

Abstract
To evaluate the crustacean gill (Na+, K+)-ATPase as a molecular marker for toxic contamination by heavy metals of estuarine and coastal environments, we provide a comprehensive analysis of the effects of Co2+ in vitro on modulation of the K+-phosphatase activity of a gill (Na+, K+)-ATPase from the blue crab Callinectes danae. Using p-nitrophenyl phosphate as a substrate, Co2+ can act as both stimulator and inhibitor of K+-phosphatase activity. Without Mg2+, Co2+ stimulates K+-phosphatase activity similarly but with a ≈4.5-fold greater affinity than with Mg2+. With Mg2+, K+-phosphatase activity is almost completely inhibited by Co2+. Substitution of Mg2+ by Co2+ slightly increases enzyme affinity for K+ and NH4+. Independently of Mg2+, ouabain inhibition is unaffected by Co2+. Mg2+ displaces bound Co2+ from the Mg2+-binding site in a concentration dependent mechanism. However, at saturating Mg2+ concentrations, Co2+ does not displace Mg2+ from its binding site even at elevated concentrations. Saturation by Co2+ of the Mg2+ binding site does not affect pNPP recognition by the enzyme. Given that the interactions between heavy metal ions and enzymes are particularly complex, their toxic effects at the molecular level are poorly understood. Our findings elucidate partly the mechanism of action of Co2+ on a crustacean gill (Na+, K+)-ATPase.
Highlights
Without Mg2+, cobalt ions stimulate the gill (Na+, K+)-ATPase
Co2+ has a 4.5-fold greater affinity for the gill (Na+, K+)-ATPase than does Mg2+
Mg2+ displaces Co2+ from the Mg2+-binding site in a concentration dependent manner
Ouabain inhibition with Co2+ or Mg2+ is identical
Saturation by Co2+ of Mg2+-binding sites does not affect substrate recognition

Graphical abstract (synopsis) Using a crab gill (Na+, K+)-ATPase, we demonstrate that Co2+ inhibits K+-phosphatase activity with Mg2+, which is stimulated without Mg2+. Mg2+ displaces Co2+ from the Mg2+-binding site but Co2+ cannot displace Mg2+. Ouabain inhibition is unaffected by Co2+, independently of Mg2+. The molecular mechanism of Co2+ toxicity is partly elucidated.
1. INTRODUCTION
Estuarine and coastal environments accumulate toxic contaminants owing to natural phenomena and/or anthropogenic activities [1]. Such pollutants include a wide variety of heavy metal ions, organic compounds and various micro/nano-particles [2–4]. Transition metals stand out specifically as the most abundant contaminants found in aquatic environments and contribute largely to toxicity [5,6]. Heavy metal ions are particularly harmful to organisms as they are not degradable and accumulate acutely or chronically in cells and tissues, altering biochemical and physiological homeostatic mechanisms, which can lead to demise at the organismal level [7].
The uptake and toxicity of heavy metals in aquatic organisms is determined by various ambient parameters like pH, temperature and salinity [8]. The biotic ligand model is the model most used to predict metal toxicity in aquatic environments [9]. A profusion of ecotoxicological studies has highlighted the importance of heavy metal toxicity in aquatic environments [e.g., 10,11,12,13]. Most studies of heavy metal toxicity in aquatic organisms concern their bioaccumulation in different tissues [1,14,15] and effects on physiological and biochemical processes like osmoregulatory capability and aerobic and oxidative stress metabolism [1,11,16]. The toxic effects of heavy metals at the molecular level are poorly known since the interactions between such metal ions and enzymes are complex [17,18]. The scant information available regarding the molecular mechanisms of heavy metal toxicity is limited mainly to fish and crustaceans [19–22].
While cobalt is a trace element indispensable for various physiological processes [21,23,24] it is toxic at high intracellular concentrations [25]. In humans, excessive exposure to cobalt results in complex health deficits involving heme oxidation, cytotoxicity, oxidative stress, apoptosis, altered membrane permeability, calcium channel blockage and DNA damage [26–28]. Active Ca2+ transport is inhibited in freshwater fish gills [22] while metabolic pathways are affected in freshwater algae [29].
Although cobalt concentrations in open oceanic waters are less than 7 ng L−1 [30], anthropogenic activities have led to progressive contamination of estuarine and coastal environments [see 25 for review]. To illustrate, mean cobalt levels measured in surface waters near an industrial plant in an Iberian Mediterranean estuarine system are ≈700 µg Co2+ L−1 and reach up to ≈2,800 µg Co2+ L−1 [31]. Cobalt titers in brachyuran crabs from marine/estuarine ecosystems range from 0.26 to 0.43 µg Co2+ g−1 soft tissue dry mass in Callinectes sapidus [32]; 0.91 to 1.2 µg Co2+ g−1 hepatopancreas dry mass in male and female Portunus segnis [33]; and 0.72 to 0.86 µg Co2+ g−1 dry mass in whole Carcinus maenas [34]. The lack of molecular information regarding harm to aquatic organisms, including the effects of Co2+, has impaired our overall comprehension of environmental damage and particularly of toxicity owing to bioaccumulation in marine organisms [16,23,25,35].
Estuarine and coastal organisms are powerful indicators of the health of the marine environment [36,37]. The portunid blue crab Callinectes danae is a crustacean species recommended as an environmental monitor of biological responses in contaminated estuarine and coastal areas [13]. The crab is a euryhaline osmoregulator, tolerant of exposure to wide range of salinities [38], and of great commercial value. It occurs from Florida (USA) to the southern Brazilian coast [39,40] and can be found in muddy estuaries and mangroves, on sandy and muddy shores, and in coastal waters up to 75 meters depth, including biotopes in which salinity varies from brackish to sea water [40,41].
