

Abstract
AlphaFold2 revolutionized structural biology with the ability to predict protein structures with exceptionally high accuracy. Its implementation, however, lacks the code and data required to train new models. These are necessary to (i) tackle new tasks, like protein-ligand complex structure prediction, (ii) investigate the process by which the model learns, which remains poorly understood, and (iii) assess the model’s generalization capacity to unseen regions of fold space. Here we report OpenFold, a fast, memory-efficient, and trainable implementation of AlphaFold2, and OpenProtein-Set, the largest public database of protein multiple sequence alignments. We use OpenProteinSet to train OpenFold from scratch, fully matching the accuracy of AlphaFold2. Having established parity, we assess OpenFold’s capacity to generalize across fold space by retraining it using carefully designed datasets. We find that OpenFold is remarkably robust at generalizing despite extreme reductions in training set size and diversity, including near-complete elisions of classes of secondary structure elements. By analyzing intermediate structures produced by OpenFold during training, we also gain surprising insights into the manner in which the model learns to fold proteins, discovering that spatial dimensions are learned sequentially. Taken together, our studies demonstrate the power and utility of OpenFold, which we believe will prove to be a crucial new resource for the protein modeling community.
1 Introduction
Predicting protein structure from sequence has been a defining challenge of biology for decades (Anfinsen 1973, Dill et al. 2008). Building on a line of work applying deep learning to co-evolutionary information encoded in multiple sequence alignments (MSAs) (Jones et al. 2015, Golkov et al. 2016, S. Wang et al. 2017, Liu et al. 2018, Senior et al. 2020, Xu et al. 2021) and homologous structures (Šali and Blundell 1993, Roy et al. 2010), AlphaFold2 (Jumper et al. 2021) has arguably solved the problem for natural proteins with sufficiently deep MSAs. The model has been made available to the public with DeepMind’s official open-source implementation, which has been used to predict the structures of hundreds of millions of proteins (Tunyasuvunakool et al. 2021, Varadi et al. 2021, Callaway 2022). This implementation has enabled researchers to optimize AlphaFold2’s prediction procedure and user experience (Mirdita, Schütze, et al. 2022) and to employ it as a module within novel algorithms, including ones for protein complex prediction (Baek 2021), peptide-protein interactions (Tsaban et al. 2022), structure ranking (Roney and Ovchinnikov 2022), and more (e.g., Baltzis et al. 2022, Bryant et al. 2022, Wayment-Steele et al. 2022).
In spite of its outstanding utility, the official AlphaFold2 implementation omits code for the model’s complex training procedure as well as the computationally expensive training data required to run it. This makes it difficult to i) investigate AlphaFold2’s learning behavior and sensitivity to changes in data composition and model architecture and ii) create variants of the model to tackle new tasks. Given the success of AlphaFold2, its many novel components are likely to prove useful for tasks beyond protein structure prediction. For instance, retraining AlphaFold2 using a dataset of protein-protein complexes resulted in AlphaFold2-Multimer (Evans et al. 2022), the state of the art model for predicting structures of protein complexes. Until recently, however, this capability has been exclusive to DeepMind.
To address this shortcoming, we developed OpenFold, a trainable open-source implementation of AlphaFold2, and OpenProteinSet, a database of five million deep and diverse MSAs that removes one of the most significant computational barriers—millions of CPU-hours—to training new protein models at the scale of AlphaFold2. We trained OpenFold from scratch using OpenProteinSet, matching AlphaFold2 in prediction quality. Apart from new training code and data, OpenFold has several advantages over AlphaFold2: (i) it runs up to three times faster on most proteins, (ii) it uses less memory, allowing prediction of extremely long proteins and multi-protein complexes on a single GPU, and (iii) it is implemented in PyTorch (Paszke et al. 2019), the most widely used machine learning framework (AlphaFold2 uses Google’s JAX (Bradbury et al. 2018)). As such, OpenFold can be readily used by the widest community of developers and interfaces with a rich ecosystem of existing machine learning software (Rasley et al. 2020, Charlier et al. 2021, Falcon et al. 2019, Charlier et al. 2021, Dao et al. 2022).
We used OpenFold to understand how the model learns to fold proteins, focusing on the geometric characteristics of predicted structures during intermediate stages of training, and identified multiple distinct phases of behavior. Specifically, by analyzing predicted structures at multiple resolutions and decomposing them into secondary and tertiary elements, we found that OpenFold learns spatial dimensions, secondary structure elements, and tertiary scales in a staggered manner. Next, taking advantage of our discovery that ~90% of model accuracy can be achieved in ~3% of training time, we retrained OpenFold multiple times on specially elided versions of the training set to quantify its ability to generalize to unseen protein folds. Surprisingly, we found the model highly robust even to large elisions of fold space, but its capacity to generalize varied based on the spatial extent of protein fragments and folds. We observed even stronger performance when training the model on more diverse but smaller datasets, some as small as 1,000 experimental structures. Taken together, these results yield fundamental new insights into the learning behavior of AlphaFold2-type models and provide new conceptual and practical tools for the development of biomolecular modeling algorithms.
2 Results
Newly trained OpenFold matches AlphaFold2 in accuracy
OpenFold reproduces the AlphaFold2 model architecture in full, without any modifications that could alter its internal mathematical computations. This results in perfect interoperability between OpenFold and AlphaFold2, enabling use of the original AlphaFold2 model parameters within OpenFold and vice versa. To verify that our OpenFold implementation recapitulates all aspects of AlphaFold2 training, we used it to train a new model from scratch. OpenFold/AlphaFold2 training requires a collection of protein sequences, MSAs, and structures. As the AlphaFold2 MSA database has not been publicly released, we generated our own database using the same MSA generation procedure described for AlphaFold2 but substituting newer versions of sequence databases, where available. Starting from approximately 15 million UniClust30 (Mirdita, Driesch, et al. 2017) MSAs, we selected approximately 270,000 diverse and deep MSAs to form a “self-distillation” set; such sets are used to augment experimental training data with high-quality predictions. We predicted protein structures for all MSAs in this set using AlphaFold2 and combined them with approximately 132,000 unique (640,000 non-unique) experimental structures from the Protein Databank (wwPDB Consortium 2018) to form the OpenFold training data set. During training on self-distillation proteins, residues with a low AlphaFold2 confidence score (< 0.5 pLDDT) were masked. Our validation set consisted of nearly 200 structures from CAMEO (Haas et al. 2018), an online repository for continuous quality assessment of protein structure prediction models, drawn over a three-month period ending on January 16, 2022. To facilitate future development of protein modeling systems, we combined the ~400,000 MSAs in our training set with ~4.6 million deep MSAs we derived from UniClust30 and released them as OpenProteinSet, the largest collection of publicly available MSAs. For more details on OpenProteinSet construction procedures, see Appendix B.
From our main training run, we selected seven snapshots to form a collection of distinct (but related) models. During prediction time, these models can generate alternate structural hypotheses for the same protein. To further increase the diversity of this collection, we fine-tuned a second set of models that we branched off from the main model. In this second branch, we disabled the model’s template pipeline, similar to the procedure used for AlphaFold2. Selected snapshots from this branch were added to the pool of final models, resulting in a total of 10 distinct models. Full training details are provided in Appendix D.
We summarize the main results of our training experiment in Figure 1. Predictions made by OpenFold and AlphaFold2 on the CAMEO validation set are assessed using the lDDT-Cα (Mariani et al. 2013) metric (Figure 1A) and show very high concordance between OpenFold and AlphaFold2, demonstrating that OpenFold successfully reproduces AlphaFold2. Figure 1C provides a visual illustration of this concordance. Tracking prediction accuracy as a function of training stage (Figure 1D) reveals the remarkable fact that OpenFold achieves ~90% of its final accuracy in just 1,500 GPU hours (~3% of training time) and ~95% in 2,500 GPU hours; total training time is approximately 50,000 GPU hours. This rapid rise in accuracy suggests that training of new OpenFold variants can be accomplished with far less compute than is necessary for full model training, facilitating rapid exploration of model architectures. We take advantage of this fact in our data elision experiments.

