, it did not provide a clear perspective of incompatibilities (Figure S7D).
Given the success of MERGE1, we examined the strategy to select double-humanized genotypes randomly from a mixture (Mix, Mate & MERGE or MERGEM&M) (Figure 3E). We inoculated single-humanized α1, α3 and α7 haploid strains of both mating-types as a mix, allowing random mating. Each double-sgRNA CRISPR-Cas9 selection enriched the corresponding paired genotype from the mated mix while converting the wild-type yeast to humanized loci (Figure 3E-iii & 3E-iv). In mixed culture, the transformation of pCas9-sgRNASc-α1,α6, a selection for a non-existing genotype, does not yield any viable genotype (Figure 3F-ii). Therefore, MERGEM&M can be scaled to obtain several paired genotype combinations of engineered loci from a mix.
MERGE is scalable to combine multiple genetically engineered loci
The CRISPR-Cas9 system enables multiplexed editing by introducing multiple sgRNAs27. To test scalability, we designed MERGEMX (MERGE, mate and multiplex) to verify if >2 genetic loci can simultaneously convert to engineered loci by building the 4-gene carotenoid biosynthesis pathway from the carotenogenic yeast Xanthophyllomyces dendrorhous into Baker’s yeast (Figure 4A). The carotenoid pathway provides a colony color readout as a proxy for pathway engineering (Figure 4A). MERGE0 generated haploid strains of opposite mating-types for each carotenoid transcription unit (Figure S5C), MERGE1 provided the double-carotenoid genotypes (Figure 4B), and MERGEMX enriched a complete homozygous carotenoid pathway genotype (Figure 4C). Furthermore, genotyping of randomly picked dark orange colonies confirmed the conversion and combination of the engineered loci (Figure 4C).

(A) Schematic shows the carotenoid pathway genes and the metabolic intermediates leading to the color colony phenotype. A complete pathway (CrtE, CrtYB & CrtI) leads to an orange colony appearance, whereas the partial assembly (CrtYB & CrtI) provides an off-white colony phenotype. MERGE1 provided single-carotenoid engineered strains in both mating types. The homozygous diploid for a complete carotenoid pathway shows an intense orange colony phenotype than the heterozygous strain. (B) Schematic showing MERGE1 combining two engineered genotypes at landing pad loci. Double-sgRNA CRISPR reagent (pCas9-sgRNA511B,FGF20) targeting two landing pad loci (511B and FGF20) quantifies the efficiency of MERGE1. The transformation of pCas9-sgRNA511B,FGF20 in double heterozygous diploid strain shows no lethality (CFUO/CFUE = ∼1). PCR- based genotyping of several colonies after MERGE1 shows the conversion to the engineered carotenoid loci. (C) Single-carotenoid gene strains of both mating-types were mixed, mated (2X) and sporulated (1X). The mixture transformed with pCas9 alone shows an equal distribution of white, off-white and orange-colored colonies. However, the triple-sgRNA pCas9-sgRNA511B,FGF20,USERX transformed mix significantly enriched the yellow colonies. PCR-based genotyping of several colonies showed MERGEMX successfully homozygosed all loci, whereas the yellow colonies on the unselected plate showed heterozygous loci. (D) Schematic shows mating haploid triple-humanized Hsα1,α2,α3 strain with a wild-type yeast enables the combination of 3 humanized loci as heterozygotes. Triple-sgRNA CRISPR reagent (pCas9-sgRNAScα1,α2,α3) targeted to the corresponding wild-type yeast alleles (CRISPR-sensitive loci) simultaneously initiates recombination at all 3 loci using the homologous chromosome with humanized CRISPR- resistant loci as a repair template. The transformation of pCas9-sgRNAScα1,α2,α3 in the wild type is lethal, suggesting ON-target activity (CFUO/CFUE = ∼0). However, the transformation of pCas9-sgRNAScα1,α2,α3 in a diploid triple heterozygous humanized strain shows no lethality (CFUO/CFUE = ∼1). PCR-based genotyping of surviving colonies after MERGEMX confirms the conversion of all 3 yeast to the humanized loci compared to before MERGEMX. The transformation of a plasmid without sgRNA (pCas9 alone) serves as a transformation efficiency control (CFUE). (E) MERGEMX similarly combined >2 loci after mating a strain with 2 humanized loci (Hsα4,α7, Mat A) with a strain carrying 1 humanized locus (Hsα6, Mat α). The transformation of pCas9-sgRNAScα4,α6,α7 in the wild type is lethal, suggesting ON-target activity (CFUO/CFUE = ∼0). However, the transformation of pCas9-sgRNAScα4,α6,α7 in a mated mix selected a diploid triple-humanized strain (CFUO/CFUE = ∼1). PCR-based genotyping of several colonies after MERGEMX shows conversion of all yeast to the humanized loci.
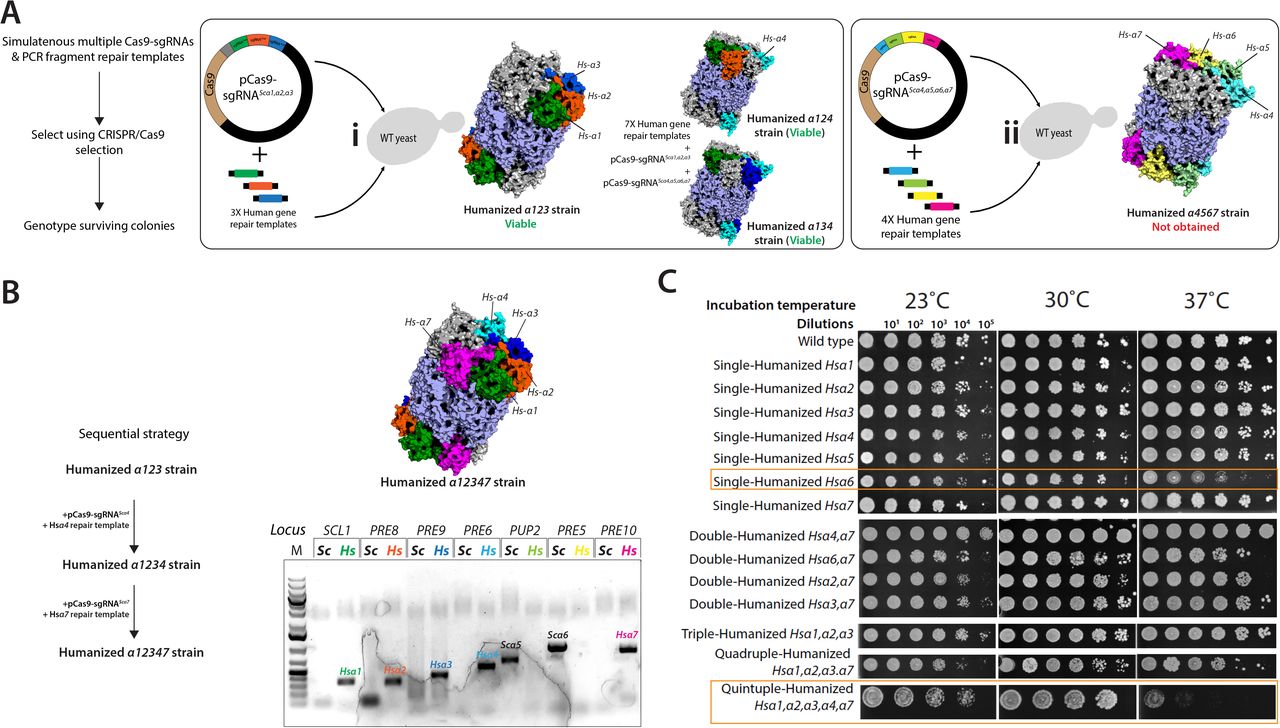
To test whether MERGEMX can perform the combination of >2 humanized α proteasome genotypes, we mated a previously obtained Hsα1α2α3 strain (MERGE1) with a wild-type type strain generating a heterozygous diploid for all three loci. The strain provided a platform to test MERGEMX by simultaneously combining 3 distinct humanized loci. The triple-sgRNA CRISPR reagent (pCas9-sgRNASc-α1,α2,α3) converted all yeast loci to human versions (7 of 9 colonies tested) (Figure 4D). Next, using MERGEMX, we tested if a triple-humanized Hsα4α6α7 genotype is viable. Transformation of triple-sgRNA, pCas9-sgRNASc-α4,α6,α7 in a mated mix of haploid Hsα4α7 (Mat A) and Hsα6 (Mat α) strains allowed simultaneous conversion of three wild-type yeast loci to humanized versions without using any diploid specific selection (Figure 4E & S11A). Thus, if a triple-humanized intermediate genotype is viable, MERGEMX can enrich and combine the specific genotype.