In crustaceans, the gills provide a selective interface between the external environment and internal fluids, contributing to osmotic, ionic, excretory, and acid-base homeostasis. They are also an important route of entry of heavy metal ions [1,2,42]. Metal toxicity can thus vary depending on osmoregulatory strategy while alterations in salinity can affect the availability of metal ion species in the water column [36,43]. In brachyuran crabs, the three posterior gill pairs are specialized in ion transport [42], exhibiting a thickened epithelium [42,44] and increased expression and activity of ion transporters, including the (Na+, K+)-ATPase [1,45,46]. The presence of the (Na+, K+)-ATPase in the gill tissue and its role in physiological homeostasis renders this enzyme a suitable bioindicator to evaluate the kinetic effects of heavy metal ions like Co2+.
The (Na+, K+)-ATPase is a transmembrane enzyme that mediates the coupled transport of three Na+ from the cytosol into the extracellular fluid and of two K+ into the cytosol per ATP molecule hydrolyzed. The enzyme consists of two main subunits: a catalytic α-subunit, responsible for ion transport driven by ATP hydrolysis, and a highly glycosylated, non-catalytic β-subunit that modulates the transport properties of the enzyme [47,48]. The subunits are often associated with an FXYD peptide or γ-subunit that modulates (Na+, K+)-ATPase activity by altering the enzyme’s apparent affinity for Na+, K+ and ATP [49]. Briefly, the catalytic cycle involves alternation between the phosphorylated E1 and E2 conformations that show high affinity for Na+ and K+, respectively [47,48]. Magnesium is also essential although the ion is not transported during the catalytic cycle. Rather, Mg2+participates as the true substrate (a Mg•ATP complex) and plays a regulatory role, interfering with the conformational transitions of the enzyme [50,51]. At high concentrations, Mg2+ can inhibit Na+ and K+ binding by occupying a second specific inhibitory site outside the α-subunit membrane domains [50–52] or binding to the protein surface near the access channel of the ion binding sites [51,53,54]. Various divalent cations like Ca2+, Mn2+, Ba2+ and Sr2+ can substitute for Mg2+ by binding to the Mg2+ regulatory site, activating the enzyme [55–57]. The presence of both ATP and Na+ induces immediate enzyme phosphorylation such that Mg2+ binding and the phosphorylation reaction cannot be examined separately [51].
While the (Na+, K+)-ATPase exhibits high specificity for ATP it also catalyzes the ouabain-sensitive hydrolysis of other nucleoside triphosphates [58] and various non-nucleotide substrates such as p-nitrophenyl phosphate, acetyl phosphate, 2,4-dinitrophenyl phosphate, β-(2-furyl)-acryloyl phosphate, O-methyl fluorescein phosphate and 4-azido-2-nitrophenylphosphate [59–64]. The activity corresponding to the hydrolysis of non-nucleotide substrates is known as the K+-phosphatase activity and requires Mg2+ and K+ but is inhibited by Na+. Such activity represents a partial reaction of the (Na+, K+)-ATPase in which the E2 form is the main conformational state involved in pNPP hydrolysis [58]. The use of such non-nucleotide substrates has disclosed important kinetic characteristics of the (Na+, K+)-ATPase and, under most experimental conditions, these are better substrates than ATP itself [59,61–63,65]. Differently from ATP, p-nitrophenyl phosphate hydrolysis by the gill enzyme involves only a single substrate binding site [61,66]. Likewise, the (Na+, K+)-ATPase is also phosphorylated by p-nitrophenyl phosphate and other acyl phosphates [56,60,67,68]. K+ occlusion does not participate in phosphatase turnover [69] although, in the absence of Na+, K+ stimulates K+-phosphatase activity [70]. The use of p-nitrophenyl phosphate as a substrate thus facilitates the study of Mg2+ binding [51].
Given the paucity of information on the molecular effects of Co2+ on aquatic crustaceans from marine environments contaminated by heavy metals, in this study we provide a comprehensive analysis of the differential effects of Co2+ in vitro on the steady state kinetic properties of the K+-phosphatase activity of a gill (Na+, K+)-ATPase from the blue crab Callinectes danae.
2. MATERIAL AND METHODS
Material
Millipore MilliQ (Merck KGaA, Darmstadt, Germany) ultrapure, apyrogenic deionized water was used to prepare all solutions. Chemicals of the highest purity commercially available were purchased from Sigma Chemical Co. (St. Louis, MO, USA) or Merck (Darmstadt, Germany). All salts were used as chlorides. The homogenization buffer consisted of 20 mmol L−1 imidazole (pH 6.8), 250 mmol L−1 sucrose and a proteinase inhibitor cocktail (1 mmol L−1 benzamidine, 5 µmol L−1 antipain, 5 µmol L−1 leupeptin,5 µmol L−1 phenyl-methane-sulfonyl-fluoride, and 1 µmol L−1 pepstatin A). Analytical estimation of the stock CoCl2 solution concentration (100 mmol L−1) was performed employing inductively coupled mass spectrometry (Perkin Elmer Avio 200 optical emission spectrometer, Boston MA, USA). NH4+ was removed from the crystalline ammonium sulfate suspensions of LDH and PK according to Fabri et al. [71]. When necessary, enzyme solutions were concentrated on YM-10 Amicon Ultra filters.
2.1. Crab collection
Adult specimens of Callinectes danae of ≈9 cm carapace width were collected at low tide using seine nets or baited hand nets from Barra Seca beach (23° 25’ 01.5” S, 45° 03’ 01.0” W), Ubatuba, São Paulo State, Brazil (SISBIO/ICMBio/IBAMA authorization 02027.002342/98-04, permit #29594-18 to John C. McNamara). The crabs were held briefly in plastic boxes containing 30 L aerated seawater from the collection site during transport to the laboratory where they were immediately anesthetized by chilling in crushed ice. After bisecting and removal of the carapace, all three posterior gill pairs (120 gills/preparation, ≈5 g wet mass) were rapidly dissected out and frozen in liquid nitrogen in homogenization buffer in Falcon tubes.
