

Abstract
Circadian rhythms play an essential role in many biological processes and surprisingly only three prokaryotic proteins are required to constitute a true post-translational circadian oscillator. The evolutionary history of the three Kai proteins indicates that KaiC is the oldest member and central component of the clock, with subsequent additions of KaiB and KaiA to regulate its phosphorylation state for time synchronization. The canonical KaiABC system in cyanobacteria is well understood, but little is known about more ancient systems that possess just KaiBC, except for reports that they might exhibit a basic, hourglass-like timekeeping mechanism. Here, we investigate the primordial circadian clock in Rhodobacter sphaeroides (RS) that contains only KaiBC to elucidate its inner workings despite the missing KaiA. Using a combination X-ray crystallography and cryo-EM we find a novel dodecameric fold for KaiCRS where two hexamers are held together by a coiled-coil bundle of 12 helices. This interaction is formed by the C-terminal extension of KaiCRS and serves as an ancient regulatory moiety later superseded by KaiA. A coiled-coil register shift between daytime- and nighttime-conformations is connected to the phosphorylation sites through a long-range allosteric network that spans over 160 Å. Our kinetic data identify the difference in ATP-to-ADP ratio between day and night as the environmental cue that drives the clock and further unravels mechanistic details that shed light on the evolution of self-sustained oscillators.
Main Text
Circadian clocks are self-sustained biological oscillators that are found ubiquitously in prokaryotic and eukaryotic organisms. In eukaryotes these systems are complex and very sophisticated, whereas in prokaryotes the core mechanism is regulated by a posttranslational oscillator that can be reconstituted in vitro with three proteins (kaiA, kaiB, and kaiC)1 and ATP. Seminal work on the KaiABC system resulted in a comprehensive understanding of its circadian clock: KaiC is the central component that auto-phosphorylates through binding of KaiA and auto-dephosphorylates upon association with KaiB2-5. The interplay between these three proteins has been shown in vitro to constitute a true circadian oscillator characterized by persistence, resetting, and temperature compensation. Consequently, the KaiABC system is considered an elegant and the simplest implementation of a circadian rhythm. The evolutionary history of kai genes established kaiC as the oldest member dating back ∼3.5 bya, with subsequent additions of kaiB and most recently kaiA to form the extant kaiBC and kaiABC clusters, respectively6,7. Interestingly, a few studies of more primitive organisms that lack kaiA hinted that the kaiBC-based systems might provide already a basic, hourglass-like timekeeping mechanism8-10. Contrary to the self-sustained oscillators found in cyanobacteria, such a timer requires an environmental cue to drive the clock and for the daily “flip” of the hourglass. The central role of circadian rhythms in many biological processes, controlled by the day and night cycle on earth, makes their evolution a fascinating topic.
Here, we investigate such a primitive circadian clock by biochemical and structural studies of the KaiBC system of the purple, non-sulfur photosynthetic proteobacterium Rhodobacter sphaeroides KD131 (RS; hereafter referred to as KaiBRS and KaiCRS). Surprisingly, the organism shows sustained rhythms of gene expression in vivo, but whether kaiBC is responsible for this observation remains inconclusive in the absence of a kaiC knockout11. A more recent study of the closely-related Rhodopseudomonas palustris demonstrated causality between the proto-circadian rhythm of nitrogen fixation and expression of the kaiC gene using a KO strain10. We discover using in vitro experiments that KaiBCRS is indeed a primordial circadian clock with a mechanism that is different from the widely studied circadian oscillator in Synechococcus elongatus PCC 7942 (hereafter referred to as KaiABCSE)2-5. We identify an environmental cue that regulates the phosphorylation state and consequently produces a 24-hour clock in vivo as the switch in ATP-to-ADP ratio between day and night. Our kinetic results combined with X-ray and cryo-EM structures of the relevant states unravel a long-range allosteric pathway that is crucial for function of the hourglass and sheds light on the evolution of self-sustained oscillators. Notably, we find a novel protein fold for KaiCRS and uncover a register shift in the coiled-coil domain spanning ∼115 Å as the key regulator in this system, which shows intriguing structural similarities to dynein signaling12.