(A) Scatter plot of lDDT-Cα values of AlphaFold and OpenFold predictions on the CAMEO validation set. (B) Average pLDDT vs lDDT-Cα of OpenFold predictions on the CAMEO set during the early stage of training. OpenFold is initially overconfident but quickly becomes underconfident, gradually converging to accurate confidence estimation. (C) Predictions by OpenFold and AlphaFold2 overlayed with an experimental structure of S. tokunonesis TokK protein (Knox et al. 2022; PDB accession code: 7KDX_A). (D) Average lDDT-Cα for OpenFold computed over the training set during the course of training. The template-free branch is shown in green, the template-utilizing one in orange, and the initial-training/fine-tuning boundary in black. Template-free accuracy is initially poor because the exponential moving average of the weights used for validation was being reinitialized.
AlphaFold2 training is broadly split into two phases, an initial training phase and a more computationally intensive fine-tuning phase. In the latter, the size of protein fragments used for training is increased to 384 residues and an additional loss function that penalizes structural violations (e.g. steric clashes) is enabled. By comparing predicted structures between the initial and fine-tuning phases, we find that the second phase has only a modest effect on overall structural quality metrics, even when considering only long proteins greater than 500 residues in length (see Appendix G.1). Instead, the primary utility of fine-tuning appears to be to resolve violations of known chemical constraints. In our training experiments, this occurs quickly after the beginning of fine-tuning, suggesting that elided fine-tuning runs can be used with minimal impact on prediction quality.
In addition to prediction accuracy, we also tracked pLDDT as a function of training stage. pLDDT is the model’s estimate of the lDDT-Cα of predicted structures and serves as its primary confidence metric. We find that pLDDT is well correlated with true lDDT early in training, albeit initially over-confident in its self-assessment and later entering a phase of under-confidence (Figure 1B). It is notable that the model is capable of assessing the quality of its own predictions early on in training, when its overall predictive capacity remains very limited.
OpenFold learns spatial dimensions sequentially
Having established the equivalency of OpenFold and AlphaFold2, we next set out to understand how these systems learn to fold proteins by analyzing structures predicted by OpenFold as it progresses through training. We focus in particular on the first five thousand steps of training, during which the model experiences rapid gains in accuracy. We find a remarkably consistent progression in the spatial dimensionality of predicted structures, summarized in Figure 2A and best visualized in animations we provide in the supplement1. At first, predictions are point-like and zero-dimensional, as the model is initialized to place all residues at the origin (using the so-called “black hole” initialization). Early on, structures extend along a single axis, remaining approximately one-dimensional. A few hundred training steps later, these tubular predictions begin to stretch in an orthogonal dimension to resemble curved, two-dimensional surfaces; a side view of one such structure is shown in Figure 2A. In this stage, flattened secondary structure elements are often clearly visible. Once the two-dimensional extent of the final structure is nearly fully realized, predicted structures begin to inflate and acquire volume along a third orthogonal dimension, arriving at reasonably accurate backbone structures. Finally, after global geometry stabilizes, secondary structure elements begin to acquire precise shape and atomic detail. Subsequent epochs make largely minor, local revisions to secondary structure elements, which we analyze in a later section. We note that this progression differs markedly from that obtained when analyzing structures predicted from intermediate layers of a fully trained model, which are all generally globular and three-dimensional in nature with at least partially-formed secondary structure elements (Jumper et al. 2021).
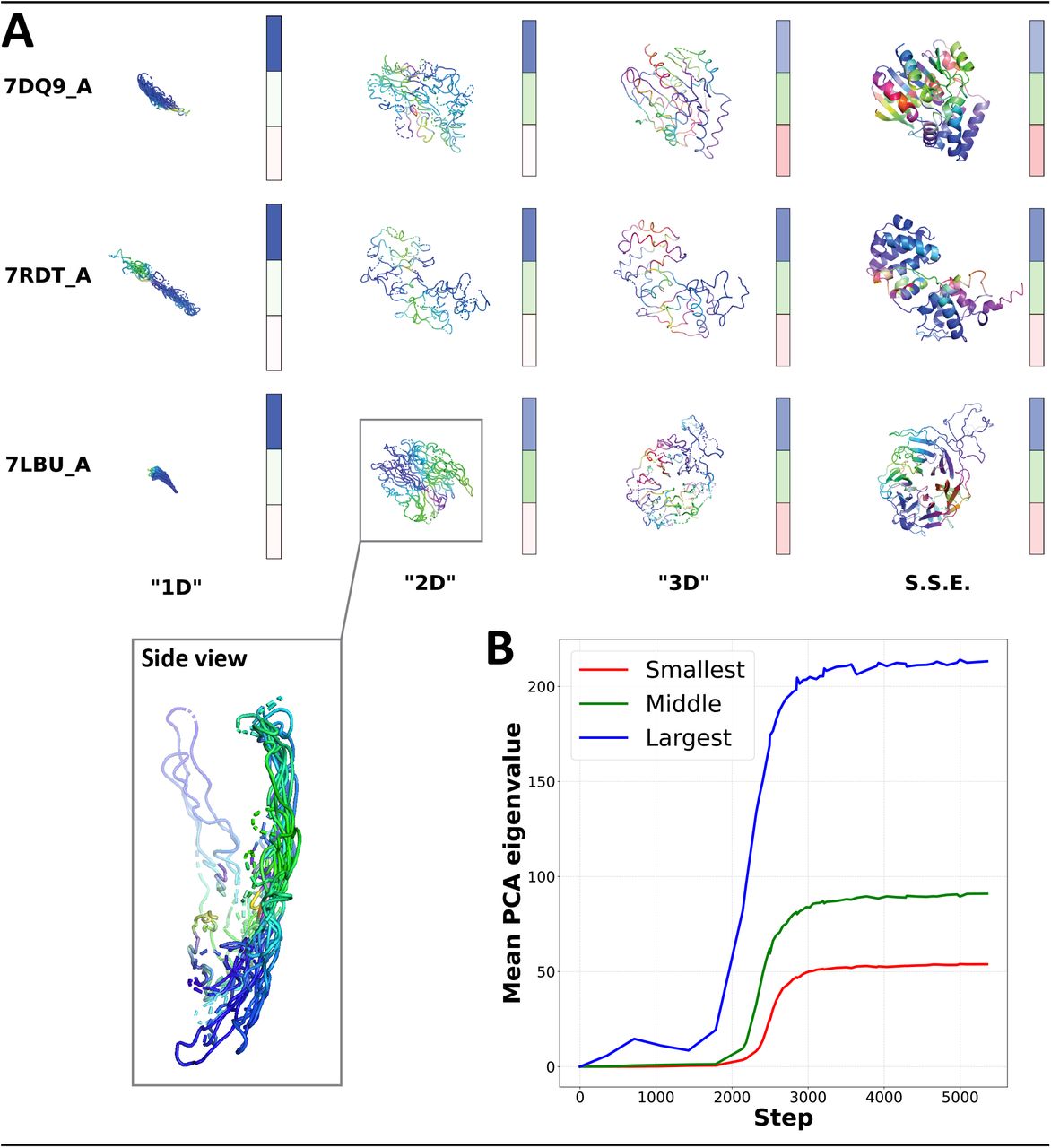
(A) Predicted structures of three validation set proteins as training progresses from left to right. The dimensionality of each prediction appears to grow in stages. Colors (red, green, blue; RGB) are used to index different dimensions by computing RGB values for each residue as the dot product of the residue’s coordinates with each of the prediction’s scaled principal components, respectively. Secondary structure elements (SSE) appear to be refined only after an accurate backbone has formed. Colorbars visualize the relative magnitudes of each structure’s three PCA eigenvalues from largest to smallest (top to bottom). (B) Mean sorted PCA eigenvalues for all proteins in the CAMEO validation set as a function of OpenFold training step.
To formally quantify this sequential learning of spatial dimensions, we apply principal component analysis (PCA) to the atomic coordinates of predicted structures during training. For each protein in our validation set, we predict its structures using partially trained models between training steps 0 and 5,000, during the critical period of rapid early improvement (for reference, full training continues for over 90,000 steps). We compute the principal components of every predicted structure along with its associated eigenvalues, which roughly quantify a structure’s flatness along each of its spatial dimensions. Intuitively, a perfectly spherical structure would have three eigenvalues of approximately equal magnitude, while a completely flat two-dimensional structure would have just two non-zero eigenvalues. We visualize PCA eigenvalues as colorbars for a few sample proteins2 in Figure 2A, where the intensities of the three colored panes shown beside each structure correspond to the magnitudes of the three (sorted) eigenvalues of that structure. We observe that during the 1D phase, the first eigenvalue is dominant, while in the 2D and 3D phases the second and third eigenvalues become discernible, respectively.
To systematically analyze this process we plot the averages, across our entire validation set, of the three eigenvalues of predicted structures in Figure 2B. We observe that the first eigenvalue begins to rise around step 1,500, corresponding to expansion of the first dimension and thus the 1D phase. By step 2,200, the first dimension has expanded substantially when the second eigenvalue begins to increase in value, and quickly thereafter (step 2,400), the third eigenvalue begins to rise while the second and first stabilize. Although it should be noted that individual proteins enter the different phases at slightly different times, the timeline is sufficiently consistent across proteins such that there are clearly visible points in time where one eigenvalue dominates, and then momentarily two, and finally three.
Low-dimensional structures are partially formed PCA projections
During our analysis of structures predicted at intermediate stages of training, we observed that predictions in the 2D phase contain patterns reminiscent of two-dimensional projections of the secondary structure elements of the final globular structure. The alpha helices of PDB structures 7DQ9_A and 7RDT_A, for example, appear as flat spirals; the beta sheets of 7LBU_A resemble wavy lines (Figure 2A). This fact contributes to a distinct impression that predictions don’t simply undergo phases of dimensionality, but that during each phase they “grow” predominantly along the new dimension corresponding to that phase. More precisely, the model appears to learn to generate successive lower dimensional PCA projections of the true 3D structures, first learning a 1D PCA projection of the final structure, then a 2D PCA projection, and finally the full 3D structure.
To investigate this learning hypothesis, we created 1D and 2D PCA projections of predicted structures at each timestep and compared them to the full 3D prediction at training step 5,000, at the end of the rapid rise in accuracy, using the translationally- and rotationally-invariant distance-based root mean square deviation (dRMSD) metric (Koehl 2001). Results are shown in Figure 3A. Before the model exits the 1D and 2D prediction phases, the full (non-projected) predictions (“3D”) and their lower dimensional projections (“1D” and “2D”) are almost indistinguishable from each other, as expected; at these stages, as we previously described, predicted structures are essentially one- or two-dimensional. Thereafter, as the predictions gain dimensions, their dRMSDs to the final structure diverge from those of the lower dimensional projections. Remarkably, much of the overall drop in dRMSD occurs before the 2D and 3D projections diverge, indicating that the model is improving its accuracy in lower dimensions before moving on to higher dimensions. It does not perfect each projection before transitioning, however. Figure 3B shows a similar experiment to 3A except low-dimensional projections for each training iteration are compared to corresponding low-dimensional projections of the final prediction at step 5,000. In the extreme, if the model were learning perfect low-dimensional PCA projections of the final 3D structure at the end of each low-dimensional training phase, the 2D projections of the intermediate and final predictions would match exactly at the end of the 2D phase. Additionally, since the 3D prediction is essentially flat at the end of the 2D phase, its dRMSD with respect to the 3D structure should be high. Such a separation is not visible in Figure 3B; instead, all three projections converge to their final counterparts at nearly the same time. This suggests that while the model learns crude approximations of low-dimensional PCA projections during each phase, its learning is largely continuous, with all spatial dimensions continuing to be refined until the end. However, the degree to which the dominant dimensions continue to be refined diminishes over time relative to less dominant ones.

(A) Mean dRMSD, as a function of training step, between low-dimensional PCA projections of predicted structures and the final 3D prediction at step 5,000 (denoted by *). Averages are computed over the CAMEO validation set. Insets show idealized behavior corresponding to unstaggered, simultaneous growth in all dimensions and perfectly staggered growth. Empirical training behavior more closely resembles the staggered scenario. (B) Low-dimensional projections as in (A) compared to projections of the final predicted structures at step 5,000. (C) Mean displacement, as a function of training step, of Cα atoms along the directions of their final structure’s PCA eigenvectors. Results are shown for two individual proteins (PDB accession codes 7DQ9_A and 7RDT_A). Shaded regions correspond loosely to the “1D,” “2D,” and “3D” phases of dimensionality.
To better assess this phenomenon and gain a finer-grained view of the progress that occurs during each phase, we analyzed the movement of atoms along the directions of the final prediction’s principal components as a function of training step. Because predicted structures and their associated principal components are in principle quite mobile over the course of training, this movement is difficult to characterize precisely. Nonetheless, we devised the following scheme to estimate it. Given n predicted structures in chronological order, for each i in {n− 1, …, 1}, we align the ith structure to the i + 1th structure sequentially. Then, for every pair of consecutive predictions, we compute the absolute value of the displacement of each Cα atom along the directions of the three principal components of the final prediction. In Figure 3C we sequentially plot these displacements for two CAMEO proteins. As before, there is not a perfect separation between phases, and discernible motion occurs in all three directions in every phase. This is compounded by the fact that predictions in the “2D phase” are generally not flat two-dimensional sheets but instead exhibit some degree of curvature and thus produce spurious movement in the third dimension. Nevertheless, both proteins show clearly differentiated spikes in each phase corresponding to rapid expansion in the phases’ respective dimensions. Furthermore, each protein nears its maximum spatial extent in each principal direction during the corresponding phase. After the 1D phase, for example, growth along the direction of the first principal component is greatly subdued.
Intuitively, the model appears to exhaust the easiest gains in the most dominant dimensions before proceeding to the less dominant ones, making relatively minor adjustments in previous dimensions thereafter.
Learning of secondary structure is staggered and multi-scale
The preceding analysis suggests that secondary structure elements (SSEs) are learned subsequent to tertiary structure. We next set out to formally confirm this observation and chronicle the order in which distinct SSEs are learned. For every protein in our validation set and every step of training, we used DSSP (Kabsch and Sander 1983) to identify residues matching the eight recognized SSE states. We treat as ground truth DSSP assignments of residues in the experimental structures, and compute F1 scores as a combined metric of the recall and precision achieved by the model for every type of SSE at various training steps (Figure 4A).