2.2. Preparation of the gill microsomal fraction
For each of the three (N= 3) gill homogenates prepared, the gills frozen in the homogenization buffer were thawed, diced and homogenized (20 mL homogenization buffer/g wet tissue) at 600 rpm in a Potter homogenizer in a crushed ice bath. The (Na+, K+)-ATPase-rich microsomal fraction was prepared by stepwise differential centrifugation (20,000 and 100,000 ×g) of the gill homogenate [38]. The resulting pellet was resuspended in 20 mmol L−1 imidazole buffer (pH 6.8) containing 250 mmol L−1 sucrose (15 mL buffer/g wet tissue). Aliquots (0.5 mL) were frozen in liquid nitrogen, stored at −20 °C and used within three-month’s storage provided that at least 95% (Na+, K+)-ATPase activity was present.
2.3. Estimation of p-nitrophenyl phosphatase activity
p-Nitrophenyl phosphate (pNPP) was used as the enzyme substrate. The p-nitrophenyl phosphatase activity (pNPPase) of the microsomal fraction was estimated continuously at 25°C, following the release of the p-nitrophenolate ion (pNP−) at 410 nm (ε 410nm, pH 7.5= 13,160 M−1 cm−1) in a Shimadzu UV-1800 spectrophotometer (Vernon Hills IL, USA) equipped with thermostatted cells. The standard incubation medium contained 50 mmol L−1 HEPES buffer, pH 7.5, and appropriate pNPP, Mg2+, K+ or NH4+ concentrations (see Results and Figure legends for substrate and specific ionic concentrations) and 9 µg alamethicin, in final volume of 1 mL. Activity was estimated with or without 7 mmol L−1 ouabain, the difference corresponding to the K+-phosphatase activity of the gill (Na+, K+)-ATPase. The reaction was always initiated by the addition of the enzyme.
Because tissue homogenization usually results in the formation of small vesicles in the microsomal preparation that can occlude the catalytic site of the enzyme, pNPPase activity also was estimated after 10 min pre-incubation with 9 µg alamethicin, a membrane pore-forming antibiotic, to verify the presence of vesicles showing ATPase activity.
Controls without added enzyme were used to estimate the spontaneous hydrolysis of the substrate under the assay conditions. The kinetic measurements were carried out in duplicate, and substrate hydrolysis was accompanied over the shortest possible period to guarantee initial velocity measurements (<5% of substrate hydrolyzed during the reaction period). One unit (U) of enzyme activity was defined as the amount of enzyme that hydrolyses 1.0 nmol of pNPP per minute at 25 °C.
2.4. Protein
Protein concentration was measured in duplicate aliquots of the microsomal preparations using the Coomassie Blue G dye-binding assay employing bovine serum albumin as a standard. Assays were read at 595 nm using a Shimadzu UV-1800 spectrophotometer [71].
2.5. Estimation K+-phosphase activity with cobalt ions
The effect of cobalt ions on pNPPase activity was estimated as above using cobalt concentrations between 10−5 and 2×10−2 mmol L−1. Our study on the effect of Co2+ on the interaction of the different ligands with the (Na+,K+)-ATPase was performed using 3 mmol L−1 Co2+ (50.8 μg L−1 Co2+) which inhibits K+-phosphatase activity by ≈50%.
2.6. Measurement of ATP hydrolysis
To provide a direct comparison with previous microsomal preparations, initial rates of ATP hydrolysis also were estimated continuously at 25 °C, using a pyruvate kinase/lactic dehydrogenase coupling system in which ATP hydrolysis is coupled to NADH oxidation [72]. The specific activity of (Na+, K+)-ATPase for ATP of 296.5 ± 10.2 nmol Pi min−1 mg−1 protein corresponding to 125.5 ± 2.3 nmol pNP− min−1 mg−1 protein of K+-phosphatase activity is comparable to our previous findings [73].
2.7. Estimation of kinetic parameters
The kinetic parameters VM (maximum velocity), K0.5 (apparent dissociation constant) and the nH value (Hill coefficient) for pNPP hydrolysis were calculated using SigrafW software ([74]; freely available from http://portal.ffclrp.usp.br/sites/fdaleone/downloads). The apparent dissociation constant of the enzyme-inhibitor complex (KI) was estimated using the Dixon plot in which the reaction rate corresponding to the K+-phosphatase activity was corrected for residual activity at high inhibitor concentrations [75]. Data points (mean ± SD) in the figures representing each substrate/ligand concentration are mean values of duplicate aliquots from the same preparation and were used to fit the saturation curves that were repeated using three different microsomal preparations (N= 3). The kinetic parameters (VM, KM, KI or K0.5) shown in the tables are calculated values and represent the mean (± SD) derived from values estimated for each of three (N= 3) microsomal preparations.
3. RESULTS
Estimation of pNPPase activity with alamethicin revealed that the gill microsomal preparation from fresh caught C. danae includes ≈25% sealed, pNPPase-containing vesicles (164.5 ± 3.8 and 122.5 ± 1.8 nmol pNP− min−1 mg−1 protein with or without alamethicin, respectively). Thus, pNPPase activity was always estimated using 9 µg alamethicin. Seven mmol L−1 ouabain decreased pNPPase activity from 164.5 ± 3.8 to 39.2 ± 1.9 nmol pNP− min−1 mg−1 protein, indicating that ≈75% of the total phosphohydrolyzing activity (125.5 ± 2.3 nmol pNP− min−1 mg−1 protein) corresponds to the K+-phosphatase activity of the gill (Na+, K+)-ATPase. Measurements using ATP as a substrate showed a (Na+, K+)-ATPase activity of 296.5 ± 10.2 nmol Pi min−1 mg−1 protein.