To further address throughput, we designed a CRISPR reagent targeting a GFP expression cassette in a GM strain17 (16 GFP loci, Figure S9A). The transformation of pCas9-sgRNAGFP resulted in a few survivors that failed to show GFP expression, likely due to mutations at GFP loci due to NHEJ (Figure S9B & S9C), suggesting successful targeting of the majority of GFP cassettes.

(A) The schematic shows a CRISPR-based strategy targeting one or many genetic loci in yeast. Transformation of a CRISPR reagent (pCas9-sgRNAGFP) targeting a GFP cassette in a Green Monster strain tests the scalability. Transformation in the wild type (no GFP) strain should show no lethality, whereas strains harboring single or multiple GFPs should show a lethal phenotype. (B) The pCas9-sgRNAGFP transformation in wild type yeast showed viable cells (CFUO/CFUE =∼1). In contrast, transformation in a strain harboring a single GFP cassette was lethal. While the Green Monster strain (GFP monster, 16 GFP cassettes) showed more viable cells than the single-GFP strain upon transformation with pCas9-sgRNAGFP. (C) Using flow cytometry, the pooled mixture of yeast cells that survive pCas9-sgRNAGFP showed significantly reduced GFP expression compared to the pCas9 alone transformed cells, suggesting that nearly every GFP cassette was successfully targeted and mutated via NHEJ.
MERGE enables fitness-driven engineering of a near entire human α proteasome core in yeast
To explore the proficiency of MERGE for testing many combinations of engineered loci, we asked if an entire yeast heptameric α proteasome ring is humanizable. As an alternate strategy, we also tested the feasibility of sequential engineering using repetitive CRISPR selections and exogenous human gene repair templates (Figure 5A). The co-transformation of a triple-sgRNA CRISPR reagent (pCas9-sgRNASc-α1,α2,α3) targeting yeast α1, α2 and α3 genes and PCR fragments of human gene repair templates was successful in obtaining a triple-humanized strain (Figure S10A-i). A similar strategy generated triple-humanized Hsα1α2α4 and Hsα1α3α4 strains but failed to obtain a quadruple-humanized Hsα4α5α6α7 (Figure S10A-ii). Thus, yeast genes are replaceable sequentially either alone or as small-scale simultaneous replacements. Using a triple-humanized genotype (Hsα1α2α3), the humanization of yeast α4 was successful. Next, we explored Hsα5, Hsα6 and Hsα7 humanizations in parallel; however, we obtained only one quadruple-humanized Hsα1α2α3α4α7 genotype (Figure 5A, Figure S10B). The functional replacement of yeast α5 or α6 was unsuccessful despite repeated attempts. The plasmid-borne expression Hsα6 in a quintuple-humanized strain (Hsα1α2α3α4α7) resulted in a toxic phenotype (no growth), suggesting that further humanizations are incompatible (Figure 5A). Overall, while the sequential strategy was partly successful, it failed to reveal if the inability to humanize yeast α core entirely was due to incompatible genotypes or inefficient genome editing, especially as hybrid human-yeast genotypes show growth defects and reduced transformation efficiencies (Figure S10C).

(A) Two multiple-sgRNA CRISPR plasmids were constructed, one expressing sgRNAs targeting yeast α subunits 1, 2, and 3 (pCas9-sgRNAScα1,α2,α3), and the other expressing sgRNAs targeting α subunits 4, 5, 6, and 7 (pCas9-sgRNAScα4,α5,α6,α7). They were transformed individually with the respective human gene repair DNA. A single colony with triple-humanized Hs-α1α2,α3 (i) was obtained. However, no clone for a quadruple humanized Hs-α4α5,α6,α7 (ii) could be obtained. A similar strategy also generated triple-humanized Hs-α1α2,α4 and Hs-α1α3,α4 genotypes. (B) Sequential strategy generated a quintuple-humanized Hs-α1α2,α3,α4,α7 strain confirmed via locus-specific PCR showing the presence of human and the absence of yeast loci. Sanger sequencing and whole-genome sequencing confirmed the genotype. (C) Spotted dilutions of double-, triple-, quadruple-, quintuple- and sextuple humanized α proteasome strains at 23°C, 30°C and 37°C show singe-humanized Hs-α6 (variant 1) and quintuple-humanized Hs- α1,α2,α3,α4,α7 strain with a temperature-sensitive growth phenotype.

The schematic shows the transition of heptameric yeast α proteasome to humanized α proteasome core. (A) A sequential strategy used a triple-humanized Hsα1,α2,α3 strain as a background to progressively humanize the rest of yeast α core genes by co-transforming a CRISPR reagent and a human gene repair template. Choices to sequentially humanize the α proteasome core were made depending on the success of the prior effort. The strategy permitted the humanization of yeast α7 followed by α4. Whereas several attempts to humanize yeast α5 and α6 failed. The expression of human Hsα6 in a quintuple-humanized Hsα1,α2α3,α4,α7 strain resulted in a lethal phenotype. (B) MERGE provided a clear readout of the fitness of combined genotypes while revealing incompatible combinations of the humanized yeast α proteasome core. Mating distinct humanized yeast combinations (show as connecting lines) followed by MERGEMX tested various triple-, quadruple-, quintuple- and sextuple-humanized α-proteasome yeast strains. MERGE generated viable triple-humanized combinations Hsα4,α5,α6 and Hsα4,α5,α7 (indicated as dashed lines in red), suggesting incompatible combinations. Similarly, while MERGE1 facilitated the generation of Hsα4,α5 and Hsα6,α7 genotypes, the subsequent MERGEMX to generate quadrupled Hsα4,α5,α6,α7 failed. Using MERGEMX followed by MERGE0 identified several quadruples, quintuples and one sextuple-humanized genotype (Hsα1,α2,α3,α4,α6,α7), revealing a fitness-driven path to the humanization of near entire yeast α-proteasome core. Whereas identifying humanized combinations (>2), comprising the Hsα5 subunit, as incompatible genotypes. Mating of Hsα1,α2,α3,α6,α7 and Hsα4,α5 strains generated a heterozygous human-yeast diploid for all 7 α proteasome core genes. Using multiplex-sgRNA CRISPR reagents permitted verifying if the entire human α-proteasome core is feasible. A triple-sgRNA CRISPR reagent (pCas9-sgRNASc-α1,α2,α3) homozygosed 3 of 7 humanized loci. However, a subsequent MERGEMX, using a quadruple-sgRNA CRISPR reagent (pCas9-sgRNASc-α4,α5,α6,α7), failed to obtain a viable genotype. Proteasome core structures were generated using Pymol and PDB- 1RYP. Colored structures show humanized α yeast subunits.