(A) F1 scores for secondary structure categories over time. The corner pane depicts the same data using a simplified 3-state assignment (details in Appendix G.3). GDT-TS and final values are also provided. (B) Corresponding counts of individual secondary structure assignments. (C) Contiguous fractions of individual helices recovered early in training.
We observe a clear sequence in which SSEs are discovered: alpha helices are learned first, followed by beta sheets, followed by less common SSEs. Unsurprisingly, this sequence roughly corresponds to the relative frequencies of SSEs in proteins (Figure 4B), with the exception of uncommon helix variants. As was previously evident, the model’s discovery of SSEs lags that of accurate global structure. For instance, the F1 score for beta sheets (‘E’) only plateaus hundreds of steps after global structural accuracy, as measured by GDT-TS (Zemla 2003). This is also clearly visible in our animations of progressive training predictions; for each protein, secondary structure is recognized and rendered properly only after global geometry is essentially finalized.
To investigate the possibility that OpenFold is achieving high alpha helical F1 scores by gradually learning small fragmentary helices, we binned predicted helices by the longest contiguous fraction of the ground-truth helix they recover and plotted the resulting histogram as a function of training step in Figure 4C. Evidently, little probability mass ever accumulates between 0.0—helices that are not recovered at all—and 1.0—helices that are completely recovered. This suggests that, at least from the perspective of DSSP, most helices become correctly predicted essentially all at once. This sudden transition coincides with most of the improvement in helix DSSP F1 scores in the early phase of training.
Based on the above observation, we reasoned that as training progresses, OpenFold may first learn to predict smaller structural fragments before larger ones, and that this may be evident on both the tertiary and secondary structure levels. Focusing first on tertiary structure, we assessed the prediction quality, at each training step, of all non-overlapping fragments of length 10, 20, and 50 residues in our validation set. Average GDT-TS values are shown in Figure 5A. Unlike global GDT-TS (pink), which improves minimally in the first 300-400 steps of training, fragment GDT-TS improves markedly during this phase, with shorter fragments showing larger gains. By step 1,000, when the model reaches a temporary plateau, it has learned to predict local structure far better than global structure (GDT-TS > 50 for 10-residue fragments vs GDT-TS < 10 for whole proteins). Soon after, at step 1,800, the accuracy of all fragment lengths including global structure begin to rise rapidly. However, the gains achieved by shorter fragments are smaller than those of longer fragments, such that the gap between 10-residue fragments and whole proteins is much smaller at step 3,000 than 1,800 (GDT-TS ~90 for 10-residue fragments vs. GDT-TS ~70 for whole proteins). This trend continues until the model is fully trained, where the gap between 10-residue fragments and whole proteins shrinks to a mere 10 GDT-TS points. Thus, while the model ultimately learns to predict global structure almost as well as local structure, it first learns to predict the latter.

(A) Mean GDT-TS and dRMSD-Cα validation scores as a function of training step for non-overlapping protein fragments of varying lengths (colorbars indicate fragment length). (B) Average contact F1 score (8Å threshold) and dRMSD for predicted alpha helices and beta sheets of varying lengths and number of strands, respectively, as a function of training step. Colorbars indicate the weighted average of the lengths and widths of helices and sheets in each bin, respectively.
Turning to secondary structure, we investigated whether the same multi-scale learning behavior is detectable when examining SSEs. As before, we treat as ground truth the DSSP classifications of experimental structures in our validation set, focusing exclusively on alpha helices and beta sheets. We bin both SSEs according to size, defined as number of residues for alpha helices and number of strands for beta sheets. As uniform binnings would result in highly imbalanced bins, we instead opt for a dynamic binning procedure. First, each SSE is assigned to a (potentially imbalanced) bin that corresponds to its size. Bins are then iteratively merged with adjacent bins, subject to the condition that no bin exceed a maximum size (in this case, 200 for helices and 30 for sheets), until no further merges can occur. Finally bins below a minimum bin size (20 for both) are unconditionally merged with adjacent bins. We compute metrics averaged over each bin independently (Figure 5B).
Similar to what we observe for tertiary structure, short helices and narrow sheets are better predicted during earlier phases of training than their longer and wider counterparts, respectively. Improvement in SSE accuracy coincides with the rapid rise in tertiary structure accuracy, albeit shifted, as we observed in Figure 4A. Notably, the final quality of predicted SSEs is essentially independent of length/width despite the initially large spread in prediction accuracies, suggesting that OpenFold ultimately becomes scale-independent in its predictive capacity, at least for secondary structure. We note that the identification of SSEs is performed by DSSP, which is sensitive to the details of their hydrogen bonding networks. It is possible that in earlier phases of training, OpenFold has already recovered aspects of secondary structure not recognized by DSSP on account of imprecise atom positioning.
OpenFold can achieve high accuracy using small training sets
AlphaFold2 was trained using ~132,000 protein structures from the PDB, the result of decades of painstaking and expensive experimental structure determination efforts. For other molecular systems for which AlphaFold2-style models may be developed, data is far more sparse; e.g., the PDB contains only 1,664 RNA structures. We wondered whether the high accuracy achieved by AlphaFold2 in fact depended on very large training sets, or if it is possible to achieve comparable performance using less data. Were the latter to be true, it would suggest broad applicability of the AlphaFold2 paradigm to molecular problems. To investigate this possibility, we performed a series of OpenFold training runs in which we used progressively less training data, assessing model accuracy as a function of training set size.
Our first set of tests randomly subsample the original training data to 17,000, 10,000, 5,000, 2,500, 2,000, and 1,000 protein chains. We used each subsampled set to train OpenFold for at least 7,000 steps, through the initial rapid rise phase to early convergence. To avoid information leakage from the full training set, we did not use self-distillation, putting the newly trained models at a disadvantage relative to the original OpenFold. We trained models with and without using structural templates. In all other regards, training was identical to that of the standard OpenFold model. Model accuracy (assessed using lDDT-Cα) is plotted as a function of training step in Figure 6A, with colors indicating size of training set used.