3.1. Effect of Co2+ on K+-phosphatase activity
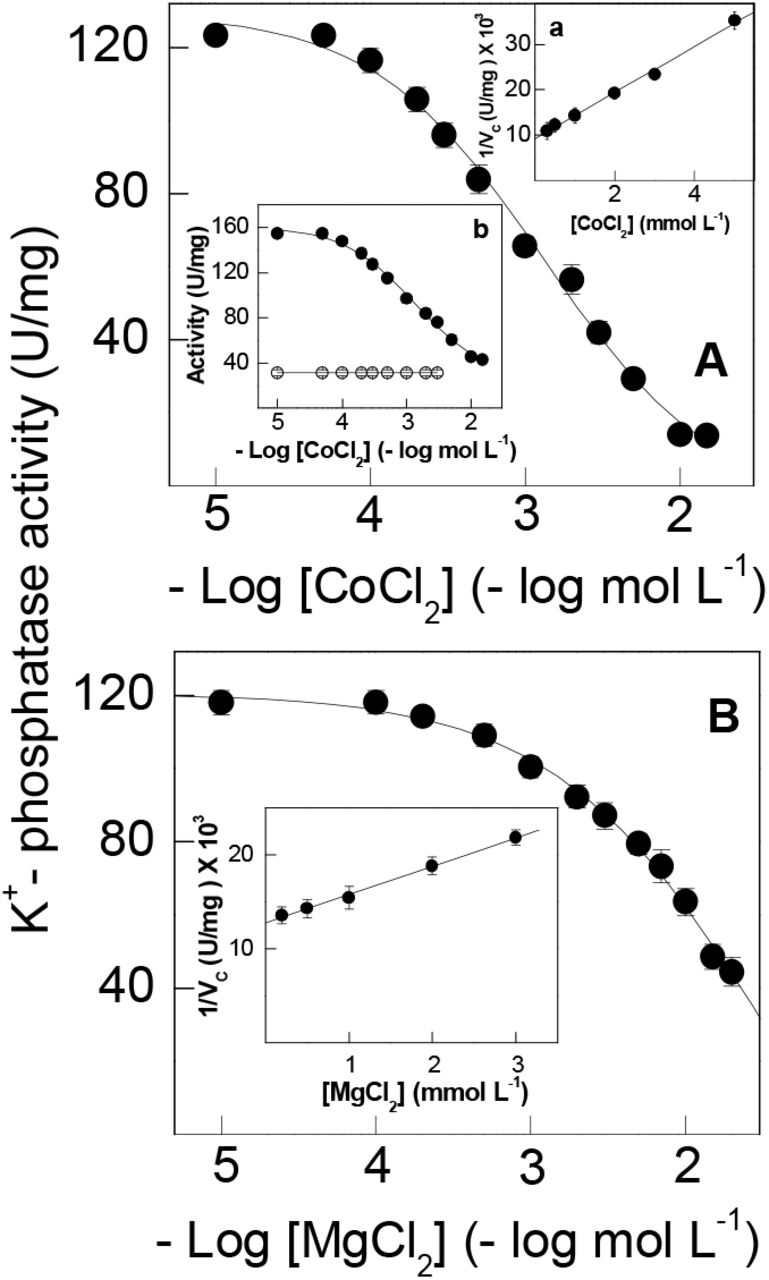
Under optimal assay conditions (see legends to Fig. 1A and 1B), increasing Co2+ concentrations from 10−5 to 2×10−2 mol L−1 in the incubation medium inhibited K+-phosphatase activity by 90% (Fig. 1A and Table 1) with KI= 2.77 ± 0.33 mmol L−1 (inset a to Fig. 1). K+-phosphatase activity decreased following a single titration curve to 13.7 ± 3.6 nmol pNP− min−1 mg−1 protein. The ouabain-insensitive pNPPase activity of ≈38 nmol pNP− min−1 mg−1 protein was unaffected by increasing Co2+ concentrations (inset b to Fig. 1). On fixing Co2+ at 3 mmol L−1, increasing Mg2+ concentrations (10−5 to 2×10−2 mol L−1) inhibited K+-phosphatase activity by ≈60% (Fig. 1B and Table 1). A KI= 4.81 ± 0.71 mmol L−1 was calculated for inhibition of K+-phosphatase activity by Mg2+ in the presence of Co2+ (inset to Fig. 1B).

Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 10 mmol L−1 pNPP, 15 mmol L−1 KCl, 9 µg alamethicin and the metal ions in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. A- with 7 mmol L−1 MgCl2. Inset a- Dixon plot for estimation of KI in which vc is the K+-phosphatase activity corrected for residual pNPPase activity found at high Co2+ concentration. Inset b- total pNPPase activity (•) and ouabain-insensitive pNPPase activity (○). B- with 3 mmol L−1 CoCl2. Inset- Dixon plot for estimation of KI in which vc is the K+-phosphatase activity corrected for residual K+-phosphatase activity found at high Mg2+ concentrations.