However, MERGEMX provided a clear readout of incompatible humanized genotypes, readily generating many combinations of humanized genotypes (Figure 5B). We first explored several triple-humanized genotypes, obtaining Hsα1α2α3 & Hsα4α6α7 strains (Figures 4D & 4E), while Hsα4α5α6 and Hsα4α5α7 genotypes are inviable (Figures 5B, S11B & S11C). By mating yeast strains with distinct humanized genotypes, we next explored many higher-order (>3) combinations, obtaining quadruple-humanized Hsα1α2α3α4 and Hsα1α2α3α7 genotypes. In comparison, the quadruple-humanized Hsα4α5α6α7 genotype is not feasible (Figure 5B, Figure S12A, S12B, S12C & S12D). Subsequent MERGE strategies generated viable quintuple-humanized genotypes, Hsα1α2α3α6α7 and Hsα1α2α3α4α6, whereas Hsα1α2α3α4α5 genotype is inviable (Figures 5B, S12E, S12F, & S12G). The following MERGEMX assay yielded a viable sextuple-humanized Hsα1α2α3α4α6α7 genotype with a delayed growth phenotype (Figure S11H). To conclusively verify if the entire yeast α proteasome core is humanizable, we mated partially humanized Hsα1α2α3α6α7 and Hsα4α5 strains, allowing the combination of all α proteasome genes as heterozygous human-yeast genotypes (Figure 13A). MERGEMX converted yeast α1,α2, and α3 loci to homozygous human alleles. However, the subsequent conversion of the remaining four yeast α4α5α6α7 to human loci using pCas9-sgRNASc-α4,α5,α6,α7 did not yield viable colonies suggesting a fully human α proteasome core is incompatible (Figure S13B). Overall, MERGE successfully tested many combined humanized genotypes. This gradual progression from yeast to humanized α proteasome core rescued the viability of specific incompatible double-humanized genotypes, suggesting that these subunits are co-humanizable when neighboring interactions are restored (Figures 3D & S14). The data reveals that the proteasome subunits α5 and α6 are not co-humanizable in yeast (Figure 5).

(A) MERGEMX (Mate and multiplex) strategy combined 3 loci after mating a strain with 2 humanized loci (Hsα4,α7, Mat A) with a strain carrying 1 humanized locus (Hsα6, Mat α) (biological replicate of data shown in Figure 4E). The transformation of pCas9-sgRNAScα4,α6,α7 in a mated mix selected a diploid triple-humanized strain (CFUO/CFUE = ∼1). PCR-based genotyping of several colonies after MERGEMX shows conversion of all yeast to the humanized loci (5 of 6). Spotted dilutions of humanized Hs-α4,α7 and Hs-α4,α6,α7 strains at 23°C, 30°C and 37°C show the triple-humanized strain with a temperature-sensitive phenotype at 37°C. The strain does not yield viable haploid spores observed as no growth on haploid-specific selection media (SD-LEU+Thialysine). (B) Double-humanized Hs-α4α5 (haploid Mat α) and single-humanized Hs-α6 (haploid Mat A, harboring a pCas9 alone or pCas9-sgRNASc-α6 vector) strains were mated, generating two distinct heterozygous diploid strains, one heterozygous for three loci and another for two with homozygous Hs-α6 locus. Each strain was transformed with double-sgRNA CRISPR plasmid (pCas9-sgRNAScα4,α5) to perform MERGE1. The transformation caused lethality in both strains (CFUO/CFUE =∼0). However, a few survivors in the context of heterozygous Hs-α6 locus showed the conversion of two loci (Hs-α4 and Hs-α7), suggesting an incompatible triple-humanized genotype but viable double-humanized genotype harboring a heterozygous yeast-human α6 locus. (C) Double-humanized haploid Hs-α4α5 (Mat α) and Hs-α7 (Mat A) strains were mated. The resulting heterozygous diploid strain was transformed with a triple-sgRNA CRISPR reagent (pCas9-sgRNAScα4,α5,α7). The transformation caused lethality with few survivors (CFUO/CFUE =∼0). Genotyping of the survivors showed the conversion of two 2 of 3 loci (Hs-α4 and Hs-α7), suggesting an incompatible triple-humanized genotype.

(A) Double-humanized haploid Hs-α4α5 (Mat α) and Hs-α6α7 (Mat α) strains were mated. The resulting heterozygous diploid strain was transformed with quadruple-sgRNA CRISPR reagent (pCas9-sgRNAScα4,α5,α6,α7) to perform MERGEMX. The transformation caused lethality with few survivors (CFUO/CFUE =∼0). The survivors showed the conversion of only 2 of 4 loci (Hs-α4 and Hs-α5), suggesting an incompatible quadruple-humanized genotype. However, the transformation of triple-sgRNA CRISPR reagent (pCas9-sgRNAScα4,α6,α7) in this strain was viable (CFUO/CFUE =∼1). The genotyping of randomly picked colonies showed the conversion of 3 yeast to human loci (Hs-α4, Hs-α6, and Hs-α7), suggesting a triple-humanized genotype is viable with a heterozygous yeast-human α5 locus. (B) Mating quintuple-humanized haploid Hs-α1α2α3α4α7 [Mat A, also harboring a triple-sgRNA CRISPR reagent (pCas9-sgRNAScα1,α2,α3)] and single-humanized Hs-α6 (Mat α) strains simultaneously combined three yeast to humanized Hsα1, Hsα2, Hsα3 loci in a diploid yeast (CFUO/CFUE =∼1). The strain harbors heterozygous yeast-human alleles for α4, α6, and α7 loci. Transformation of the diploid strain with (C) pCas9-sgRNAScα4 or (D) pCas9-sgRNAScα6 generated viable quadruple-humanized strains (Hs-α1α2α3α4 and Hs- α1α2α3α6) (CFUO/CFUE =∼1). Genotyping of randomly picked colonies confirmed the homozygosity of the loci. (E) Using the quadruple-humanized strain Hs-α1α2α3α4 as a background, the transformation of pCas9-sgRNAScα6 yielded a viable quintuple-humanized (Hs- α1α2α3α4α6) genotype (CFUO/CFUE =∼1) confirmed using locus-specific PCR. (F) Double-humanized haploid Hs-α6α7 (Mat α) and triple-humanized Hs-α1,α2,α3 [Mat A, also harboring a triple-sgRNA CRISPR reagent (pCas9-sgRNAScα1,α2,α3)] strains were mated. MERGEMX simultaneously combined three yeast to humanized Hsα1, Hsα2, and Hsα3 loci in the diploid background (CFUO/CFUE =∼1). The next transformation, using pCas9-sgRNAScα6,α7 also homozygosed the Hsα6 and Hsα7 loci, generating a quintuple-humanized strain (CFUO/CFUE =∼1). However, post sporulation, the mix plated on Mat α selection failed to produce any viable humanized haploid strains. (G) Triple-humanized Hs-α1α2α3 [haploid Mat A, also harboring a triple-sgRNA CRISPR reagent (pCas9-sgRNAScα1,α2,α3)] and double-humanized Hs-α4α5 (haploid Mat α) strains were mated generating a heterozygous diploid strain for 5 loci. The resulting diploid strain simultaneously combined three yeast to humanized Hsα1, Hsα2, and Hsα3 loci while harboring two yeast-human heterozygous loci (α4 and α5) (CFUO/CFUE =∼1). The subsequent MERGE0 strategy (using pCas9-sgRNAScα4) generated a viable quadruple-humanized strain (Hs-α1α2α3α4) with heterozygous yeast-human α5 locus (CFUO/CFUE =∼1). The locus-specific PCR confirmed the homozygosity for most of the colonies. However, the subsequent CRISPR selection (using pCas9-sgRNAScα5) did not produce a viable strain (CFUO/CFUE =∼0), suggesting an incompatible quintuple-humanized (Hs-α1α2α3α4α5) genotype. (H) Using the quintuple-humanized diploid strain Hs-α1α2α3α4α6 harboring a yeast-human heterozygous α7 locus as a background, the transformation of pCas9-sgRNAScα7 yielded a viable sextuple-humanized (Hs-α1α2α3α4α6α7) genotype (MERGE0, CFUO/CFUE =∼1) after incubation for 8 days. The genotype of the viable colonies confirmed the homozygosity and humanization of the α7 locus.