(A) Validation set lDDT-Cα as a function of training step for models trained on elided training sets (10k random split repeated 3x). (B) Same as (A) but for CATH-stratified dataset elisions. Validation sets vary across stratifications and are not directly comparable. (C) Experimental structures (orange) and mainly alpha-trained (purple) and mainly beta-trained (green) predictions of largely helical Lsi1 (top) and beta sheet-heavy TMED1 (bottom).
We find that merely 10, 000 protein chains—about 7.6% of all training data (yellow curves)—suffice to reach essentially the same initial lDDT-Cα as a model trained on the full training set (pink curve). After 20,000 steps (not pictured), the full data model reaches a peak lDDT-Cα of 0.83, while after 7,000 steps, the 10,000-sample model has already exceeded 0.81 lDDT-Cα. Although performance gradually degrades as training set size decreases further, we find that all models are surprisingly performant, even ones trained on our smallest subsample of 1,000 protein chains, corresponding to just 0.76% of the full training set. In fact, this model reaches an lDDT-Cα of 0.64, exceeding the median lDDT-Cα of 0.62 achieved at CASP13 by the first AlphaFold, the best performing model at the time.
Comparing the accuracies of models trained with and without templates, we find that templates on average contribute little to prediction quality even in the low-data setting. This is consistent with the original AlphaFold2 ablation studies which showed that templates have a minimal effect except when MSAs are shallow or entirely absent.
OpenFold generalizes to unseen regions of fold space
Randomly subsampling the OpenFold training set, as in the preceding analysis, reduces the quantity of the training data used but not necessarily its overall diversity. In molecular modeling tasks, the data available for training often does not reflect the underlying diversity of the molecular system being modeled, due to biases in the scientific questions pursued, experimental assays available, etc. To assess OpenFold’s capacity to generalize to out-ofdistribution data, we subsample the training set in a structurally stratified manner such that entire regions of fold space are excluded from training but retained for model assessment. Multiple structural taxonomies for proteins exist, including the hierarchical CATH (Orengo et al. 1997, Sillitoe et al. 2021) and SCOP (Andreeva et al. 2020) classification systems. For this task we use CATH, which assigns protein domains—in increasing order of specificity— to a (C)lass, (A)rchitecture, (T)opology, and (H)omologous superfamily. Domains with the same homologous superfamily (H) classification may differ superficially but have highly similar structural cores. Our preceding analysis can be considered to structurally stratify data at the H level. For the present analysis, we stratify data further, holding out entire topologies (T), architectures (A), and classes (C).
We start by filtering out protein domains that have not been classified by CATH, leaving ourselves with ~440,000 domains spanning 1,385 topologies, 42 architectures, and 4 classes. For the topology stratification, we randomly sample 100 topologies and remove all associated chains from the training set. We construct a validation set from the held out topologies by sampling one representative chain from each. We also construct successively smaller training sets from shrinking fractions of the remaining topologies, including a training set that encompasses all of them. We follow an analogous procedure for architectures except that in this case, the validation set consists of 100 chains randomly selected from 5 architectures (20 per architecture). For class-based stratification, the validation set comprises domains that are neither in the mainly alpha nor mainly beta classes, hence enriching for domains with high proportions of both SSEs. For training, we construct two sets, one corresponding exclusively to the mainly alpha class and another to the mainly beta class—this enables us to ascertain the capability of models trained largely on either alpha helices or beta sheets to generalize to proteins containing both. For all stratifications, we train OpenFold to early convergence (~7,000 steps) from scratch. To prevent leakage of structural information from held out categories, all runs are performed without templates. We plot model accuracies as a function of training step in Figure 6B, with colors indicating the fraction of categories retained during training for each respective level of the CATH hierarchy.
As expected, removing entire regions of fold space has a more dramatic effect on model performance than merely reducing the size of the training set. For example, retaining 10% of topologies for training (green curve in Figure 6B, topology split), which corresponds to ~6,400 unique chains, results in a model less performant than one containing 5,000 randomly selected chains (green curve in Figure 6A, no templates). However, even in the most severe elisions of training set diversity, absolute accuracies remain unexpectedly high. For instance, the training set containing 5% of topologies (2,000 chains) still achieves an lDDT-Cα near 0.6, comparable again to the first AlphaFold, which was trained on over 100,000 protein chains. Similarly, the training set for the smallest architecture-based stratification only contains domains from one architecture (out of 42 that cover essentially the entirety of the PDB), yet it peaks near 0.6 lDDT-Cα. Most surprisingly, the class-stratified models, in which alpha helices or beta sheets are almost entirely absent from training, achieve very high lDDT-Cα scores of >0.7 on domains containing both alpha helices and beta sheets. These models likely benefit from the comparatively large number of unique chains in their training sets—15,400 and 21,100 for alpha helix- and beta sheet-exclusive sets, respectively. It should also be noted that the mainly alpha and mainly beta categories do contain small fractions of beta sheets and alpha helices, respectively (see Supplementary Figure 8). Despite these caveats, the model is being tasked with a very difficult out-of-distribution generalization problem in which unfamiliar types of SSEs (from the perspective of the training set) have to essentially be inferred with minimal quantities of corresponding training data. Taken together, these results show that the AlphaFold2 architecture is capable of remarkable feats of generalization.
To better understand the behavior of class-stratified models, we analyzed the structures of two protein domains, one composed almost exclusively of alpha helices (rice Lsi1 aquaporin domain (Saitoh et al. 2021)) and another of beta sheets (human TMED1 domain (Mota et al. 2022)), as they are predicted by models trained on the mainly alpha or mainly beta datasets. In the top row of Figure 6C, we show an experimental structure (orange) for Lsi13 along with predictions made by the mainly alpha-trained model (purple) and mainly betatrained model (green). In the bottom row we show similar figures for TMED14. Predictably, the mainly alpha-trained model accurately predicts the alpha helices of Lsi1 but fails to properly form beta sheets for TMED1 and incorrectly adopts a small alpha helix in part of the structure. The mainly beta-trained model has the opposite problem: its Lsi1 prediction contains poorly aligned helices and an erroneous beta sheet, but TMED1 is reasonably well predicted. Notably, however, neither fails catastrophically. Regions corresponding to the beta sheets of Lsi1 are predicted by the mainly alpha model with approximately the right shape, except that their atomic coordinates are not sufficiently precise to enable DSSP to classify them as beta sheets.
Generalization capacity is scale-dependent
OpenFold’s surprising capacity for generalization across held out regions of fold space suggests that it is somewhat indifferent to the diversity of the training set at the global fold level. Instead, the model appears to learn how to predict protein structures from local patterns of MSA/sequence-structure correlations—fragments, secondary structure elements, individual residues, and so on—rather than from global fold patterns captured by CATH. This raises the possibility that the model’s capacity for generalization depends on the spatial scale of the prediction task. To directly test this hypothesis, we assessed model accuracy on protein fragments of increasing length using both the GDT_TS and lDDT-Cα metrics as a function of the fraction of the training set retained for topology- and architecture-stratified models (Figure 7). Note that lDDT-Cα is less sensitive to global fit than GDT_TS. To make results directly comparable between different stratifications, we used a common validation set derived from CAMEO. This validation set likely contains domains from all CATH categories and may thus overestimate accuracies.
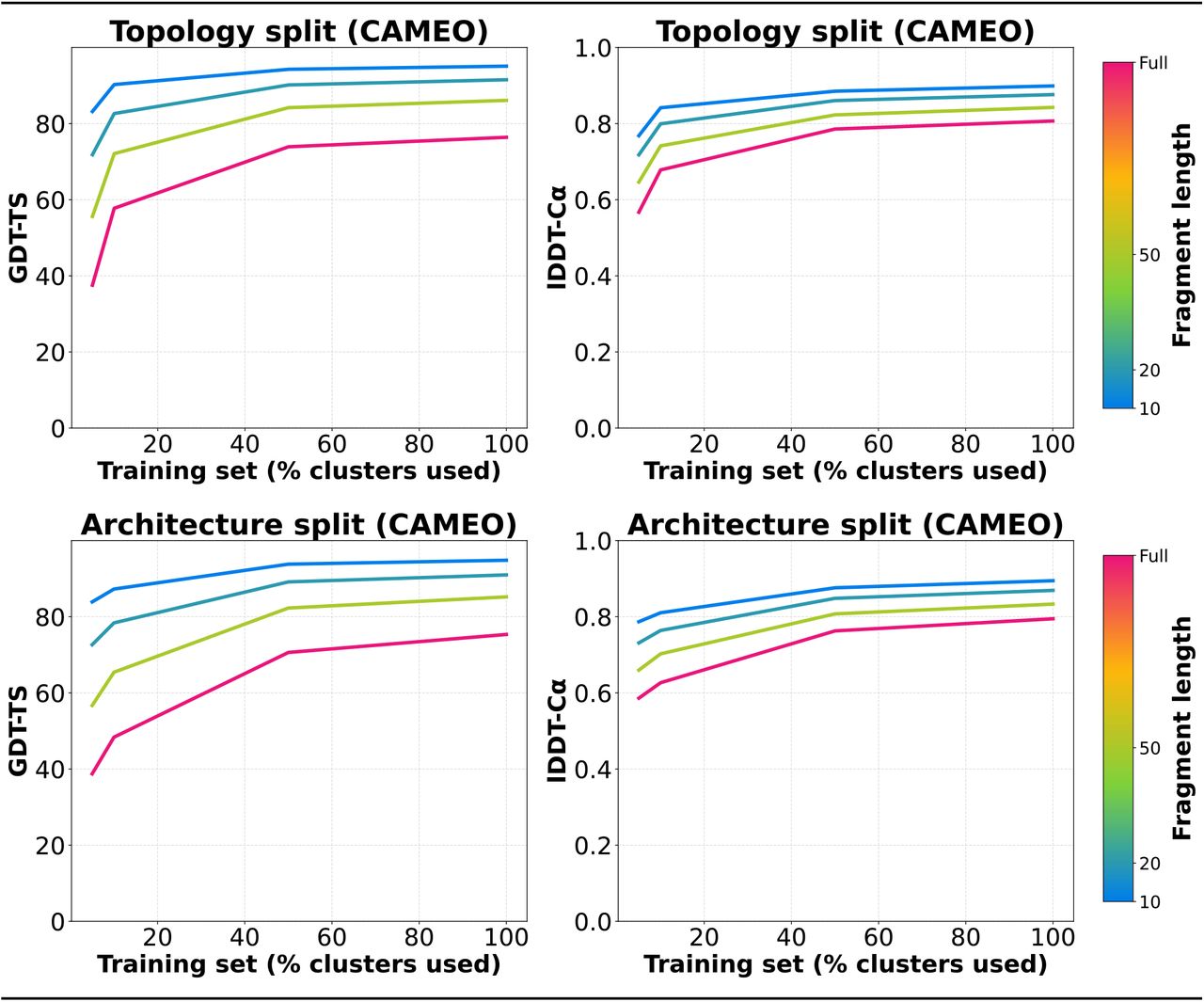
Mean GDT-TS and lDDT-Cα of non-overlapping protein fragments from CAMEO validation set as a function of the percentage of CATH clusters in elided training sets. Data for both topology and architecture elisions are included. Fragmenting procedure is the same as that described in Figure 5A.
We observe that fragments of all lengths are better predicted when more data is used for training. However, the relative gains seen by larger fragments and whole domains (pink) far exceed those seen by smaller fragments (blue), consistent with the hypothesis that generalization capacity is length-dependent. In particular, it indicates that local structure can in fact be robustly learned from highly elided data sets, while global structure is more dependent on broad representation of fold diversity in the training set.
OpenFold is more efficient and trains more stably than AlphaFold2
While the OpenFold model we used in all of the above experiments perfectly matches the computational logic of AlphaFold2, we have additionally implemented a number of changes that minimally alter model characteristics but improve ease of use and performance when training new models and performing large-scale predictions.
First, we made several improvements to the data preprocessing and training procedure, including a low-precision (“FP16”) training mode that facilitates model training on commercially available GPUs. Second, we introduced a change to the primary structural loss, FAPE, that enhances training stability. In the original model, FAPE is clamped—i.e., limited to a fixed maximum value—in a large fraction of training batches. We find that in the dynamic early phase of training, this strategy is too aggressive, limiting the number of batches with useful training signal and often preventing timely convergence. Rather than clamping entire batches in this fashion, we instead clamp the equivalent fraction of samples within each batch, ensuring that each batch contains at least some unclamped chains. In doing so, we are able to substantially improve training stability and speed up model convergence (see Figure 8 and Appendix C.2).