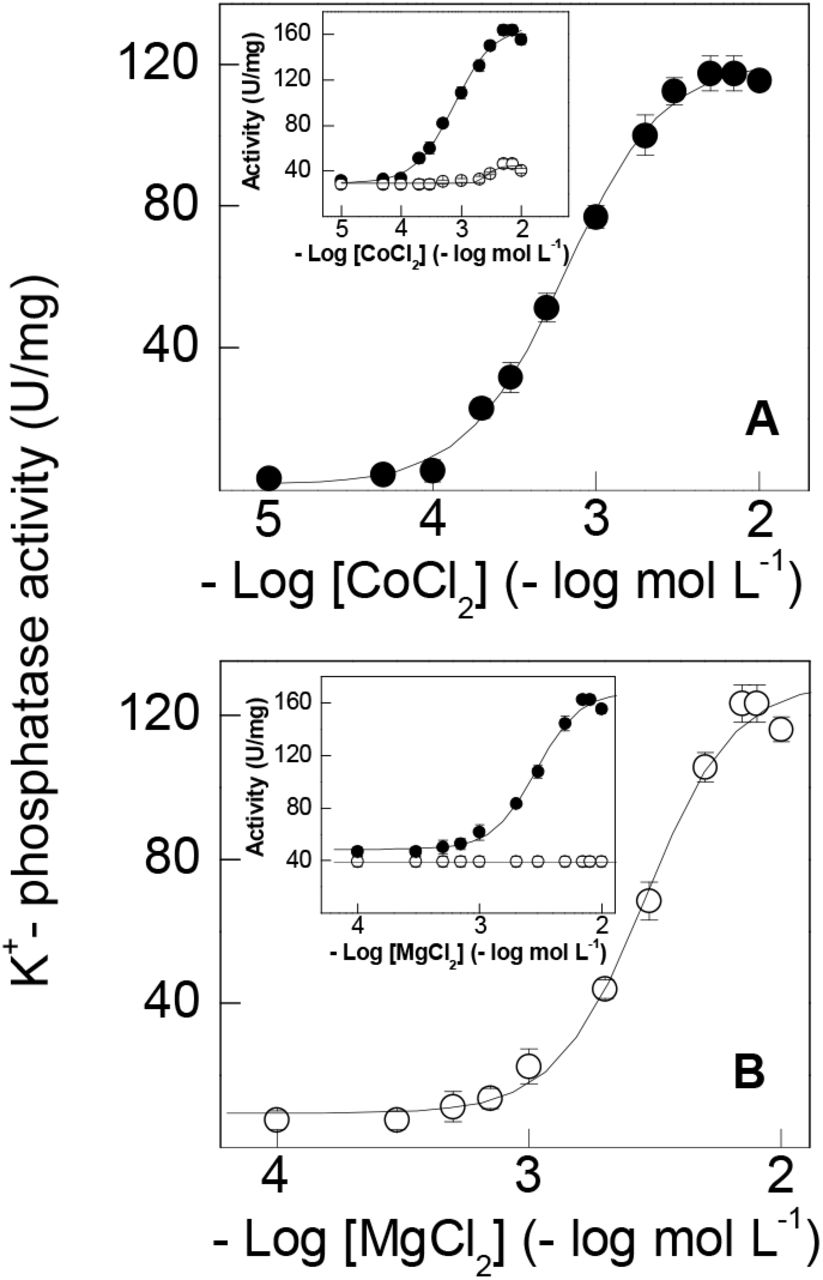
Cobalt ions can substitute for Mg2+, stimulating gill K+-phosphatase activity (Fig. 2A and Table 1). Under optimal assay conditions (see legends to Fig. 2A and 2B) without Mg2+, Co2+ stimulated K+-phosphatase activity to a maximum rate of VM= 122.5 ± 3.1 nmol pNP− min−1 mg−1 protein with K0.5= 0.69 ± 0.23 mmol L−1, showing a single saturation curve obeying cooperative kinetics (nH= 1.4). Ouabain-insensitive pNPPase activity was stimulated to 46.4 ± 3.7 nmol pNP− min−1 mg−1 protein over the same Co2+ concentration range (inset to Fig. 2A). K+-phosphatase activity was inhibited by Co2+ concentrations above 10−2 mol L−1. In the absence of Co2+, Mg2+ (10−4 to 2×10−2 mol L−1) stimulated K+-phosphatase activity to a maximum rate of VM= 135.1 ± 5.0 nmol pNP− min−1 mg−1 protein with K0.5= 2.98 ± 0.59 mmol L−1 and cooperative kinetics (nH= 2.2), following a single saturation curve (Fig. 2B and Table 1). Stimulation by Mg2+ of ouabain-insensitive pNPPase activity was negligible over the concentration range used (inset to Fig. 2B). The ≈4.5-fold lower K0.5 suggests that Co2+ binds more tightly to the Mg2+-binding sites than does Mg2+ itself.

Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 10 mmol L−1 pNPP, 15 mmol L−1 KCl, 9 µg alamethicin in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. A- with cobalt ions. B- with magnesium ions. Inset to figures-total pNPPase activity (•); ouabain-insensitive pNPPase activity (◯).
3.2. Effect of Co2+ on pNPP hydrolysis
Under optimal assay conditions (see legends to Fig. 2A and 2B), increasing pNPP concentrations stimulated K+-phosphatase activity to a maximum rate of VM= 138.1 ± 4.2 nmol pNP−1 min−1 mg−1 protein with K0.5= 1.76 ± 0.49 mmol L−1, following cooperative kinetics (nH= 1.3) and showing a single saturation curve (Fig. 3A and Table 1). Substitution of Mg2+ with 3 mmol L−1 Co2+ also gave a single saturation curve, overlapping that for Mg2+, showing a maximum rate of VM= 128.2 ± 4.4 nmol pNP− min−1 mg−1 protein with K0.5= 1.15 ± 0.61 mmol L−1 (Fig. 3A). With Mg2+, the ouabain-insensitive pNPPase activity of ≈30 nmol pNP− min−1 mg−1 protein was not affected by increasing pNPP concentrations (not shown). However, with 3 mmol L−1 Co2+ this activity was stimulated by ≈35% over the same pNPP concentration range (inset to Fig. 3A). With both Mg2+ and Co2+, under the same saturating ionic and substrate concentrations, K+-phosphatase activity decreased to a maximum rate of VM= 63.1 ± 3.8 nmol pNP− min−1 mg−1 protein with K0.5= 0.92 ± 0.28 mmol L−1 also obeying cooperative kinetics (Fig. 3B and Table 1). This ≈50% inhibition of K+-phosphatase activity was accompanied by a 2-fold decrease in K0.5 compared to Mg2+ (Table 1). The ouabain-insensitive pNPPase activity of ≈20 nmol pNP− min−1 mg−1 protein was stimulated ≈60% over the same pNPP concentration range (inset to Fig. 3B).

Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 15 mmol L−1 KCl, 9 µg alamethicin and the metal ion (7 mmol L−1 Mg2+ or 3 mmol L−1 Co2+) in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. A- with magnesium (○) or cobalt ions (•). B- with both magnesium and cobalt ions. Inset to figures- total pNPPase activity (•,◼); ouabain-insensitive pNPPase activity (○,□) for Co2+only.
3.3. Effect of Co2+ on Mg2+ stimulation
Magnesium can displace Co2+ bound to the gill (Na+, K+)-ATPase below 2 mmol L−1 Co2+ (Fig. 4). Increased Mg2+ (10−5 to 5×10−2 mol L−1) displaces bound Co2+ (0.5 and 1 mmol L−1), stimulating K+-phosphatase activity to a maximum rate of ≈130 nmol pNP− min−1 mg−1 protein with similar K0.5 (Fig. 4 and Table 1). However, at 2 mmol L−1 Co2+ and above no displacement was seen and increasing Mg2+ concentrations inhibited K+-phosphatase activity with KI= 4.41 ± 0.69 and 4.81 ± 0.71 mmol L−1 for 2 and 3 mmol L−1 Co2+, respectively (Table 1).

Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 10 mmol L−1 pNPP, 15 mmol L−1 KCl and 9 µg alamethicin in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. (◼) without Co2+. (◯) 0.5 mmol L−1 Co2+. (▴) 1 mmol L−1 Co2+. (□) 2 mmol L−1 Co2+. (•) 3 mmol L−1 Co2+.
3.4. Effect of Co2+ on K+ stimulation
Under saturating ionic and substrate concentrations (see legends to Fig. 5A and 5B) in the absence of Co2+, increasing K+ stimulated K+-phosphatase activity to a maximum rate of VM= 134.2 ± 4.5 nmol pNP− min−1 mg−1 protein with K0.5= 9.60 ± 2.04 mmol L−1 (Fig. 5A and Table 1) following cooperative kinetics (nH= 2.2). Substitution of Mg2+ by 3 mmol L−1 Co2+ also gave a single saturation curve overlapping with that for Mg2+ and showing a maximum rate of VM= 133.3 ± 3.3 nmol pNP− min−1 mg−1 protein with K0.5= 6.00 ± 1.50 mmol L−1 (Fig. 5A and Table 1). Ouabain-insensitive pNPPase activity was not stimulated by either metal ion over the concentration range used (inset to Fig. 5A). With Co2+ plus Mg2+, K+-phosphatase activity decreased to VM= 59.5 ± 4.0 nmol pNP− min−1 mg−1 protein with K0.5= 2.79 ± 0.41 mmol L−1 following a single titration curve (Fig. 5B and Table 1), obeying cooperative kinetics (nH= 1.5). K+-phosphatase activity at K+ concentrations be low 10−4 mol L−1 was ≈15 nmol pNP− min−1 mg−1 protein. Ouabain-insensitive pNPPase activity was stimulated to ≈22 nmol pNP− min−1 mg−1 protein over the same K+ concentration range (inset to Fig. 5B). K+-phosphatase activity was not synergically stimulated by K+ plus NH4+ (not shown).

Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 10 mmol L−1 pNPP, 9 µg alamethicin and the metal ion (7 mmol L−1 Mg2+ or 3 mmol L−1 Co2+) in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. A- K+-phosphatase activity with Mg2+ (◯) or Co2+ (•). B- K+-phosphatase activity with both Mg2+ and Co2+. Insets to figures- total pNPPase activity ( ) and ouabain-insensitive pNPPase activity (•) Co2+.
3.5. Effect of Co2+ on NH4+ stimulation
Under saturating ionic and substrate concentrations (see legends to Fig. 6A and 6B) in the absence of Co2+, a maximal rate of VM= 122.2 ± 5.2 nmol pNP− min−1 mg−1 protein with K0.5= 9.02 ± 2.51 mmol L−1 was estimated for NH4+ concentrations increasing from 10−4 to 5×10−2 mol L−1 (Fig. 6A and Table 1). Substitution of Mg2+ by 3 mmol L−1 Co2+ also gave a single saturation curve with a maximum rate of VM= 127.9 ± 4.2 nmol pNP− min−1 mg−1 protein with K0.5= 6.00 ± 1.10 mmol L−1, overlapping with that for Mg2+ (Fig. 6A and Table 1). Stimulation of ouabain-insensitive pNPPase activity by Mg2+ and Co2+ was negligible over the NH4+ concentration range used (inset to Fig. 6A). With Co2+ plus Mg2+, K+-phosphatase activity decreased to VM= 61.9 ± 3.7 nmol pNP− min−1 mg−1 protein with K0.5= 5.46 ± 0.64 mmol L−1 (Fig. 6B and Table 1). Stimulation of the ouabain-insensitive pNPPase activity was <10% with Co2+ plus Mg2+ (inset to Fig. 6B). K+-phosphatase activity was not stimulated synergically by K+ plus NH4+ (not shown).

Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 10 mmol L−1 pNPP, 9 µg alamethicin and the metal ion (7 mmol L−1 Mg2+ or 3 mmol L−1 Co2+) in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. A- K+-phosphatase activity with Mg2+ (◯) or Co2+ (•). B- K+-phosphatase activity with both Mg2+ and Co2+. Insets to figures- total pNPPase activity (•) and ouabain-insensitive pNPPase activity (◯) Co2+.
3.6. Effect of Co2+ on inhibition by Na+ and ouabain
Cobalt does not affect inhibition of pNPPase activity by Na+ or ouabain (Fig. 7) Sodium concentrations from 10−3 to 0.2 mol L−1 inhibited pNPPase activity by ≈70% either with or without Co2+ (Fig. 7A and Table 1). The IC50 estimated for Na+ inhibition of pNPPase activity was ≈16 mmol L−1 (Table 1). Over the ouabain concentration range from 10−6 to 10−2 mol L−1, the Na+ and ouabain inhibition curves overlapped independently of Co2+ (Fig. 7B and Table 1). Their monophasic behavior with very similar inhibition constants (KI= 2.27 ± 0.62 mmol L−1 and KI= 2.13 ± 0.97 mmol L−1 for Mg2+ and Co2+ respectively) (inset to Fig. 7B) suggests a single ouabain binding site.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Activity was assayed continuously at 25 °C in 50 mmol L−1 HEPES buffer, pH 7.5, containing 10 mmol L−1 pNPP, 15 mmol L−1 KCl and 9 µg alamethicin in a final volume of 1 mL. The mean activity of duplicate aliquots of the same microsomal preparation (≈15 µg protein) was used to fit the saturation curve which was repeated using three different microsomal preparations (± SD, N= 3). Where lacking, error bars are smaller than the symbols used. A- sodium ion. B- ouabain. Inset to Fig. 8B-Dixon plot for estimation of KI in which vc is the pNPPase activity corrected for residual pNPPase activity found at high ouabain concentrations. (○) with 7 mmol L−1 Mg2+. (•) with 3 mmol L−1 Co2+.