(A) Mating quintuple-humanized haploid Hs-α1α2α3α6α7 (Mat α) and double-humanized Hs-α4α5 (Mat A) strains generated a diploid strain harboring yeast-human heterozygous alleles for 7 α-proteasome loci. Locus-specific PCR confirmed the heterozygous nature of the loci. (B) The resulting diploid strain was transformed with a triple-sgRNA CRISPR reagent (pCas9-sgRNAScα1,α2,,α3), resulting in a viable genotype homozygous for triple-humanized Hs-α1α2α3 loci (CFUO/CFUE =∼1) with yeast-human heterozygous alleles for α4, α5, α6, and α7 loci. The subsequent transformation of a quadruple-sgRNA CRISPR reagent (pCas9-sgRNAScα4,α5,α6,α7) in the diploid strain was lethal, suggesting an incompatible genotype (CFUO/CFUE =∼0).
Proteasome biogenesis is a highly regulated process aided by several assembly chaperones28–30. In the case of α proteasome core, particularly α5 and α6, subunits interact with assembly chaperones, enabling ordered assembly31–33. Furthermore, the β core assembly immediately follows the α core, and the incompatible interface may now require human β subunits in yeast28, 34. The heterologous expression of human assembly chaperones or human β subunits in a humanized α core strain may permit the synthesis of a fully human catalytic core particle in yeast. Given the complex assembly and architecture of the proteasome35, it is challenging to know if there are a limited number of ‘paths’ to engineer a fully-humanized 20S core particle, due to a rapidly accumulating number of assays to perform (as in Figure S1).
The sporulation failure observed in genomically-replaced strains can lead to dead ends while performing MERGE (associated with all humanized Hsα7 genotypes, Figures S11A, S12A & S12F). We propose two solutions: One, by allowing the strains with heterozygous engineered loci to sporulate without MERGE, followed by using CELECT to enrich combined haploid genotypes (Figure S15A). Alternatively, a sequential strategy can engineer viable genotypes in a haploid strain (Figures S12F & S15B). Furthermore, using CRISPR reagents to generate multiple DSBs at several yeast loci could potentially have OFF-target effects. Therefore, we performed whole-genome sequencing of singly humanized (Hs-α1) and quintuple-humanized strains (Hsα1α2α3α4α7), ruling out OFF-target DSBs and mutations (Figure S16).

Inviable double-humanized genotypes, Hsα1/α6 and Hsα4/α6, become viable when the neighboring interactions are restored in higher-order humanized strains (indicated as green lines). However, incompatible paired genotypes comprising Hsα5/α6 and Hsα5/α7 could not be rescued (marked as red lines). Proteasome core structures were generated using Pymol and PDB-1RYP. Colored structures show humanized α yeast subunits.

(A) Mating single-humanized Hs-α6 (Mat α, with pCas9 alone or pCas-sgRNASc-α6) and Hs-α7 (Mat A) strains generated two diploid strains; one harboring yeast-human heterozygous allele at α6 locus and another with homozygous humanized α6 locus. MERGE0(using strategy pCas-sgRNASc-α7) converted the heterozygous yeast-human α7 locus to a homozygous human allele at ∼100% efficiency (CFUO/CFUE =∼1). Locus-specific PCR confirmed the humanized homozygous alleles. However, the diploid strains cannot sporulate. However, keeping the α7 locus as heterozygous rescues the sporulation defect. The haploid-specific selection (SD-HIS+CAN; Mat A) allowed haploid yeast to grow, followed by CELECT (using pCas-sgRNASc-α7) or simply genotyping the haploids to identify homozygous double-humanized Hs-α6α7 genotype. (B) The diploid quintuple-humanized Hs-α1α2α3α6α7 strain shows a sporulation defect as in Figure S12F. However, using a sequential CRISPR strategy, a haploid quadruple-humanized Hs-α1α2α3α6 strain enables the generation of haploid quintuple-humanized Hs-α1α2α3α6α7 strain to be used for subsequent MERGE experiments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
WGS analysis of quintupled-humanized Hs-α1α2α3α4α7 shows humanized loci aligned to a reference S288C genome with replaced human coding sequences. The human genes reside on different yeast chromosomes, replacing their corresponding native yeast loci (Chr. 7, 13, and 15). Green lines indicate the read lengths with a 200 base range, yellow is below, and blue is above the range.
Conclusions
In conclusion, MERGE offers highly scalable multi-locus genome engineering in diploid yeast cells by using a high-efficiency CRISPR-based gene-drive-like strategy to overcome the independent assortment of unlinked loci. MERGE has the potential to allow systematic functional genomic analysis in other systems lacking sophisticated tools and drive synthetic and systems biology research from engineering heterologous systems to performing multi-site and genome-wide combinatorial editing in yeast, as we demonstrated by engineering 89 independent sites along Chromosome 1, a complete four gene carotenoid biosynthesis pathway, 16 GFP insertions within the same strain, and a majority of the α proteasome genes. By engineering 6 of 7 human α proteasome core genes in yeast, our work also demonstrates the remarkable degree of functional conservation in the proteasome complex despite over a billion years of evolutionary divergence, extending from a single gene to nearly an entire module. The data confirm our previous observations that humanization seems to be driven by modules of physically or functionally interacting proteins being similarly replaceable24. Further characterization of the incompatibilities should reveal novel orthogonal functions or interactions in diverged species. However, pursuing a combinatorial strategy with MERGE along with a sequential strategy in parallel allows one to inform the other about simultaneous replacements that are likely to work. Humanizing all or multiple members of a protein complex will allow a novel approach to learning human biology, including complex assembly, biogenesis, and variant effects on function, investigations of their contributions to disease, and the possibility of seeking therapies for these diseases in the simplified context of a yeast cell.