lDDT-Cα and dRMSD-Cα on CAMEO validation set as a function of training step for five independent training runs with (orange) and without (blue) new FAPE clamping protocol. Runs using old protocol exhibit substantial instability with two rapidly converging runs, two late converging runs, and one non-converging run. In contrast, all 15 independent runs using the new protocol converge rapidly. Runs using the new protocol also reach high accuracy faster.
Third, we made optimizations that improve memory efficiency during training, when model weights are continually updated to optimize model behavior for prediction, and inference, when the model is used to make new predictions. In AlphaFold2, the computational characteristics of these two modes vary greatly. To save memory at training time, which requires storing intermediate computations during the optimization procedure, AlphaFold2 and OpenFold are evaluated on short protein fragments ranging in size from 256 to 384 residues.
At inference time, intermediate computations need not be stored, but input sequences can be more than ten times longer than the longest fragments encountered during training. Since the model’s memory usage naively grows cubically with input length, inference-time prediction stresses modules that are not necessarily bottlenecks at training time. To satisfy both sets of desiderata and enhance model efficiency, we implemented a number of training- and inference-specific optimizations. These optimizations create trade-offs between memory consumption and speed that can be tuned differently for training and inference. They include advanced implementations of neural network attention mechanisms (Vaswani et al. 2017) with favorable properties for unusually short and long sequences (Rabe and Staats 2021, Dao et al. 2022), module refactoring for lower memory usage, optional approximations of certain computations that reduce the memory burden, and specialized low-level code customized for GPU hardware. For technical details see appendices F.1 and F.2.
Taken together, these optimizations result in a substantially more efficient implementation than AlphaFold2. We report OpenFold runtimes in Table 1. During inference, OpenFold is up to three times faster than AlphaFold2 for proteins shorter than 1,100 residues. AlphaFold is faster than OpenFold for proteins between 1,100 and 2,400 residues in length, but thereafter AlphaFold2 crashes on single GPUs due to memory constraints. OpenFold runs successfully on longer proteins and complexes, with single-GPU predictions reaching up to 4,700 residues. OpenFold training speed matches or improves upon that of AlphaFold2, as reported by other researchers using OpenFold (Cheng et al. 2022, Li et al. 2022).
Prediction runtimes in seconds on a single A100 NVIDIA GPU for OpenFold and AlphaFold2 on proteins of varying lengths. OpenFold is faster than AlphaFold2 for proteins shorter than 1,100 residues and is able to predict longer proteins than AlphaFold2 on the same hardware.
3 Discussion
We have developed OpenFold, a complete open-source reimplementation of AlphaFold2 that includes training code and data. By training OpenFold from scratch and matching the accuracy of AlphaFold2, we have demonstrated the reproducibility of the AlphaFold2 model for protein structure prediction. Furthermore, the OpenFold implementation introduces technical advances over AlphaFold2, including markedly faster prediction speed. It is built using PyTorch, the most widely used deep learning framework, facilitating incorporation of OpenFold components in future machine learning models.
OpenFold immediately makes possible two broad areas of advances: (i) deeper analyses of the strengths, weaknesses, and learning behavior of AlphaFold2-like models and (ii) development of new (bio)molecular models that take advantage of AlphaFold2 modules. In this work, we have focused on the former. First, by analyzing predicted structures of partially trained models, we discovered that AlphaFold2-like models learn spatial dimensions sequentially. This behavior has implications for the design of model architectures and training regimens. For example, integrating physical priors into machine learning models is an area of outstanding scientific interest (Karniadakis et al. 2021). Efforts at such syntheses have had mixed results, and, indeed, AlphaFold2 serves as a seminal example of a highly successful model that is almost entirely devoid of physical priors. Its learning behavior illustrates why incorporating such priors would be difficult—during the collapsed 1D and 2D phases of learning, all predicted structures exhibit gross violations of basic chemical laws with numerous steric clashes. Forbidding such violations however would drastically alter AlphaFold2’s learning behavior. In fact, in the original AlphaFold2 paper, it is observed without further elaboration that enabling a violation loss to penalize steric clashes and non-physical bond lengths destabilizes training. Our observation of the spatially collapsed learning phase provides an explanation for this observation. The solution that AlphaFold2 adopts for this problem, namely to penalize against physical violations only in later stages of training, suggests a broader strategy to tackle the incorporation of physical priors: a curriculum learning approach in which models are first free to extract information and learn from data, after which more complex physical priors can be gradually introduced to boost the model’s capacity for generalization. Analyzing learning trajectories, as we have done for OpenFold, provides a concrete timeline for when such priors can be injected into the training process.
Second, we observed that the spatially collapsed phases correspond to imperfect lower-dimensional PCA projections of the final predicted structure. Why this occurs is not a priori obvious, given that other end-to-end differentiable protein structure models do not exhibit the same behavior (see e.g., AlQuraishi 2019). Although we do not have direct evidence, we suspect that aspects of the AlphaFold2 architecture—specifically the FAPE loss function— likely drive this phenomenon. We speculate that the PCA-like progression allows the model to greedily minimize error by solving problems with the biggest payoff to the FAPE loss first, which by definition lie along the largest principal component of the ground-truth structure. Once solved, the model moves on to smaller problems lying along other, lower-dimensional projections. Were this to be the case, the staggering of spatial dimensions during learning would be contingent on the geometry of proteins in the training set. The extreme case of a training set composed entirely of long, slim, tubular proteins would produce even more dramatically staggered phases. Conversely, a training set composed of perfectly spherical proteins would exhibit even growth along all spatial dimensions. This behavior would be a function of the overall training set and would not necessarily get reflected in individual proteins. For instance, the “sphere”-like type-A feruloyl esterase protein5 shown in Figure 2 undergoes staggered dimensional expansion, consistent with our training set being broadly representative of protein fold space. Regardless, these observations suggest that it may be possible to deliberately simplify other difficult problems in molecular modeling with a learning curriculum in which “toy” models are first trained to predict lower-dimensional projections of target molecules (or more generally, geometric objects) before being tasked to predict their fully realized instantiations.
Third, we assessed OpenFold’s capacity to learn from training sets substantially reduced in size. Remarkably, we found that even a 100-fold reduction in dataset size (0.76% models in Figure 6A) results in models more performant than the first version of AlphaFold. Stated differently, the architectural advances introduced in AlphaFold2 enable it to be 100x more data efficient than its predecessor, which at the time of its introduction set a new state of the art. These results demonstrate that architectural innovations can have a more profound impact on model accuracy than larger datasets, particularly in domains where data acquisition is costly or time-consuming, as is often the case in (bio)molecular systems. However, it merits noting that AlphaFold2 in general learns MSA-structure, not sequence-structure, relationships. MSAs implicitly encode a substantial amount of structural knowledge, as evidenced by early co-evolution-based structure prediction methods which were entirely unsupervised, making no use of experimental structural data (Marks et al. 2011, Sułkowska et al. 2012). Hence, the applicability of the AlphaFold2 architecture to problems that do not exhibit a co-evolutionary signal remains undemonstrated.
Our data elision results can be interpreted in light of recent work on large transformer-based language models that has revealed broadly applicable “scaling laws” that predict model accuracy as a simple function of model size, compute utilized, and training set size (Kaplan et al. 2020, Hoffmann et al. 2022). When not constrained by any one of these three pillars, models benefit from investments into the other two. These observations have largely focused on transformer-based architectures, of which AlphaFold2 is an example, but more recent work has revealed similar behavior for other architectures (Tay et al. 2022). Although determining the precise scaling properties of AlphaFold2 is beyond the scope of the present study, our results suggest that it is hardly constrained by the size or diversity of the PDB, motivating potential development of larger instantiations of its architecture.
OpenFold lays the groundwork for future efforts aimed at improving the AlphaFold2 architecture and repurposing it for new molecular modeling problems. Since the release of our codebase in November 2021, there have been multiple efforts to build upon and extend OpenFold. These include the ESMFold method for protein structure prediction (Lin et al. 2022), which replaces MSAs with protein language models (Alley et al. 2019, Chowdhury et al. 2022, Wu et al. 2022), and FastFold, a community effort that has implemented significant improvements including fast model-parallel training and inference (Cheng et al. 2022). We expect future work to go further by disassembling OpenFold to attack problems beyond protein structure prediction. For instance, the evoformer module is a general purpose primitive for reasoning over evolutionarily related sequences. DNA and RNA sequences also exhibit a co-evolutionary signal, with efforts aimed at predicting RNA structure from MSAs fast materializing (e.g., Singh et al. 2022, Baek et al. 2022, Pearce et al. 2022). It is plausible that even more basic questions in evolutionary biology, such as phylogenetic inference, may prove amenable to evoformer-like architectures. Similarly, AlphaFold2’s structure module, and in particular the invariant point attention mechanism, provide a general purpose approach for spatial reasoning over polymers, one that may be further extendable to arbitrary molecules. We anticipate that as protein structures and other biomolecules shift from being an output to be predicted to an input to be used, downstream tasks that rely on spatial reasoning capabilities will become increasingly important (e.g., McPartlon, Lai, et al. 2022, McPartlon and Xu 2022). We hope that OpenFold will play a key role in facilitating these developments.
Code availability
OpenFold can be accessed at https://github.com/aqlaboratory/openfold
It is available under the permissive Apache 2 Licence.
Data availability
OpenProteinSet and OpenFold model parameters are hosted on the Registry of Open Data on AWS (RODA) and can be accessed at https://registry.opendata.aws/openfold/
Both are available under the permissive CC BY 4.0 license.
Author contributions
G.A. wrote and optimized the OpenFold codebase, generated data, trained the model, performed experiments, and maintained the GitHub repository. S.K. wrote data preprocessing code. G.A., N.B., and M.A. conceived of and managed the project, designed experiments, analyzed results, and wrote the manuscript. G.A., B.Z., Z.Z., N.Z., and A.N. ran ablations. All authors read and approved the manuscript. The Flatiron Institute provided compute for ablations, all data generation, and our main training experiments. NVIDIA performed training stability experiments, fixed critical bugs in the codebase, added new model features, and provided compute for ablations. StabilityAI provided compute for ablations.
Competing interests
M.A. is a member of the Scientific Advisory Boards of Cyrus Biotechnology, Deep Forest Sciences, Nabla Bio, Oracle Therapeutics, and FL2021-002, a Foresite Labs company. P.K.S. is a member of the Scientific Advisory Board or Board of Directors of Glencoe Software, Applied Biomath, RareCyte, and NanoString and is an advisor to Merck and Montai Health.
Acknowledgements
We would like to thank the Flatiron Institute, OpenBioML, Stability AI, the Texas Advanced Computing Center, and NVIDIA for providing compute for experiments in this paper and Amazon Web Services for hosting OpenProteinSet. Individually, we would like to thank Milot Mirdita, Martin Steinegger, and Sergey Ovchinnikov for providing valuable support and expertise. This research used resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper. N.B. is supported by DARPA PANACEA program grant HR0011-19-2-0022 and NCI grant U54-CA225088. B.Z. and Z.Z. are supported by grants NSF OAC-2112606 and OAC-2106661.
Appendix
A Related work
We first released the trainable and PyTorch-based OpenFold codebase in November 2021. In December 2021, UniFold (Li et al. 2022) released JAX-based AlphaFold2 training code, and in August 2022, the same team released a PyTorch-based implementation derived from OpenFold along with weights, training data, and new training code for AlphaFold-Multimer. FastFold (Cheng et al. 2022), a modification of OpenFold that speeds up training and permits inference across multiple GPUs, was released in March 2022. There have additionally been a number of AlphaFold2 reimplementations using less widely used frameworks. MEGA-Protein (MindSpore 2022) and HelixFold (G. Wang et al. 2022), both released in July 2022, are implemented using MindSpore and PaddlePaddle (Ma et al. 2019), respectively. HelixFold claims to reduce AlphaFold and OpenFold training times by approximately 40%. With the exception of UniFold, none of the other projects have released model parameters or training data. The version of OpenFold (v1.0.1) released with this manuscript contains new inference and training optimizations that were previously unavailable.
B OpenProteinSet details
OpenProteinSet consists of nearly 5 million unique MSAs, making it the largest publicly available MSA database. For ~400,000 of those entries, constituting the OpenFold training set, we provide additional MSAs—computed using Mgnify (Mitchell et al. 2020), Uni-Clust30 (Mirdita, Driesch, et al. 2017), BFD (Jumper et al. 2021), and UniRef90 (Suzek et al. 2013)—and template hits retrieved from PDB70 (Steinegger et al. 2019). These were generated with multiple sequence databases and alignment tools, as in the AlphaFold2 paper. JackHMMer (Johnson et al. 2010) was used to search MGnify and UniRef90; HHblits-v3 was used to search BFD and Uniclust30. Templates were computed using HHSearch (Remmert et al. 2012) run on the UniRef90 MSA and then realigned, if necessary, using Kalign (Lassmann et al. 2009).
As in the original AlphaFold2 procedure, we changed some of the default options for the MSA generation tools. For JackHMMer, we used
For HHBlits, we used
To generate MSAs for the ~270,000 protein chain self-distillation set, we performed an all-against-all search on UniClust30 using HHblits-v3 with the same parameter settings as before. This yielded approximately 15 million MSAs. Using the first sequence in each cluster as a representative, we iteratively removed MSAs whose representative chains appeared in the greatest number of other MSAs until each representative chain appeared only in its own MSA. We then removed clusters whose representative sequences were longer than 1,024 residues or shorter than 200. Finally, we removed clusters whose corresponding MSAs contained fewer than 200 sequences, leaving just 270,262 MSAs in total. Template hits were again computed using HHsearch against PDB70. To speed up the expensive training process, we generated structures for the self-distillation set using OpenFold run with AlphaFold2 weights rather than a pretrained version of OpenFold.
The remainder of OpenProteinSet consists of ~4.85 million Uniclust30 MSAs with depth >50 that were filtered from the core distillation set by this process.
C Differences between OpenFold and AlphaFold2
In this section we describe additions and improvements we made to OpenFold subject to the constraint that the weights of the two models should be interchangeable. We also describe our design decisions in the handful of cases where the AlphaFold2 paper was ambiguous.
C.1 Changes to the data pipeline
Template trick
During AlphaFold2 training, structural template hits undergo two successive rounds of filtering. Between these two rounds, the dataloader parses the template structure data. The top 20 template hits to pass both filters are shuffled uniformly at random. Finally, the dataloader samples a number of templates uniformly at random in the range [0, 4] and draws that many samples from the shuffled pool of valid hits. These are then passed as inputs to the model. This subsampling process is intended to lower the average quality of templates seen by the model during training. We note that preemptively parsing structure files for each template hit during the filtering process is an expensive operation and, for proteins with many hits, considerably slows training. For this reason, we replace the original algorithm with an approximation. Instead of sampling hits from the top 20 template candidates from the pool of templates that pass both sets of filters, we use the top 20 hits to pass the first filter. These are then shuffled and subsampled as before. Only when a hit is drawn is it passed through the second filter; hits that fail to pass the second filter at this point are discarded and replaced. If not enough hits in the initial 20-sample pool pass the second filter, we continue drawing candidates from the top hits outside that pool, without further shuffling. This procedure has the disadvantage that, if too many hits pass the first filter but not the second, the hits used for the model are not shuffled. Even in cases where only x of the initial hits fail to pass the second filter, OpenFold effectively only shuffles the top (20 − x) proteins to pass both filters, strictly increasing the expected quality of template hits relative to those used by AlphaFold2. However, in most cases, this approximation allows the dataloader to parse only as many structure files as are needed, speeding up the process by a factor of at least 5. In practice, the vast majority of invalid template hits are successfully detected by the first filter, suggesting that the difference in final template quality between the two procedures is marginal.
Self-distillation training set filtering
The 270,262 MSAs yielded by our self-distillation procedure is smaller than the 355,993 reported by DeepMind, despite having started with the same database. We suspect that the discrepancy arises due to the first step of the filtering process, of which the description in the AlphaFold2 paper is somewhat ambiguous.
Zero-centering target structures
We find that centering target structures at the origin slightly improves the numerical stability of the model, especially during low-precision training.
C.2 Plateaus and phase transitions
During training of the original version of OpenFold, we and third party developers observed two distinct training behaviors. The large majority of training runs are almost identical to the training curves shown in Figure 1; after a few thousand training steps, validation lDDT-Cα rapidly rises to ~0.83 and improves only incrementally thereafter. Occasionally, such runs exhibit a “double descent,” briefly improving and then degrading in accuracy before finally converging in the same way. In a fraction of training runs, however, lDDT-Cα plateaus between 0.30 and 0.35 on the same set (see Figure 8A). Anecdotally, these values appear to be consistent across environments, OpenFold versions, architectural modifications, and users. If the runs are allowed to continue long past the point where lDDT-Cα would otherwise have stopped improving (>10k training steps), they eventually undergo a phase transition, suddenly exceeding 0.8 lDDT-Cα and then continuing to improve much as normal runs do. We have not been able to determine whether this phenomenon is the result of an error in the OpenFold codebase or if it is a property of the AlphaFold2 algorithm.
Since running the experiments described in the main text of this paper, we have discovered a workaround that deviates slightly from the original AlphaFold2 training procedure but that appears to completely resolve early training instabilities. In the original training configuration, for each AlphaFold2 batch, backbone FAPE loss, the model’s primary structural loss, is clamped for all samples in the batch with probability 0.9. This practice is potentially problematic during the volatile early phase of training, when FAPE values can be extremely large and frequent clamping zeroes gradients for most of the residues in each crop. We find that clamping each sample independently, i.e., clamping approximately 90% of the samples in each batch rather than clamping 90% of all batches, eliminates training instability and speeds up convergence to high accuracy by about 30%. We show before-and-after data in Figure 8B.
D Training details
Our main OpenFold model was trained using the abridged training schedule outlined in Table 4 of the AlphaFold2 Supplementary Materials rather than the original training schedule in Table 5. Specifically, it was trained for three rounds: the initial training phase, the finetuning phase, and the predicted-TM (Zhang and Skolnick 2004) fine-tuning stage. During the initial training phase, sequences were cropped to 256 residues, MSA depth was capped at 128, and extra MSA depth was capped at 1,024. During fine-tuning, these values were increased to 384, 512, and 5,120, respectively. The second phase also introduced the “violation” and “experimentally resolved” losses, which respectively penalize non-physical steric clashes and incorrect predictions of whether atomic coordinates are resolved in experimental structures. Next, we ran a short third phase with the predicted TM score loss enabled. The three phases were run for 10 million, 1.5 million, and 0.5 million protein samples, respectively. We trained the model with PyTorch v1.10, DeepSpeed (Rasley et al. 2020) v0.5.10, and stage 2 of the ZeRO redundancy optimizer (Rajbhandari et al. 2019). We used Adam (Kingma and Ba 2015) with β1 = 0.9, β2 = 0.99, and ϵ = 10−6. We warmed up the learning rate linearly over the first 1,000 iterations from 0 to 10−3. After approximately 7 million samples, we marginally decreased the learning rate to 9.5 * 10−4. This decrease had no noticeable effect on model training. For the latter two phases, the learning rate was halved to 5 * 10−4. All model, data, and loss-related hyperparameters were identical to those used during AlphaFold2 training. We also replicated all of the stochastic training-time dataset augmentation, filtering, and resampling procedures described in the original paper.
During the initial fine-tuning and subsequent predicted-TM fine-tuning phases, we manually sampled checkpoints at peaks in the validation lDDT-Cα (Mariani et al. 2013). These checkpoints were added to the pool of model checkpoints used in the final model ensemble. Training was run on a cluster of 44 NVIDIA A100 GPUs, each with 40GB of DRAM. The model was trained in a data-parallel fashion, with one protein per GPU. In order to simulate as closely as possible the batch size of 128 used in training AlphaFold2, we performed threeway gradient accumulation to raise our effective batch size from 44 to 132. Supplementary Figure 1 contains additional data from the training run.
As in the original paper, CAMEO chains longer than 700 residues were removed from the validation set.
E Inference details
Runtime benchmarks were performed on a single 40GB A100 GPU. Times correspond to the intensive ‘model_1_ptm’ config preset, which uses deep MSAs and the maximum number of templates.
For proteins shorter than 1,000 residues, we take advantage of OpenFold’s TorchScript tracing capability and FlashAttention. For longer proteins, both tools become unstable and so we disable them. We plan to address this shortcoming in the future.
For runtime benchmarks we upgraded to PyTorch v1.12 for its improvements to Torch-Script. AlphaFold was run with JAX v. 0.3.13.
For reference, we include the distribution of lengths of our 132,000 PDB chains in Supplementary Figure 2.
F Additional model optimizations and features
F.1 Training-time optimizations and features
Despite its relatively small parameter count (~93M), AlphaFold2 manifests very large intermediate activations during training, resulting in peak memory usage—along with floating point operation counts—much greater than that of state-of-the-art transformer-based models from other domains (G. Wang et al. 2022). In AlphaFold2, peak memory usage during training grows cubically as a function of input sequence length. As a result, during the second phase of training when the inputs are longest, the model manifests individual tensors as large as 12GB. Intermediate activation tensors stored for the backward pass are even larger. This bottleneck is exacerbated by several limitations of the PyTorch framework. First, PyTorch is run eagerly and doesn’t benefit from the efficient compiler used by JAX models that improves runtime and reduces memory usage. Second, even on GPUs that in principle have sufficient memory to store all intermediate tensors used during the forward pass, suboptimal allocation patterns frequently result in memory fragmentation, preventing the model from utilizing all available memory. For this reason, among others, a preliminary version of OpenFold naively modeled after the official JAX-based implementation frequently ran out of memory despite having allocated as little as 40% of total available memory. To ameloriate these problems, we introduced several features that reduce peak memory consumption during training.