C-terminal tail as a primitive regulatory moiety
To gain insight into the evolution of the kaiBC cluster, we constructed a phylogenetic tree of kaiC after the emergence of the kaiB gene (Fig. 1a, Extended Data Fig. 1a). The first obvious question is how KaiCRS and other members in the clade can auto-phosphorylate despite having no KaiA, which is known to be crucial for this function in the canonical KaiABC system at its optimum temperature. We observe a large clade that exhibits a C-terminal tail about 50 amino acids longer compared to kaiC in other clades (Extended Data Fig. 1b). This C-terminal extension near the A-loop is predominantly found in the kaiC2 subgroup, which was previously annotated as having two serine phosphorylation sites instead of the Thr/Ser pair found in kaiC1 and kaiC3 subgroups (Extended Data Fig. 1b)13-15. In Synechococcus elongatus, the binding of KaiASE to the A-loop of KaiCSE tethers them in an exposed conformation16 that activates both auto-phosphorylation and nucleotide exchange17. Given the proximity of the extended C-terminal tail to the A-loop we conjectured that it could serve as the “primitive” regulatory moiety made redundant concomitantly with the appearance of KaiA.
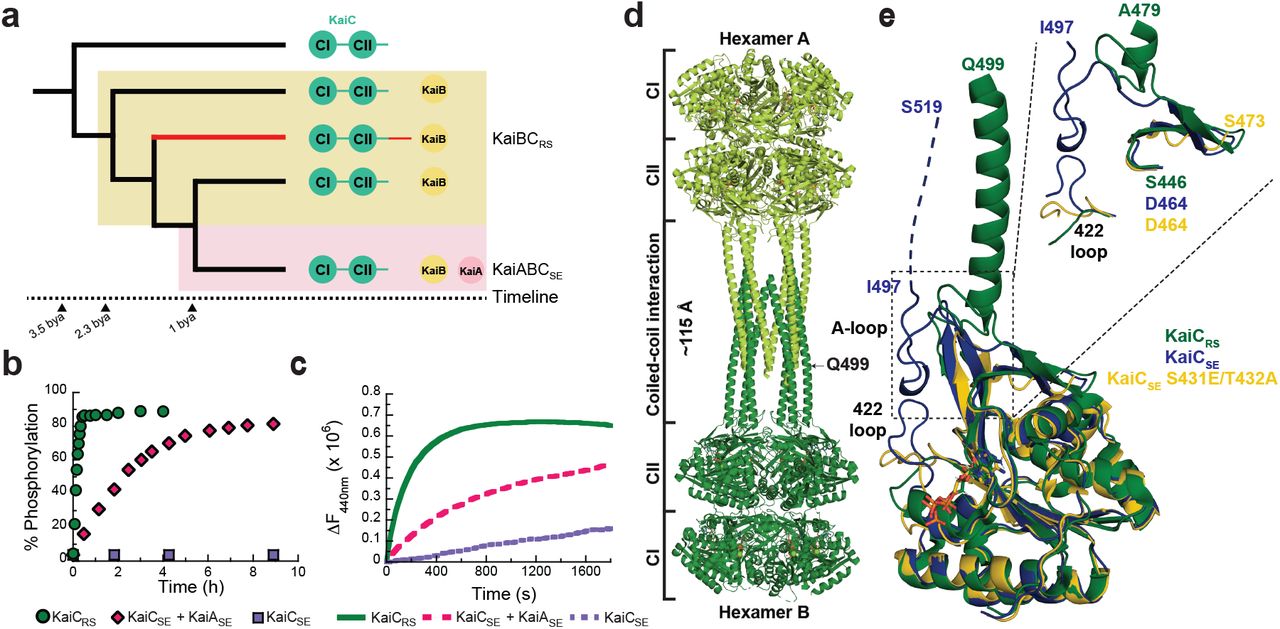
(a) Schematic phylogenetic tree of kaiC showing the appearance of kaiB and kaiA during evolution. The kaiC clade with an approximately 50 amino acids C-terminal extension is labeled in red and a timeline was predicted as previously reported6 (b) Phosphorylation over time of KaiCRS (green circles, 6.5 ± 1.0 h-1) and KaiCSE in the presence (pink circles, 0.40 ± 0.02 h-1) or absence (purple circles) of KaiASE at 30 °C. The standard deviation in reported parameters are obtained from the fitting. (c) Nucleotide exchange between ATP and mant-ATP in KaiCRS alone (green trace, 18.0 ± 1.5 h-1) compared to KaiCSE in the presence (pink trace, 4.7 ± 0.3 h-1) and absence of KaiASE (purple trace, 0.08 ± 0.04 h-1) measured by fluorescence at 30 °C. Representative traces are shown and the fitted parameters (mean ± S.D) were obtained from three replicate measurements. (d) X-ray structure of dodecameric KaiCRS (PDB 8dba) colored by hexamer A (light green) and hexamer B (dark green). The CI, CII, and coiled-coil domains are indicated in (a), and the A-loop is labeled in (e). (e) Superposition based on alignment of the CII domain of KaiCRS (green, PDB 8dba, chain B), KaiCSE (purple, PDB 1tf7, chain B)36, and KaiCSE S431E/T432A (yellow, PDB 7s65, chain A)19 shows that KaiCRS has an extended A-loop orientation that no longer forms the inhibitory interaction with the 422-loop (KaiCSE numbering). The conformation of the 422-loop in KaiCRS resembles the one seen in the cryo-EM structure of the phosphomimetic KaiCSE S431E/T432A (yellow, PDB 7s65)19. No electron density is observed for the C-terminal part of wild-type KaiCSE and S431E/T432A mutant due to flexibility, and the missing 22 residues for wild-type KaiCSE (46 for S431E/T432A) are represented by a dashed line for wild-type KaiCSE (not shown for the mutant).