Funding
This research was funded by grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery grant) [RGPIN-2018-05089], CRC Tier 2 [NSERC/CRSNG-950-231904], Canada Foundation for Innovation and Québec Ministère de l’Économie, de la Science et de l’Innovation (#37415), and FRQNT Research Support for New Academics to A.H.K., fellowship support from School of Graduate Studies (SGS), Faculty of Arts and Sciences, Concordia University to M.A., B.M.G and SynBioApps program sponsored by NSERC-CREATE to M.V. E.M.M. acknowledges support from the Welch Foundation (F-1515), Army Research Office (W911NF-12-1-0390), and National Institutes of Health (R35 GM122480). The authors declare no competing interests.
Contributions
M.A., B.M.G., J.M.L., A.H.K., and E.M.M. designed the research. M.A., B.M.G., J.M.L., and M.V. performed the experiments. A.H.K. and E.M.M. supervised the research. M.A., B.M.G. A.H.K., and E.M.M. wrote the manuscript with editing help from J.M.L., and M.V.
Abbreviations
- CFU
- Colony Forming Units
- DSB
- Double-Strand Break
- HDR
- Homology-directed DNA Repair
- HR
- Homologous Recombination
- CELECT
- CRISPR-Cas9-based selection to enrich genotypes
- MERGE
- Marker-less Enrichment and Recombination of Genetically Engineered loci
- SGA
- Synthetic Genetic Array
Materials & methods
1. Targeting sgRNA design
CRISPR-Cas9 targeting sequences consist of a 20 bp recognition sequence preceding an ‘NGG’ sequence motif (PAM). To design our targeting sequences, we used the built-in guide RNA design tool in version 11 of the Geneious software36, using a recent version of the yeast genome (available from http://www.yeastgenome.org/strain/S288C/overview) to screen for possible off-target sequences13, 14. We chose two high-scoring guide sequences to target each gene, requiring that they be near the 5’ end of the gene so any NHEJ repair would likely result in an early frameshift. The guides were ordered as complementary oligos with overhangs according to the sgRNA template described in the Yeast Tool Kit15 (Table S1).
2. Plasmid construction using Yeast Tool Kit (YTK)
Entry vectors for each guide RNA sequence were constructed by the BsmbI-mediated Golden Gate (GG) assembly (Thermo) into plasmid pYTK050 from the Yeast ToolKit15. The targeting sequences were ordered (IDT) as two single-stranded DNA oligos with a complementary region and unique overhangs according to the ‘sgRNA’ template described in Lee, ME et al15. Briefly, the two oligos for each guide were annealed by slow cooling from 95°C to 4°C (1-5 hrs) in a thermal cycler, and 1μl of the annealed product was added to the entry vector GG reaction. Entry Vectors were sequence-verified using custom primers.‘Transcriptional unit’ (TU) vectors (pYTK095) were constructed by a BsaI-mediated GG assembly (Thermo). The entry vector for a particular guide RNA was combined with a left connector part plasmid (ConLX) and right connector part plasmid (ConRX) into the AmpR-ColE1 (pYTK095) backbone plasmid13, 14. To create single-sgRNA TU vectors, we assembled the sgRNA entry vector with left connector 1 (ConL1, pYTK003) and right connector E (ConRE, pYTK072). To create TU vectors for the multi-sgRNA knockout, an entry vector was combined with the appropriate connectors to enable proper subsequence knockout vector assembly (i.e. a triple-sgRNA vector would have one TU vector with ConL1 and R2, one with ConL2 and R2, and the third with ConL3 and RE). ‘Knockout’ (KO) vectors were constructed by BsmbI-mediated GG assembly (Thermo). The appropriate TU vectors were assembled into the CEN6-URA3 backbone and the Cas9-TU1 vector to create a KO vector. Each GG reaction was performed in a 10μl volume, with approximately 20 fmol of each starting DNA molecule, along with 0.5μl each of the appropriate restriction enzyme, 0.5μl of T7 DNA ligase (NEB), and 1μl of 10X T4 ligase buffer (NEB). A thermocycler was used to cycle between 16°C and 37°C each for (5 minutes). 3μl of the reaction volume was transformed into competent DH5α E. coli and plated on appropriate selective media for the backbone (i.e. chloramphenicol for entry vectors, ampicillin for TU vectors, and kanamycin for KO vectors). Each backbone plasmid is a ‘GFP-dropout’ vector, so correct clones were selected by screening for non-fluorescent colonies when viewed by blue light and verified by sequencing.
3. Cas9-TU1
The Cas9-TU1 vector was constructed by BsaI GG assembly of YTK parts pYTK002 (ConLS), 011 (PGK1 promoter), 036 (Cas9 coding sequence), 055 (ENO2 terminator, 067 (ConR1), and 095 (AmpR-ColE1 GFP dropout backbone).
4. CEN6-URA3 expression vector
The CEN6-URA GFP dropout backbone vector was constructed by BsaI GG assembly of YTK parts pYTK008 (ConLS’), pYTK047 (GFP dropout), pYTK073 (ConLE’), pYTK074 (URA3), pYTK081 (CEN6/ARS4), pYTK084 (KanR-ColE1 RFP dropout vector).
5. An alternate strategy to directly clone annealed sgRNAs in a yeast expression vector
To perform faster direct sgRNA-Cas9 plasmid construction without requiring three independent cloning strategies (as described in section 2), we constructed pCas9-GFPdo to directly clone the annealed sgRNA primers into yeast expression vectors. The sgRNA expression unit from the pYTK050 plasmid was amplified via PCR using primers with BsaI sites that generated overhangs similar to ConL1 (forward primer) and ConRE (reverse primer) to clone in CEN6-URA or CEN6-KanMX yeast expression vectors. Finally, using the GG reaction protocol, the sgRNA TU PCR was assembled into the CEN6-URA3 or CEN6-KanMX backbone with the Cas9-TU1 vector to create a direct knockout (pCas9-GFPdo) vector.
6. An alternate strategy to directly clone multiple sgRNAs TUs in a yeast expression vector for multiplexing in MERGEMX
The YTK kit allows cloning multiple sgRNA expression cassettes in a yeast expression vector. However, the strategy involves three cloning steps. To build >1 sgRNAs TUs directly into the yeast expression vector without generating intermediate connector vectors, we designed the pCas9-GFPMX (CEN6-URA3) vector. Primers were designed to PCR amplify the GFP expression cassette (for E. coli expression, pYTK047) with GG enzyme sites to clone GFP (BsaI for cloning the GFP insert, the sites are lost after GG cloning). The BsmBI sites, with overhangs similar to ConLS and ConRE, designed in the primers, allow the PCR-based sgRNA TU cloning (obtained by ligating annealed sgRNA primers in pYTK050) in CEN6-URA3 yeast expression vector (Table S1). The primers used to amplify the sgRNA expression units now harbor connector overhangs to clone >1 sgRNA expression units in tandem, eliminating the need to individually make each connector clone.