Mean correlation between lDDT-Cα and pLDDT over validation set chains as a function of training step in the early stage of training. Mean values for both metrics are superimposed.
In-place operations
We refactored the model by replacing element-wise tensor operations with in-place equivalents wherever possible to prevent unnecessary allocation of large intermediate tensors.
Custom CUDA kernels
We implemented custom CUDA kernels for the model’s “MSA row attention” module, the multi-head attention operation where the aforementioned 12GB tensor is allocated. Modified from optimized softmax kernels from FastFold (Cheng et al. 2022), which are in turn derived from OneFlow kernels (Yuan et al. 2021), our kernels operate entirely in-place. This is made possible partly by a fusion of the backward passes of the softmax operation and the succeeding matrix multiplication. Overall, only a single copy of the quadratic attention logit tensor is allocated, resulting in peak memory usage 5 and 4 times lower than equivalent native PyTorch code and the original FastFold kernels, respectively.

Binned sequence lengths of the 132,000 chains in the PDB training set.
DeepSpeed
OpenFold is trained using DeepSpeed. Using its ZeRO Redundancy optimizer (Rajbhandari et al. 2019) in the “stage 2” configuration, the model partitions gradients and optimizer states between GPUs during data-parallel training, further reducing peak memory usage.
Half-precision training
By default, as a memory-saving measure, AlphaFold2 is trained using bfloat16 floating point precision. This 16-bit format trades the large precision of the classic half-precision format (FP16) for the complete numerical range of full-precision floats (FP32), making it well-suited for training deep neural networks of the type used by AlphaFold2, which is not compatible with FP16 training by default. However, unlike FP16, bfloat16 hardware support is still limited to relatively recent NVIDIA GPUs (Ampere and Hopper architectures), and so the format remains out of reach for academic labs with access to older GPUs that are otherwise capable of training AlphaFold2 models (e.g., V100 GPUs). We address this problem by implementing a stable FP16 training mode with more careful typecasting throughout the model pipeline, making OpenFold training broadly accessible.
F.2 Inference
We also introduce several inference-time optimizations to OpenFold. As previously mentioned, these features trade off memory usage for runtime, contributing to more versatile inference on chains of diverse lengths.
FlashAttention
We incorporate FlashAttention (Dao et al. 2022), an efficient fused attention implementation that tiles computation in order to reduce data movement between different levels of GPU memory, greatly improving peak memory usage and runtime in the process. We find it to be particularly effective for short sequences with 1,000 residues or less, on which it contributes to an OpenFold speedup of up to 15% despite only being compatible with a small number of the attention modules in the network.
Low-memory attention
Separately, OpenFold makes use of a recent attention algorithm that uses a novel chunking technique to perform the entire operation in constant space (Rabe and Staats 2021). Although enabling this feature marginally slows down the model, it nullifies attention as a memory bottleneck during inference.
Refactored triangle multiplicative attention
A naive implementation of the triangle multiplicative update manifests 5 concurrent tensors the size of the input pair representation. These pair representations grow quadratically with input length, such that during inference on long sequences or complexes, they become the key bottleneck. We refactored the operation to reduce its peak memory usage by 50%, requiring just 2.5 copies of the pair representation.
Template averaging
AlphaFold2/OpenFold create separate pair embeddings for each structural template passed to each model, then reduce them to a single embedding at the end of the template pipeline with an attention module. For very long sequences, or very many templates, this operation can become a memory bottleneck. AlphaFold-Multimer (Evans et al. 2022) avoids this problem by computing a running average of template pair embeddings. Although we trained OpenFold using the original AlphaFold2 (non-multimer) procedure, we find that the newer approach can be adopted during inference without a noticeable decrease in accuracy. We thus make it available as an optional inference-time memory-saving optimization.
In-place operations
Without the requirement to store intermediate activations for the backward pass, OpenFold is able to make more extensive use of in-place operations during inference. We also actively remove unused tensors to mitigate crashes caused by memory fragmentation.
Chunk size tuning
AlphaFold2 offsets extreme inference-time memory costs with a technique called “chunking,” which splits input tensors into “chunks” along designated, module-specific sub-batch dimensions then runs those modules sequentially on each chunk. In AlphaFold2’s case, the chunk size used in this procedure is a model-wide hyperparameter that is manually tuned. OpenFold, on the other hand, dynamically adjusts chunk size values for each module independently, taking into account the model’s configuration and the current memory limitations of the system. Although the profiling runs introduced by this process incur a small computational overhead, the modules do not need to be recompiled for each run, unlike their AlphaFold2 equivalents, and said profiling runs are only necessary the first time the model is run; once computed, the optimal chunk sizes are cached and reused until conditions change. We find this to be a robust way to seamlessly improve runtimes in a variety of settings.
Tensor offloading
Optionally, OpenFold can aggressively offload intermediate tensors to CPU memory, temporarily freeing additional GPU memory for memory-intensive computations at the cost of a considerable slowdown. This feature is useful during inference on extremely long sequences that would otherwise not be computable.
TorchScript tracing
Specially written PyTorch programs can be converted to Torch-Script, a JIT-compiled variant of PyTorch. We use this feature during inference to speed up parts of the Evoformer module. Although TorchScript tracing and compilation do introduce some overhead at the beginning of model inference, and lock the model to a particular sequence length similar to JAX compilation, we find that using TorchScript achieves overall speedups of up to 15%, especially on sequences shorter than 1,000 residues. This feature is particularly useful during batch inference, where sequences are grouped by length to avoid repeated re-compilations and take maximal advantage of faster inference times.
AlphaFold-Gap implementation
OpenFold currently supports multimeric inference using AlphaFold-Gap (Baek 2021), a zero-shot hack that allows inference on protein complexes using monomeric weights. Although it falls short of the accuracy of AlphaFold-Multimer6 it is a capable tool, especially for homomultimers. Since complexes manifest in the model as long sequences, OpenFold-Gap in particular benefits from the memory optimizations discussed earlier.
G Extended analysis
G.1 Effect of fine-tuning on long proteins
In Supplementary Figure 3, we illustrate the effect of fine-tuning on the lDDT-Cα of long chains. Though we observe a larger increase here than is seen for all proteins in Figure 1D, it is still less than half a point.
G.2 Dimensionality of output structures
Here we provide additional analyses of the staged learning of dimensionality. First, supplementary Figure 4 provides a visual aid for the projection operations used to produce Figure 3A and 3B. Second, because the precise timing of the phases of dimensionality differ slightly for individual proteins, we include for reference Supplementary Figure 5, which shows individual eigenvalues for all proteins in the validation set.