4. DISCUSSION
We provide a comprehensive analysis of the effects of Co2+ on the modulation in vitro of the K+-phosphatase activity in a gill (Na+, K+)-ATPase from the blue crab Callinectes danae. Depending on Mg2+, Co2+serves as both stimulator and inhibitor of K+-phosphatase activity. Without Mg2+, Co2+ stimulates activity as does Mg2+, although with a ≈4.5-fold greater affinity. With Mg2+, activity is almost completely inhibited by Co2+, while ouabain inhibition is unaffected. Substitution of Mg2+ by Co2+ slightly increases enzyme affinity for K+ and NH4+. Mg2+ displaces bound Co2+ from the Mg2+-binding site in a concentration dependent manner; however, Co2+ does not displace bound Mg2+ even at elevated concentrations. Saturation by Co2+ of the Mg2+-binding site does not affect substrate recognition by the enzyme.
K-phosphatase activities estimated with Mg2+ or Co2+ are similar and their overlapping pNPP saturation curves show comparable cooperative effects; their similar K0.5 values suggest that Co2+ saturation of the Mg2+-binding site does not affect substrate recognition. The high stability constants for the Co2+-pNPP (130 mol L−1, [76]) and Mg2+-pNPP (170 mol L−1, [77]) complexes suggest that negligible free metal ions are present at millimolar metal ion concentrations, i.e., the metal-pNPP complex is the true enzyme substrate. The lower apparent dissociation constant for Co2+ (K0.5= 1.15 ± 0.61 mmol L−1), close to that for Mg2+ (K0.5= 1.76 ± 0.49 mmol L−1), is comparable to the enzyme from Cancer pagurus axonal membranes despite its 2-fold greater maximum pNPP hydrolysis rate [78].
Millimolar Mg2+ concentrations are required for K+-phosphatase activity of the C. danae (Na+, K+)-ATPase and, like the mammalian enzyme [58,79], no detectable activity can be measured without this ion. The millimolar Mg2+ or Co2+ concentrations necessary for maximum K+-phosphatase activity exclude the likelihood of metal binding other than Mg2+ or Co2+ to the Mg2+-binding site during the catalytic cycle [56]. The inhibition by free Mg2+ or Co2+ of K+-phosphatase activity may result from competition with K+ for the Mg2+-binding site [56,61,80] or to excess Mg2+ bound during the E2K conformation, decreasing affinity for pNPP [52,56,81].
Co2+ can substitute for Mg2+, stimulating K+-phosphatase activity more efficiently (K0.5 ≈4.5-fold lower). However, Co2+ does not displace Mg2+ from the Mg2+-binding site of the C. danae enzyme. Inhibition by excess Co2+ likely results from Co2+ binding at a site different from the Mg2+-binding site. Two distinct Mg2+ binding sites are known for crustacean [73] and mammalian [50] (Na+, K+)-ATPases, although only one is relevant for pNPPase and ATPase activities [82]. Like Co2+, Mn2+ also stimulates sheep kidney (Na+, K+)-ATPase activity, the KD= 0.88 μmol L−1 for Mn2+ binding being very similar to the kinetic constant for ATP hydrolysis [83]. Co2+ stimulates dog kidney outer medulla (Na+, K+)-ATPase activity by substituting for Mg2+ [20]. Differently from Co2+, Cu2+ inhibits the gill (Na+, K+)-ATPase activity of rainbow trout Oncorhynchus mykiss [84], and the K+-phosphatase and (Na+, K+)-ATPase activities of rabbit kidney [85] by directly interfering with Mg2+ binding, affecting Mg•ATP hydrolysis.
The similar VM and K0.5 values for stimulation by K+ or NH4+ of K+-phosphatase activity with Mg2+ or Co2+ suggests that NH + binds to the same site as K+ during the catalytic cycle, independently of Mg2+ or Co2+ bound to the Mg2+-binding site. While K+-phosphatase activity is inhibited to same degree by Mg2+ and Co2+, the 2-fold greater K0.5 for NH4+ (5.46 mmol L−1) compared to K+ (2.79 mmol L−1) suggests that Co2+ binding to a different Mg2+-binding site affecting enzyme interaction with NH +. Without Na+, stimulation by K+ of K+-phosphatase activity involves two K+ binding sites: one that regulates pNPP access to the phosphatase site, the other increasing catalytic activity [86]. ATP binding to the low-affinity substrate binding site induces a conformational change in the cytoplasmic domain of the enzyme attributed to the E2 to E1 transition; the subsequent binding of Mg2+ to the enzyme ATP complex induces a new conformational change that facilitates the E1 to E2 transition [87]. Like Mg2+, Co2+ may induce a conformational change, stimulating the enzyme during pNPP hydrolysis.
K+-phosphatase activity can be stimulated by Tl+, Rb+ or NH + to rates similar to K+ stimulation while Cs+ and Li+ exhibit lower stimulation (5-30%) [88,89]. For 21‰ (low salinity)-acclimated C. danae, Rb+ stimulates gill pNPPase activity by 1.5-fold compared to K+ [63]. NH4+ may sustain ATP hydrolysis by replacing K+ [89,90] and is actively transported by crustacean and vertebrate enzymes [91,92]. Together with K+, NH4+ synergically stimulates ATP hydrolysis by the C. danae (Na+, K+)-ATPase through an additional increment, strongly influenced by Mg2+ and Na+, underlying ammonia excretion in crustaceans [90]. The E2 conformation is the main state responsible for pNPP hydrolysis [58] and is likely the reason that K+-phosphatase activity is not synergically stimulated by K+ and NH4+. Synergic stimulation using pNPP as a substrate is known exclusively for the shelling crab C. ornatus [63]. When using ATP as a substrate, species-specific synergic stimulation is found in various crustaceans [73].