7. Repair design and construction
Repair DNA was designed to be a linear DNA molecule that contained the human coding sequence, from the start codon to stop, with at least 100bp of flanking homology to the yeast genome immediately upstream and downstream of the native yeast start and stop codons (Figure S3B). They were constructed by PCR using human ORFeome37 or MGC38 clones as templates, using primers with long extensions providing the homology sequence. Repair template PCRs were performed with Accuprime Pfx (Thermo) as multiple 100μl reactions according to the manufacturer’s protocol, combined and purified using the Zymo DNA Clean or Qiagen PCR Cleanup and Concentrator −25 kit. Approximately 5μg of repair DNA was included in the transformation mix during sequential replacement transformations.
8. Humanization procedure
Yeast transformations were performed using the Frozen-EZ Yeast Transformation II Kit from Zymo Research (Cedarlane). In short, cells were grown to mid-log phase, washed with kit Solution 1, resuspended in kit Solution 2 and either used directly to transform or frozen at −80 C for future use. In general, we transformed approximately 1μg of the KO plasmid along with 5μg of repair DNA, which yielded anywhere from single to dozens of colonies, depending on transformation efficiency and the number of simultaneous molecules necessary. In general, nearly all screened clones were successful. For initial single replacement in the wild-type strains, we tested two sgRNA sequences for each gene. These were tested in parallel, together with a no-repair control transformation to assay for sgRNA effectiveness. Successful sgRNAs show zero or very few colonies in the no-repair control transformation (CFUO/CFUE =∼0, Figure 1A & S4A).
9. Engineering Carotenoid genes at the landing pad loci
Single-sgRNA CRISPR reagents targeting three landing pad loci (511B, USERX, FGF20)39 were generated as described in section 1. The repair templates were generated as previously described in section 7. Specifically, carotenoid gene transcription units were amplified from plasmids harboring the transcription units using the primers with 80bp homology to the landing pad loci40 (Table S1). Each landing pad locus was edited in the SGA strain (Mat A) to harbor a carotenoid gene transcription unit (511B for CrtE, USERX for CrtI, and FGF20 for CrtYB) driven by strong constitutive yeast promoters. The transformation was carried out using the Zymogen frozen EZ yeast transformation kit (Cedarlane), and colonies were confirmed by PCR using primers specific to the landing pad or the carotenoid gene loci as described below (Figure S4B & Table S1).
10. Clone verification
Clones were initially screened by colony PCR using a rapid DNA isolation method and colony PCR41. Forward primers for PCR screening were designed such that the upstream primer would bind in the yeast genome approximately 150bp - 500bp upstream of the yeast ORF or the landing pad loci, to ensure it was outside of the homology region used for repair. Two reverse primers were designed for each locus, one binding into the yeast and the other within the human or carotenoid gene sequence. To facilitate multiplex PCR screening, each pair amplifies different-sized bands for the yeast and human or carotenoid genes. Following plasmid loss, clones were further verified by directly sequencing a PCR product using Sanger sequencing.
11. Plasmid loss to perform sequential editing of loci
Successful clones were subjected to a plasmid loss procedure to alleviate any stress incurred by the constitutive expression of the Cas9 protein and allow further knockout plasmids to be transformed. To avoid any potential stress-induced defects, we rarely used 5-FOA counter-selection to force loss of the URA3 plasmid, instead relying on spontaneous plasmid loss. One two methods were used to identify spontaneous plasmid loss. Clones were grown overnight in YPD and then again either spread onto YPD (100μl of 1:1000 dilution) and replica plated on SC-Ura and YPD or patch plated (typically 6-12 colonies) to both minimal media lacking uracil and YPD. This procedure resulted in around 10-60% of colonies losing the plasmid (estimated). For a plasmid harboring URA3 selection, the strains that lost the plasmid were identified using 5-FOA selection.
12. Growth curves
Cells were diluted to approximately 0.01-0.02 OD660 (∼2-5 x105 cells) in 150μl YPD across 3-4 replicates. They were grown and read in a BioTek Synergy H1 or Sunrise Tecan microplate reader, with continuous double orbital shaking at 30℃ or 37℃. Reads were taken while stationary every 10-20 minutes, and experiments were run for at least 24-36 hours. For spot assays, strains were grown overnight in YPD at 30℃ and spotted with serial dilutions on YPD agar. The plates were incubated for 2-3 days at 23℃, 30℃ and 37℃.
13. CRISPR-based selection (CELECT) to identify and enrich unique genotypes
To test the efficiency of CRISPR selection, a subset of engineered strains (Haploid Mat A) were mixed as a pool. Each strain was cultured overnight in YDP individually and back-diluted into a pool the next day at equal OD and grown to mid-log phase. The strains used in the mix were all of the same mating types to avoid random mating events during the experiment. Competent cells were prepared and transformed with various pCas9-sgRNAlocus plasmids. We performed the experiments as biological and technical replicates using different genotypes in the mix. The same competent cell mix was either transformed with pCas9 alone or with various pCas9-sgRNAWT locus vectors, each harboring a different yeast-specific sgRNA expression unit. Competent cells were generated using Zymo Research Frozen EZ kit transformation protocol. Each transformation was plated on SD-URA. pCas9 alone served as a positive control allowing the growth of all genotypes in the mixture. pCas9-sgRNAADE2 was used to visually observe and quantify the efficiency of the selection. The plasmid will eliminate all the other genotypes except the ade2Δ::kanMX (red colonies due to the disruption of the Ade2 gene). pCas9-sgRNARPT5was used as a negative control as the yeast Rpt5 gene is present in all the strains such that yeast cells should survive this transformation. Colonies from each Petri plate were picked randomly and genotyped by locus-specific PCR to confirm the CRISPR reagent exclusively selected a unique genotype as described in section 10. (Figure 1C).
14. Mating and MERGE0 assays
The mating assays were carried out in the following ways:
14.1. The engineered strains with single gene modification in BY4741 (Haploid Mat A) backgrounds were mated with SGA-haploid strains harboring haploid specific markers (SGA Mat A; can1Δ::STE2pr_Sp_his5his3Δ1leu2Δ0ura3Δ0; select on SD-His+Canavanine & SGA Mat α; lyp1Δ::STE3pr_LEU2 his3Δ1leu2Δ0ura3Δ0; select on SD-Leu+Thialysine). Each parental strain was transformed with two different empty vector plasmids with distinct selectable markers URA3 and KanMX, and mated diploids were selected on SD-Ura+G418. The haploid strains were grown overnight in 5ml selection media (SC-Ura or YPD +G418). The strains were mixed in a rich medium and incubated at 30°C for 3-4 hrs. with shaking. 500µl of the mated mixture was washed with distilled water, and 10µl of the mix was plated at different dilutions on solid agar media using SD-URA+G418 selection and incubated for 2-3 days at 30°C. Plasmids were cured using a previously described strategy in section 11. The heterozygous colonies were confirmed by PCR genotyping using primers specific to the wild-type and engineered loci as described in section 10. The confirmed heterozygous strains for all proteasome and carotenoid strains were transformed with CRISPR plasmids targeting wild-type loci and converting the parent heterozygous strain into homozygous diploid strain. Colonies were picked randomly from the plate and genotyped for homozygosity using the same set of primers, confirming the loss of yeast and the presence of humanized or carotenoid loci.