Mean lDDT-Cα over validation proteins with at least 500 residues as a function of fine-tuning step.
Third, for an additional perspective on the low-dimensionality phenomenon, we consider the radius of gyration, a popular measure of the compactness of a structure (Hinsen et al. 2013), given by
 where K is the number of atoms in the protein and P ∈ ℝK×3 is a coordinate vector. Real proteins obey known protein-phase-specific radius scaling laws (see e.g. Krokhotin et al. 2012), and we wish to determine exactly how and when OpenFold begins to produce plausible structures that do the same. To accomplish this, we compute the radius of gyration of each of the experimental structures in our validation set and compare them to the radii of gyration of model predictions as a function of training step. Results are shown in Supplementary Figure 6.
where K is the number of atoms in the protein and P ∈ ℝK×3 is a coordinate vector. Real proteins obey known protein-phase-specific radius scaling laws (see e.g. Krokhotin et al. 2012), and we wish to determine exactly how and when OpenFold begins to produce plausible structures that do the same. To accomplish this, we compute the radius of gyration of each of the experimental structures in our validation set and compare them to the radii of gyration of model predictions as a function of training step. Results are shown in Supplementary Figure 6.
The plots correspond approximately to the phases of dimensionality illustrated in 2. In the first two panels, before the model enters the three-dimensional phase, the radii of gyration of predicted structures are indeed systematically smaller than those of experimental structures. Immediately thereafter, the radii of gyration are largely correct, and only minor adjustments are made in the final panel.

A “2D vs. 3D” comparison (left, corresponding to Figure 3A) and a “2D vs. 2D” comparison (right, corresponding to Figure 3B). The blue, two-dimensional structure on the right is the 2D PCA projection of the 3D structure on the left. The red structure in both images is the same 2D PCA projection of a prediction from the two-dimensional phase.
G.3 DSSP state reduction
We reduce the 8-state DSSP assignment to 3 states using the following mapping:
 where ‘C’ denotes “coil” and ‘E’ denotes “strand”.
where ‘C’ denotes “coil” and ‘E’ denotes “strand”.
G.4 Additional secondary structure data
In Supplementary Figure 7, we provide another view of the sheet data in Figure 5B by distinguishing between small-(S: 6Å), medium-(M: 12Å) and long-(L: 24Å) range contacts, as in e.g., (Xu et al. 2021).

Sorted PCA eigenvalues for all proteins in the CAMEO validation set as a function of OpenFold training step. The values shown were used to generate Figure 2B.
G.5 Data elision validation using CAMEO
In order to properly assess the model’s generalization capacity, we evaluated each set of CATH ablations on a corresponding validation set in the main text. I.e., the T ablations were evaluated on held-out topologies, the A ablations on held-out architectures, and so on. As a result, different data elision experiments cannot not be compared directly. For a more consistent picture of the relative final accuracies of each set of data elision experiments, we reevaluate the final checkpoints of each model on our standard CAMEO validation set in Supplementary Table 1.
Note that a potential confounding factor is that CATH classifications are not yet available for proteins in the CAMEO validation set, making it difficult to determine the degree of overlap in fold space between the training set of each data elision and the validation set. If the CAMEO validation set happens to contain chains with architectures in the training sets of the smaller A ablations, for example, the values in Supplementary Table 1 would overestimate the accuracies of the corresponding models.

Radii of gyration for proteins in the CAMEO validation set (orange) as a function of sequence length over training time, plotted on a log-log scale against experimental structures (blue). Legends show equations of best fit curves, computed using non-linear least squares. The training steps chosen correspond loosely to the four phases of dimensional growth.

Binned contact F1 scores (8Å threshold) for beta sheets of various widths as a function of training step at different residue-residue separation ranges (SMLR ≥ 6 residues apart; LR ≥ 24 residues apart, as in Xu et al. 2021). Sheet widths are weighted averages of sheet thread counts within each bin, as in Figure 5B.
G.6 Secondary structure recovery of class-stratified models
In Figure 6C, we show predictions of two class-stratified models for two CAMEO chains. For a more comprehensive picture, we report mean reduced-state DSSP recall and F1 over the entire CAMEO validation set for both models in Supplementary Table 2.
G.7 Characteristics of class-stratified training sets
We note in the main text that domains in the class used to train the “Mainly alpha” class elision still contain some beta sheets, and vice versa. To quantify this, in Supplementary Figure 8 we show the distribution of alpha helices and beta sheets of different sizes in the two class elision training sets based on 1,000 randomly chosen samples.
H Known issues during training
During and after the primary OpenFold retraining experiment, we discovered a handful of minor implementation errors that, given the prohibitive cost of retraining a full model from scratch, could not be corrected. In this section, we describe these errata and the measures that we have taken to mitigate them.
Rows correspond to CATH elisions reported in Figure 6, except evaluations reported here are based on the CAMEO validation set.
Recall and F1 scores for reduced secondary structure categories derived using DSSP. Results are shown for the two class-stratified models from the final panel of Figure 6B, here evaluated on the CAMEO validation set. The reduced secondary state scheme described in Appendix G.3 is used.

Counts for alpha helices and beta sheets in the mostly alpha and mostly beta CATH class-stratified training sets from Figure 6, based on 1,000 random samples. Counts are binned by size, defined as the number of residues for alpha helices and number of strands for beta sheets.
H.1 Distillation template error
As described in the main text, OpenFold/AlphaFold2 training consists of three phases, of which the first is the longest and most determinative of final model accuracy. During this first phase of the main OpenFold training run, a bug in the dataloader caused distillation templates to be filtered entirely, such that OpenFold was only presented with templates for PDB chains, which constitute ~25% of training samples, and no self-distillation set chains. The issue was corrected for later phases, which were run slightly longer than usual to compensate.
Although the accuracy of the resulting OpenFold model matches that of the original AlphaFold2 in holistic evaluations, certain downstream tasks that exploit the template stack (Ovchinnikov 2022) do not perform as well as the original AlphaFold2. There is evidence, for instance, that OpenFold disregards the amino acid sequence of input templates while AlphaFold2 does not. However, after the bug was corrected, follow-up experiments involving shorter training runs showed template-usage behavior at parity with AlphaFold2. Thus any current and future OpenFold-based training runs will not be affected by this issue. Furthermore, OpenFold can be run with the original AlphaFold2 weights in cases where templates are expected to be important, to take advantage of the new inference characteristics without diminution of template-related performance.
H.2 Gradient clipping
OpenFold, unlike AlphaFold2, was trained using per-batch as opposed to per-sample gradient clipping (first noted by the UniFold team (Li et al. 2022)). UniFold experiments show that models trained using the latter clipping technique achieve slightly better accuracy.
H.3 Training instability
Our primary training run was performed before we introduced the changes described in Appendix C.2. While we have no reason to believe that the instabilities we observed there are a result of a bug in the OpenFold codebase, as opposed to an inherent limitation of the AlphaFold2 architecture, the former remains a possibility. It is unclear how potential issues of this kind may have affected runs that—like our primary training run—appeared to converge at the expected rate.
Footnotes
Author list and affiliations cleaned; animations added; acknowledgements and author contributions list updated; figures adjusted
https://figshare.com/articles/media/Folding_animations/21561939
↵1 Predicted structure animations for diverse validation proteins can be accessed [here]. Residues are color-coded according to the 3-state secondary structure of the accompanying experimental fold.
↵2 PDB accession codes 7DQ9_A (Wei et al. 2021), 7RDT_A (Carroll et al. 2021), and 7LBU_A (Yu et al. 2021)
↵3 PDB accession code 7CJS_B (Saitoh et al. 2021)
↵4 PDB accession code 7RRM_C (Mota et al. 2022)
↵5 PDB accession code 7DQ9_A (Wei et al. 2021)
↵6 For a comparison of the two techniques, see (Evans et al. 2022).
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵Ovchinnikov, S. (2022). Twitter post: Weekend project!
 So now that OpenFold weights are available. I was curious how different they are from AlphaFold weights and if they can be used for AfDesign evaluation. More specifically, if you design a protein with AlphaFold, can OpenFold predict it (and vice-versa)? (1/5). https://twitter.com/sokrypton/status/1551242121528520704.
So now that OpenFold weights are available. I was curious how different they are from AlphaFold weights and if they can be used for AfDesign evaluation. More specifically, if you design a protein with AlphaFold, can OpenFold predict it (and vice-versa)? (1/5). https://twitter.com/sokrypton/status/1551242121528520704. - ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵

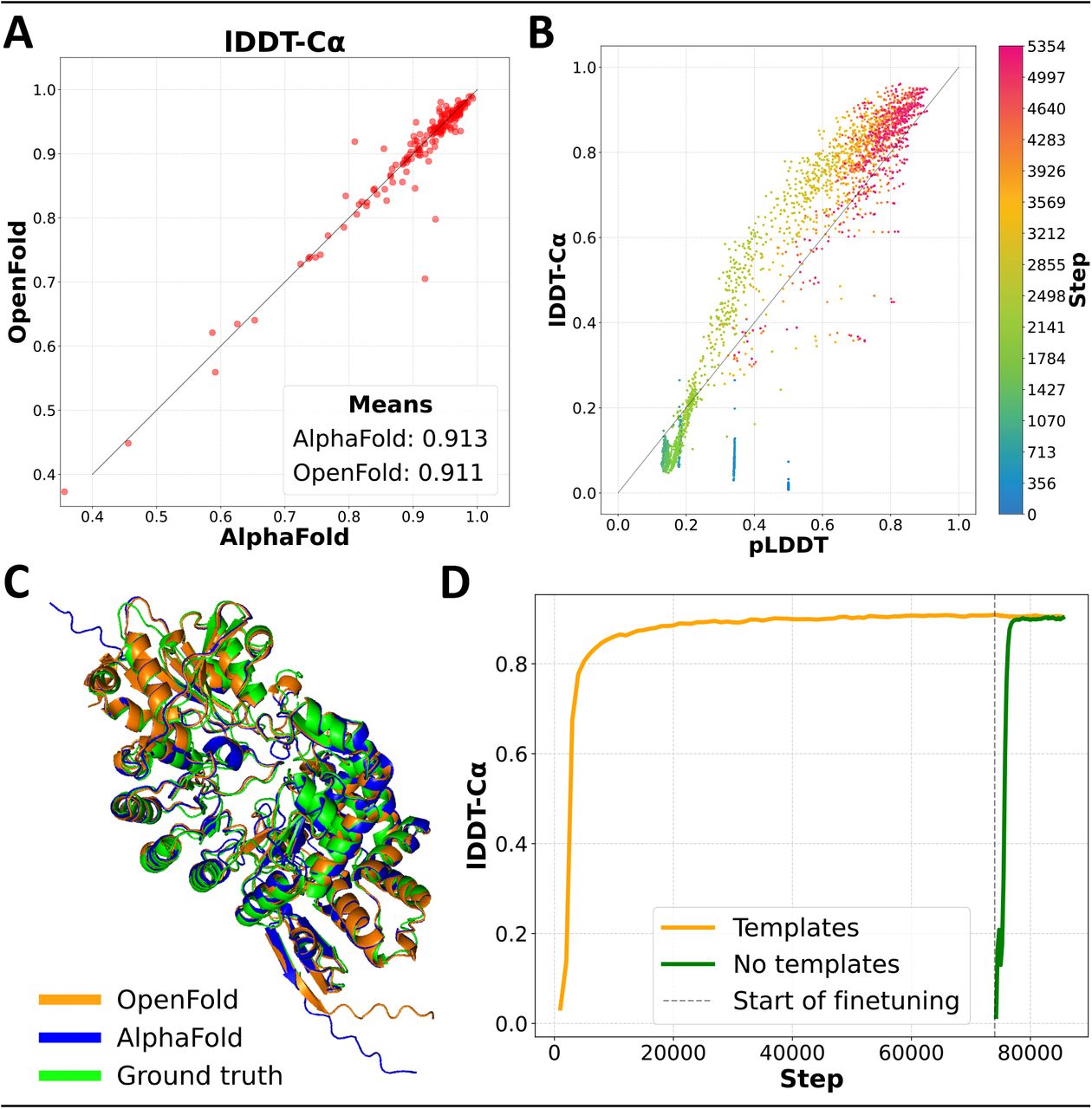{kind=link}
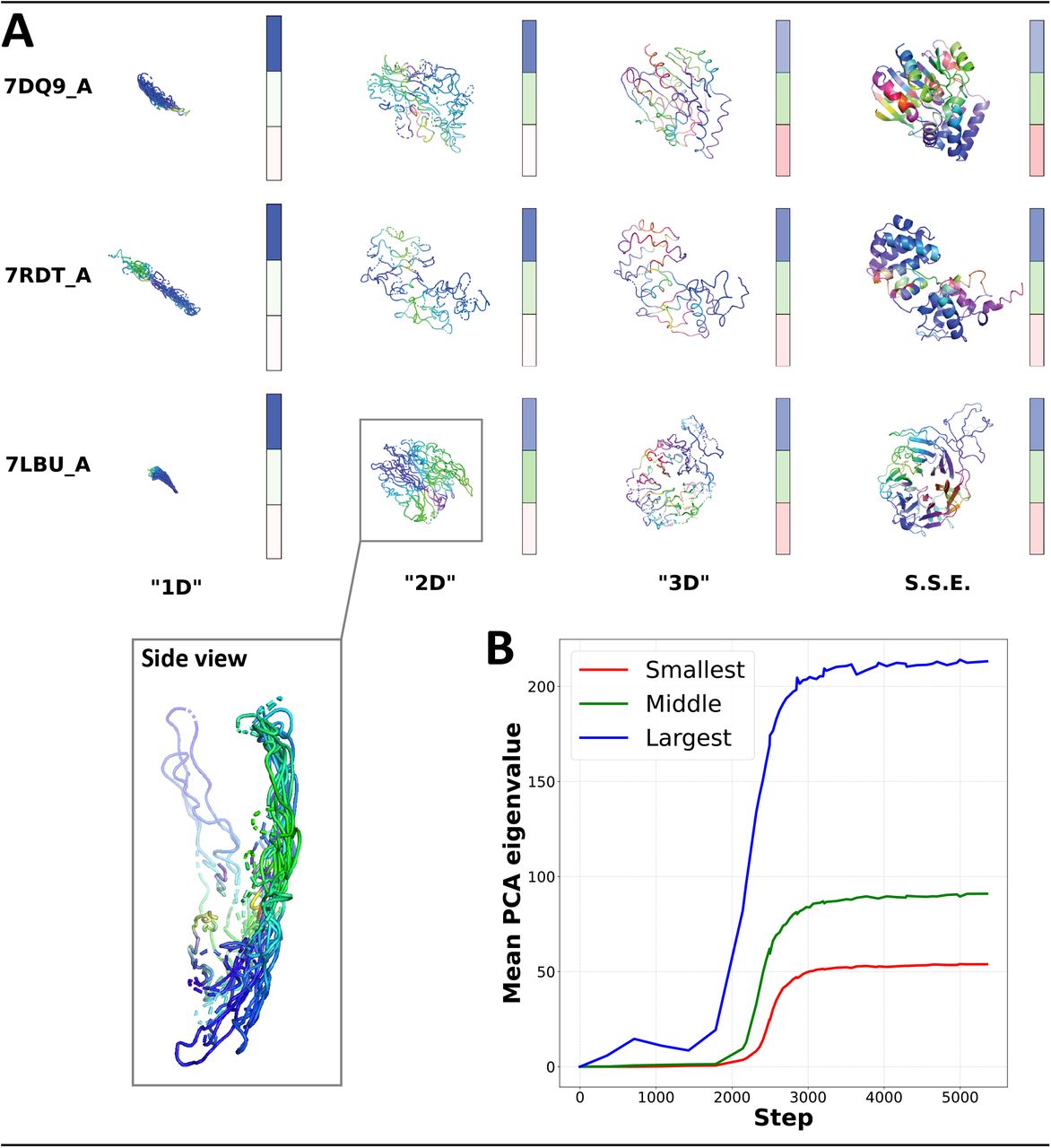{kind=link}
{kind=link}
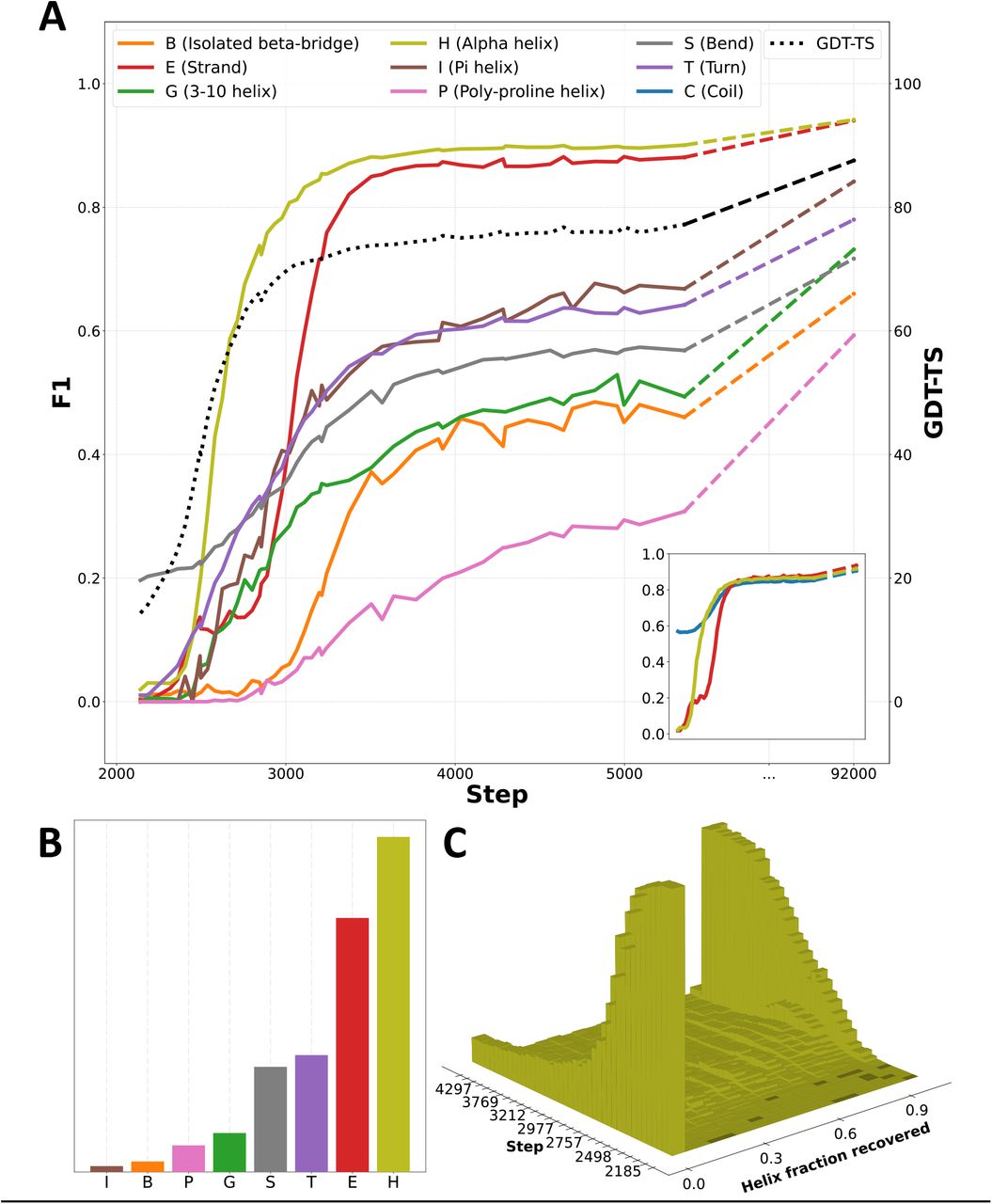{kind=link}
{kind=link}
{kind=link}
{kind=link}
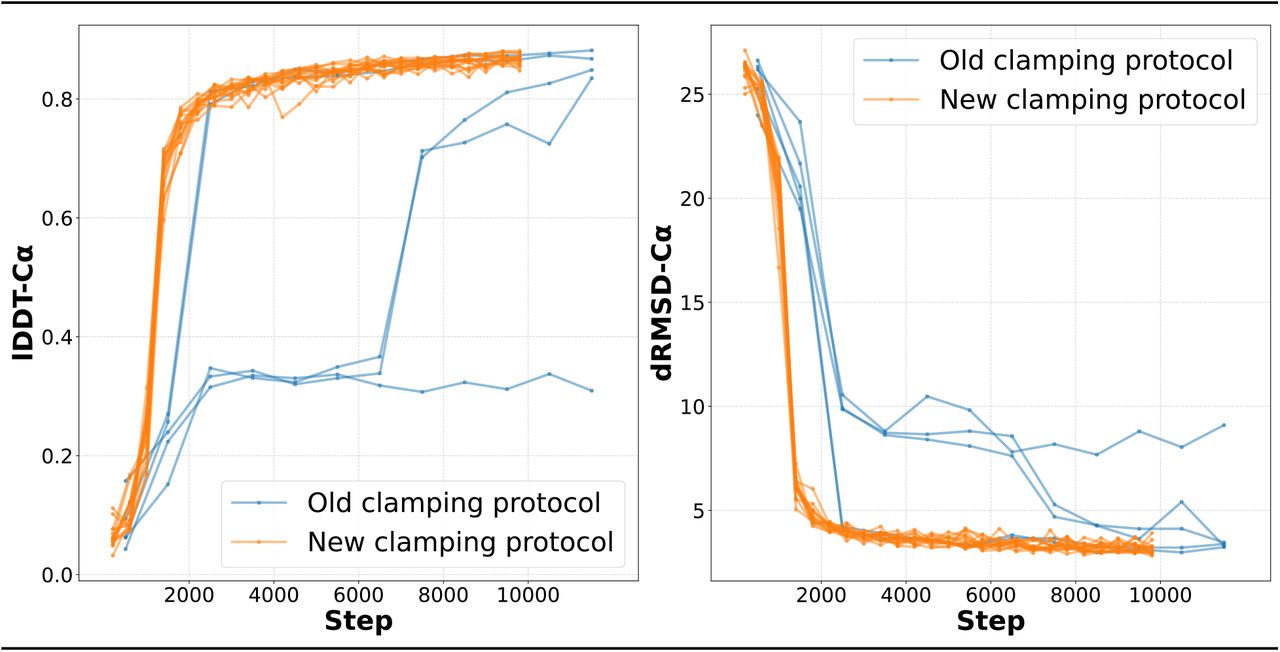{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