To test our hypothesis, we first measured the auto-phosphorylation and nucleotide exchange rates in KaiCRS that both depend heavily on the presence of KaiA in the KaiABCSE system. We observe an auto-phosphorylation rate for KaiCRS that is ∼16-fold higher than for KaiCSE activated by KaiASE (6.5 ± 1.0 h-1 versus 0.40 ± 0.02 h-1, respectively; Fig. 1b and Extended Data Figs. 2a-e). Similarly, the nucleotide exchange is faster in KaiCRS compared to KaiCSE even in the presence of KaiASE (18.0 ± 1.5 h-1 compared to 4.7 ± 0.3 h-1, respectively; Fig. 1c and Extended Data Fig. 2f). Our data clearly show that KaiCRS can perform both auto-phosphorylation and nucleotide exchange on its own and, in fact, does so faster than its more recently evolved counterparts.
Coiled-coil interaction assembles KaiCRS dodecamer
To assess mechanistically how KaiC in the kaiA-lacking systems accomplishes auto-phosphorylation we turned to structural biology. The crystal structure of KaiCRS, unlike KaiC from cyanobacteria, reveals a homododecamer consisting of two homohexameric domains joined by a twelve-helical coiled-coil domain that is formed by the extended C-terminal tail (PDB 8dba; Fig. 1d and Extended Data Table 1). A closer inspection of the CII domains in KaiCRS and KaiCSE/TE showed an obvious difference in A-loop orientations: an extended conformation in KaiCRS versus a buried orientation in KaiCSE/TE (Fig. 1e). The existence of such an extended conformation upon binding of KaiA was conjectured earlier based on the perceived hyper- and hypo-phosphorylation upon removing the A-loop or disrupting KaiA binding, respectively18. Importantly, a recent cryo-EM structure of the nighttime phosphomimetic KaiCSE S431E/T432A in its compressed state was solved and directly showed a disordered A-loop that no longer interacts with the 422-loop19, similar to the extended A-loop conformation we observe in KaiCRS (Fig. 1e). The loss of interaction between the A-loop and 422-loop (just 10 residues apart from the phosphorylation sites), results in closer proximity between the hydroxyl group of Ser431/Thr432 and the Ψ-phosphate of ATP, thereby, facilitating the phosphoryl-transfer step20. Furthermore, the sequence similarity between KaiCRS and KaiCSE is less than 30% for the A-loop and residues considered important for stabilization of this loop in its buried orientation (i.e., 422-loop and residues 438-444; Fig. 1e). Taken together, our structural and kinetic data support the idea that an exposed A-loop is key for KaiA-independent enhancement of nucleotide exchange and hence auto-phosphorylation in KaiCRS and perhaps other KaiBC-based systems.