14.2. Alternatively, instead of an empty vector, the engineered humanized or carotenoid strains were transformed with a CRISPR reagent for which the corresponding strain harbors a CRISPR-resistant locus. The subsequent mating with an opposite mating-type strain with an empty vector (pCas9 alone or a backbone plasmid) results in the loss of heterozygosity and conversion of single loci (MERGE0). In each scenario, the number of colonies obtained on plates transformed with CRISPR reagent (CFUO) were compared with empty vector transformations (CFUE) to calculate the efficiency of MERGE0. Each experiment was performed at least 3 times.
15. Sporulation of diploid and selection of haploid yeast strains
Diploid strains were sporulated in a media containing 0.1% potassium acetate and 0.005% zinc acetate for 4-7 days. For Tetrad dissection, spores were spun down at 5000rpm for 5 minutes, resuspended in 200μl of 20mg/ml Zymolyase and incubated at 37°C for 25-30 minutes. The mix was then incubated at −20°C to stop the reaction. Cells were thawed on ice, and 20ul of the mix was plated into YDP for Tetrad dissection. Tetrad Dissection was performed using Spore play (Singer Instruments for every engineered proteasome spore on YPD and then replicated on SGA selection. For carotenoid and humanized loci in SGA background strain, the sporulation mix was directly plated on SGA selection. Locus-specific PCR verified the haploid strains for the presence or absence of engineered loci.
16. Estimating the efficiency of MERGE0 using yeast ADE2 locus as a readout
The yeast ADE2 locus was used to estimate MERGE0 using a CRISPR-resistant allele ade2Δ::kanMX on the homologous chromosome as a repair template. The sgRNAs were designed using Geneious version 11 as described in section 1 (Table S1). The pCas9-sgRNAADE2 vector showed ON-target activity causing lethality in wild-type haploid or diploid strains. Occasional colonies show a red colony phenotype suggesting mutations via NHEJ at the ADE2 locus. To estimate MERGE0 efficiency, the pCas9-sgRNAADE2 was transformed in a diploid hetKO ADE2/ade2Δ::kanMX obtained from the yeast “Magic Marker’’ hetKO collection26. To further verify that the red colonies observed after MERGE0 are not due to the mutations in the ADE2 locus (NHEJ) and instead due to conversion to ade2Δ::kanMX locus (HDR), the heterozygous diploid (ADE2/ade2Δ::kanMX) strain transformed with a control (pCas9 alone) and pCas9-sgRNAADE2 plasmid were sporulated followed by tetrad dissection (Figure S5A). Haploid spores were selected on YPD or YPD + G418 (200µg/ml). Spores harboring a control plasmid showed the expected 2 : 2 (red : white phenotype) and 2 : 0 (G418 resistant, G418 sensitive) phenotype compared to the pCas9-sgRNAADE2 transformed cells that show 4 : 0 (red, white) colonies and 4 : 0 (G418 resistant, G418 sensitive) colonies phenotype, respectively, suggesting the conversion to ade2Δ::kanMX rather than the mutation of Ade2 locus. In contrast, the co-transformation of pCas9-sgRNAADE2 and oligo as a repair template harboring (100bp homology, 5X molar excess than the plasmid) to the 5’ and 3’ UTRs of ADE2 locus in haploid wild-type yeast cells, shows resulted in significantly fewer survivors (Figure 2C; % CFUO/CFUE= 21.6+/-SD; N=4). However, this method is still far less-efficient than MERGE0(Figure 2C; ∼100% vs 21.6%).
17. Using MERGE0 to perform one-step gene essentiality assays in yeast
HetKO diploid strains for 7 α proteasome core genes were obtained from the yeast “Magic Marker’’ hetKO collection26. The strains were transformed with either a single-sgRNA CRISPR reagent targeting the corresponding yeast α proteasome genes or the empty vector control and selected on SD-URA+G418. CRISPR reagent transformed plates caused lethality in 6 of 7 α proteasome hetKO strains, except in the case of non-essential α3. A similar assay in the hetKO ADE2/ade2Δ::kanMX strain showed viable homozygous null cells for a non-essential ADE2 locus.
18. Testing MERGE0 efficiency for all yeast loci on Chromosome I
All the strains harboring heterozygous knockout diploid loci from the yeast “Magic Marker” collection on chromosome I were arrayed in a 96-well format26 (Figure S6A). A2 is the locus close to the left telomere, F1 near the centromere, and G10 close to the right telomere (Figures 2D & S6A). The sgRNA targeting the KanMX cassette was designed using the strategy mentioned earlier (Table S1). Since the bacterial selection for the previously described (section 4) yeast shuttle vector harbors a KanR cassette identical to the KanMX cassette in the case of hetKO strains, we designed a new expression vector with AmpR selection. The plasmid showed a lethal phenotype in the strains harboring the KanMX allele (haploid or homozygous diploid) in the genome (Figure 2D). Approximately 1µg of pCas-sgRNAKanMX was transformed for each transformation.
First, we performed a pilot assay using 5 hetKO strains with genes located at various regions on chromosome I (Figure S6B). These strains showed no lethality (CFUo/CFUE of ∼1). Each strain was inoculated in 800µl of YDP + His(50 mg/L) + G418 (200 µg/ml) overnight in 96-well format. The following day, cultures were back-diluted and grown to the mid-log phase. Competent cells were generated in a 96-well format using the Gietz yeast transformation protocol (Gietz, 2007). Each transformation mix was transformed with either 1µg of pCas9 alone or 1µg of the pCas9-sgRNAKanMx plasmid. An equal amount of the transformed cells were plated onto SD-URA and SD-URA + G418. All of the yeast loci (except for A10- CNE1) on chromosome I show MERGE0 mediated conversion of KanMX to wild-type yeast locus irrespective of the position of the gene along the chromosome (CFUO/CFUE =∼1) and simultaneously lost the KanMX cassette (Figure S6A).
19. Performing MERGE1 and MERGEMX at 2 or more loci using humanized proteasome and carotenoid strains
CRISPR plasmids expressing 2 or more sgRNA cassettes in tandem were designed and constructed as described in sections 1 - 6. Each single-humanized α proteasome (BY4741, Mat A) was mated with SGA strain (Mat α) as described in section 14. Heterozygous strains from each mating mix were confirmed by locus-specific PCR genotyping as in section 10. MERGE0 enabled the conversion to humanized loci. The next round of mating in SGA strain (Mat A) followed by MERGE0 generated humanized strains in both mating-type SGA backgrounds. Carotenoid strains were directly generated using SGA compatible strains described in section 9, followed by MERGE0 to obtain strains in both mating-types.
Each double-sgRNA CRISPR reagent was lethal in singly engineered strains (Figure S7B). Mating between each distinct single-engineered genotype combines the loci as heterozygotes. Transformation of 0.5-1µg of double-sgRNA CRISPR reagent in heterozygous diploid strains either showed no lethality (viable genotype, CFUO/CFUE =∼1) or a lethal phenotype (inviable genotype, CFUO/CFUE =∼0) (Figures 3B, 3C &S7C). Homozygosity was confirmed by randomly picking colonies after MERGE1 and performing a locus-specific PCR confirming the presence or absence of yeast or engineered genotype.