The inhibition by Na+ of C. danae pNPPase activity independently of Mg2+ or Co2+ is seen also for (Na+, K+)-ATPases from various sources [73], and reflects competition by Na+ for the cytoplasmic K+-binding sites, favoring the E1 conformation [50,56,58,93–95]. Na+ inhibition also may involve events other than the simple binding of the ion. To illustrate, the synergistic stimulation by Na+ and K+ (3%) of pNPPase activity in the electric organ of Electrophorus electricus [86] suggests that both ions bind to different sites. Both 10 mmol L−1 Na+ and 15 mol L−1 K+ inhibit the C. danae pNPPase activity by ≈40%, as seen in the freshwater shrimp Macrobrachium olfersii [61] and crabs Cancer pagurus [95] and C. ornatus [63] under identical assay conditions. Na+ inhibits pNPPase activity allosterically in various mammalian and crustacean (Na+, K+)-ATPases [61,63,86,89,95] including M. olfersii [61] and C. pagurus [95]. Thus, considering a two K+-binding site model [86], at low (10-fold less than K+) concentrations, Na+ competes for the high-affinity K+ binding site, leading to allosteric effects. At high concentrations (similar to K+), Na+ competes for the low-affinity K+-binding site, reducing maximum hydrolysis rate.
Co2+ does not affect ouabain binding to the C. danae gill (Na+, K+)-ATPase, the single inhibition curve and KI being very similar to Mg2+. Most species, except the red river crab Dilocarcinus pagei [96], exhibit a single ouabain inhibition curve independently of substrate [62,63,72,97,98]. The KI for ouabain inhibition with Co2+ lies in the range for various (Na+, K+)-ATPases [73]. It should be noted that only for the cerebromicrovascular (Na+, K+)-ATPase, Pb2+ and Al3+ caused selective alterations in ATP hydrolysis inhibiting and stimulating ouabain binding, respectively [99].
Our findings reveal that without Mg2+, Co2+ stimulates the gill (Na+, K+)-ATPase to levels similar to Mg2+. Without Mg2+, Co2+ stimulates K+-phosphatase activity similarly although with a ≈4.5-fold greater affinity than with Mg2+, which almost completely inhibits K+-phosphatase activity. Mg2+ displaces Co2+ from the Mg2+-binding sites in a concentration dependent manner. Ouabain inhibition is identical with Co2+ or Mg2+. Saturation by Co2+ of the Mg2+-binding sites does not affect substrate recognition by the enzyme. Given the complex interactions between heavy metal ion contaminants and enzymes, their toxic effects at the molecular level are poorly understood. Our findings contribute to elucidate partly the mechanism of action of Co2+ on a crustacean gill (Na+, K+)-ATPase.
Funding information
This investigation was financed by research grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2016/25336-0 and 2019/21899-8), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG APQ-01893-16), Conselho de Desenvolvimento Científico e Tecnológico (CNPq 458246/2014-0), and in part by INCT ADAPTA II (CNPq 465540/2014-7) and the Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM 062.1187/2017).
FAL (302072/2019-7), and JCM (303613/2017-3) received Excellence in Research scholarships from CNPq. LMF, CMM and MICC received scholarships from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance code 001).
Author contributions
Francisco A. Leone: Conceptualization, Formal analysis, Resources, Funding acquisition, Methodology, Supervision, Project administration. Writing original draft, Review & Editing. Leonardo M. Fabri: Methodology, Investigation, Conceptualization, Writing original draft, Review & Editing. Cintya M. Moraes: Methodology, Investigation, Writing original draft. Maria I. C. Costa: Methodology, Investigation, Writing original draft. Daniela P. Garçon: Conceptualization, Methodology, Formal analysis, Funding acquisition, Writing original draft, Review & Editing. John C. McNamara: Conceptualization, Methodology, Formal analysis, Writing original draft, Review & Editing.
Data availability
The datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request.
Declaration of competing interests
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Ethical approval studies in animals
This investigation complies with all local, state, federal and international guidelines as regards the use of invertebrate animals in scientific research. This study also complies with the ARRIVE guidelines.
Acknowledgements
The authors thank the Instituto Chico Mendes de Conservação da Biodiversidade, Ministério do Meio Ambiente (ICMBio/MMA) for authorizing collecting permit #29594-18 to JCM, and INCT-ADAPTA II (Instituto Nacional de Ciência e Tecnologia para Adaptações da Biota Aquática da Amazônia, ADAPTA-II) with which FAL’s laboratory is integrated, and the Amazon Shrimp Network (Rede de Camarão da Amazônia).
REFERENCES
- [1].↵
- [2].↵
- [3].
- [4].↵
- [5].↵
- [6].↵
- [7].↵
- [8].↵
- [9].↵
- [10].↵
- [11].↵
- [12].↵
- [13].↵
- [14].↵
- [15].↵
- [16].↵
- [17].↵
- [18].↵
- [19].↵
- [20].↵
- [21].↵
- [22].↵
- [23].↵
- [24].↵
- [25].↵
- [26].↵
- [27].
- [28].↵
- [29].↵
- [30].↵
- [31].↵
- [32].↵
- [33].↵
- [34].↵
- [35].↵
- [36].↵
- [37].↵
- [38].↵
- [39].↵
- [40].↵
- [41].↵
- [42].↵
- [43].↵
- [44].↵
- [45].↵
- [46].↵
- [47].↵
- [48].↵
- [49].↵
- [50].↵
- [51].↵
- [52].↵
- [53].↵
- [54].↵
- [55].↵
- [56].↵
- [57].↵
- [58].↵
- [59].↵
- [60].↵
- [61].↵
- [62].↵
- [63].↵
- [64].↵
- [65].↵
- [66].↵
- [67].↵
- [68].↵
- [69].↵
- [70].↵
- [71].↵
- [72].↵
- [73].↵
- [74].↵
- [75].↵
- [76].↵
- [77].↵
- [78].↵
- [79].↵
- [80].↵
- [81].↵
- [82].↵
- [83].↵
- [84].↵
- [85].↵
- [86].↵
- [87].↵
- [88].↵
- [89].↵
- [90].↵
- [91].↵
- [92].↵
- [93].↵
- [94].
- [95].↵
- [96].↵
- [97].↵
- [98].↵
- [99].↵