For MERGEMX to simultaneously target >2 loci, the CRISPR reagents were generated as described in sections 2-4 and 6. The haploid strains harboring combined engineering loci were mated with either a wild-type or strains carrying a different set of engineered loci. The diploids were transformed with either an empty vector or a multiplex-sgRNA CRISPR reagent. The genotypes were confirmed by locus-specific PCR as described in section 10.
20. Performing MERGEM&M to combine 2 loci from the mated mix of single-humanized proteasome genes
Single-humanized α proteasome strains Hsα1, Hsα3, and Hsα7 of both mating-types were allowed to mate randomly in a mix. Each strain was inoculated at 0.3 OD in 2 ml YPD overnight, then added to a mix at equal OD and incubated on a shaking incubator for 4-6 hrs 30°C. 500µl of the mix was further cultured overnight at starting 0.3 OD, followed by competent cell preparation. The mixture was transformed with either an empty vector or double-sgRNA CRISPR reagent. Several colonies were observed post MERGEM&M for Hsα1α7, and Hsα3α7 paired genotypes. PCR-based genotyping confirmed the combination as a homozygous double-humanized diploid.
21. Testing the potential of CRISPR reagent to target multiple loci using the Green Monster strain
To address the scalability of targeting multiple yeast loci, we used the Green Monster (GM) strain17. The sgRNA target sequence was designed in GFP (S65T) variant using Geneious version 11 as described in section 1. pCas9-sgRNAGFP was constructed using the method of direct cloning strategy as described in section 5 using a backbone with AmpR (E. coli) and KanMX (yeast) selection. The Mat α mating-type Green Monster was used MATα lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 can1Δ::GMToolkit-α [CMVpr-rtTA NatMX4 STE3pr-LEU2]. The OFF-target activity of pCas9-sgRNAGFP was tested in a haploid WT strain with no GFP gene showing a CFUo/CFUE =∼ 1. The ON-target activity was measured transformation in natively tagged C-terminal fusion of GFP to BRO1 gene (Bro1-GFP) obtained from the yeast GFP collection42.
To estimate the loss-of-function mutations at the GFP locus, cells from an empty vector and pCas9-sgRNAGFP transformed GM yeast cells were subjected to flow cytometry (BD Accuri™ C6 Plus Flow Cytometer). Cells were grown in 96-well format in YDP overnight and back diluted in SC media with 10µg/ml of doxycycline for ∼48 hrs. The culture was diluted 1 in 10 in water and passed through the flow cytometer. Using BY4741 yeast with no GFP as a control, the cells showed mostly background fluorescence as expected. GM cells transformed with an empty vector show high fluorescence as this strain harbors 16 GFP cassettes serving as a positive control. Finally, random colonies were picked and pooled from haploid GM cells transformed with pCas-sgRNAGFP, and the mixture passed through the flow cytometer. The cells show little GFP expression similar to the wild type, suggesting ON-target NHEJ-mediated mutations at nearly all GFP loci (Figures S8).
22. Performing MERGEMX to assemble an entire Carotenoid pathway in yeast using mating and sporulation cycles
SGA strains of both mating-types harboring single-carotenoid transcription units at the landing pad loci were generated in sections 9 and 14. The strains were cultured independently overnight, followed by mixing at a similar 0.3 OD and cultured on a shaking incubator for 5-6 hours. A hole was made on the lid of the Eppendorf tube to allow aeration, and the mixture was incubated at 30℃ for 12-16 hrs. The formation of diploids was confirmed by light microscopy. Next, 500µl of culture was centrifuged at 3500 rpm for 5 minutes. The supernatant was removed and resuspended in 2 - 5ml of sporulation media, followed by incubation on a rotating shaker for 5-8 days at room temperature. Next, 500 ul of sporulation mix was centrifuged at 3500 rpm for 5 min and treated with Zymolyase. After the first sexual cycle, the cells were centrifuged, washed with water, and resuspended in rich media YPD. The cycle was repeated to generate a second set of diploids. The mixture was incubated for 4-6 hours in a shaking incubator and incubated at 30℃ for 2 days. The confirmation and appearance of most diploids were again confirmed with a light microscope, and the 100µl of culture was inoculated overnight to make competent cells. The competent cells were transformed with either an empty vector or a triple-sgRNA CRISPR reagent pCas9-sgRNA511B,USERX,FGF20. For the most part, we followed a GFP monster protocol. However, we did not use haploid or diploid-specific selection43. Instead, CRISPR selection was sufficient to enrich unique combined genotypes.
23. Genomic DNA isolation & Whole Genome sequencing, and SNP analysis
Genomic DNA was purified from single-humanized Hsα1 and quintuple-humanized Hsα1,α2,α3,α4,α7 strains using the Monarch Nucleic acid purification kit according to the manufacturer’s protocol (NEB). Spheroplasts were obtained before the genomic DNA extraction for a high-quality DNA prep. Wild type and engineered strains were sequenced using Illumina MiSeq 2×150 at 30x coverage using 150-bp paired-end reads. The Geneious Pro Software [PMID: 22543367] and its included tools were utilized for pairing paired-end sequences, trimming ends and adapters based on quality using the BBDuk tool, and reference mapping using the Geneious Read Mapper algorithm at medium-low sensitivity, iterated 5 times. Reads were mapped to a reference BY4741 strain (for the engineered strain, the same reference sequence was used, but replacing the sequences of the 5 engineered genes with their human ortholog to ensure alignment with the humanized loci reads) using the Geneious Prime Software 2020.2.4 algorithm. Mapping was run at medium sensitivity: word length to allow for matching was set to 18 with a maximum permitted mismatch of 20% of the read length, maximum mismatches per read were set to 20%, a minimum 80% overlap was required, and reads with errors were set to accurately be mapped to repeat regions - this was iterated 5 times to give the final mapping. For analysis, low coverage regions of below 2 standard deviations from the mean were excluded from SNP-calling, and only SNPs with a variant frequency of 0.90 or higher were considered. SNPs were called with a minimum variant frequency of 0.25, and low coverage regions below 2 standard deviations from the mean were excluded. SNPs that were unique to the engineered strain (i.e. not in the mappings of the wild-type strain) were marked. To ensure SNPs were not introduced by off-target CRISPR effects, 300bp up- and down-stream of each SNP was searched for CRISPR-gRNA alignment at 75% sequence similarity.
Acknowledgements
The authors would like to thank Riddhiman Garge and Dan Boutz (University of Texas at Austin) for many suggestions and critical feedback on gene drives and proteasome biochemistry, Charles Boone (University of Toronto) for sharing the SGA-compatible yeast strains, Frederick Roth (University of Toronto) and Harvard University for sharing Green monster strains, Jef Boeke (New York University) and Grant Brown (University of Toronto) for sharing the carotenoid gene yeast expression plasmids, and Maitreya Dunham (University of Washington) for insightful discussions and feedback on performing MERGE across the entire yeast chromosome.
Footnotes
Figure S10
References