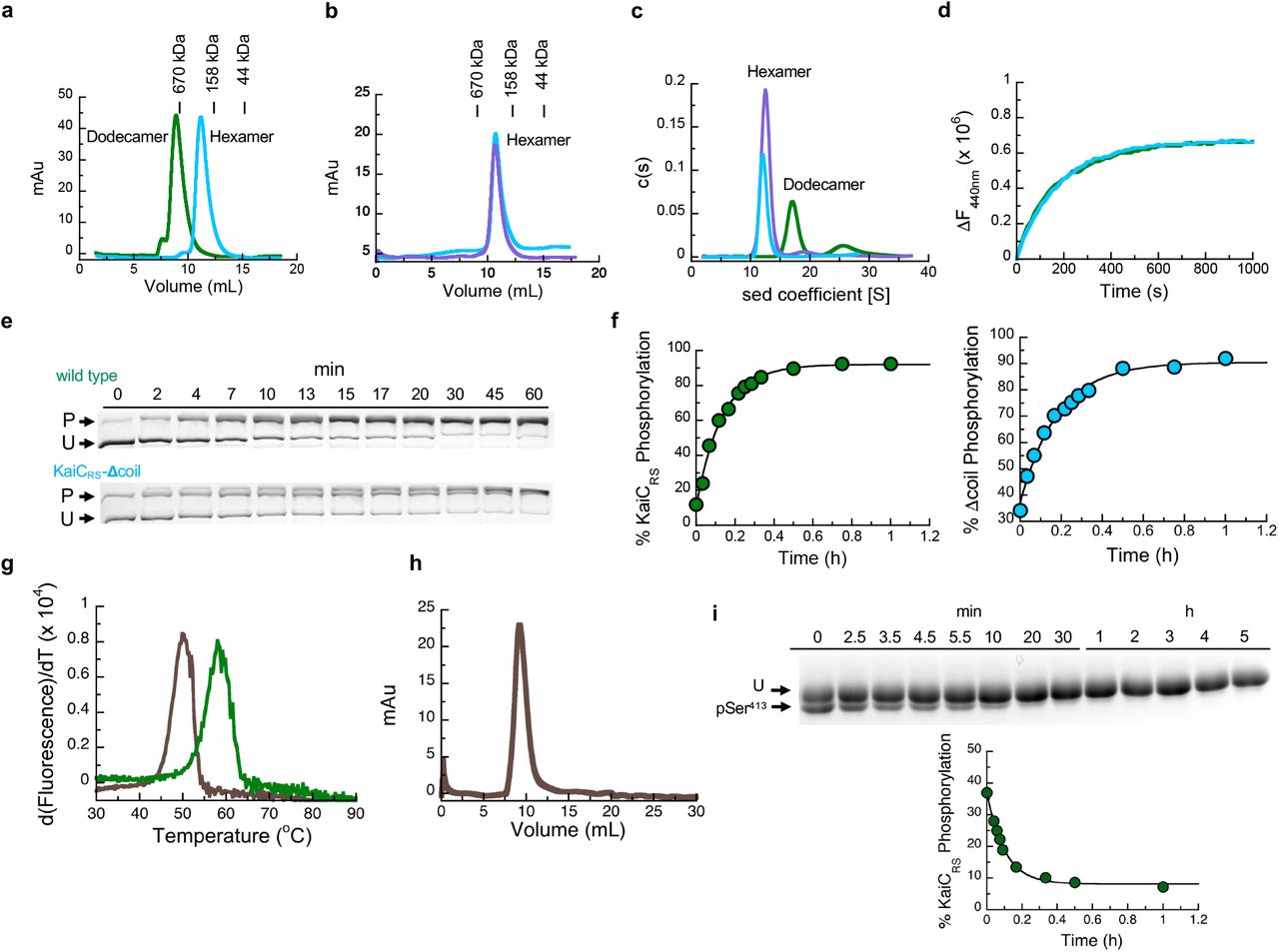
Is the purpose of the coiled-coil domain merely to “pull up” the A-loop or does it actively participate in nucleotide exchange and auto-phosphorylation of KaiC? To further understand its role, we generated a truncation at residue Glu490 based on the phylogenetic tree and crystallographic information (KaiCRS-Δcoil; Extended Data Fig. 1b) to disrupt the coiled-coil interaction between the two hexamers. Indeed, the crystal structure of KaiCRS-Δcoil (PDB 8db3; Fig. 2a,b and Extended Data Table 1) and its size-exclusion chromatogram and analytical ultracentrifugation profile (Extended Data Fig. 3a-c) show a hexameric structure with no coiled-coil interaction. Nucleotide exchange rates in the CII domain for KaiCRS-Δcoil and wild type are comparable (19.1 ± 0.8 h-1 and 18.0 ± 1.5 h-1, respectively; Extended Data Fig. 3d) as are the phosphorylation rates (5.5 ± 0.4 h-1 and 7.4 ± 0.3 h-1 for KaiCRS-Δcoil and wild type, respectively; Extended Data Fig. 3e,f). These results indicate that the extended A-loop and not the coiled-coil interaction plays a pivotal role in nucleotide exchange and auto-phosphorylation in KaiCRS, potentially explaining auto-phosphorylation in other KaiBC-based systems lacking a coiled-coil bundle. Notably, the coiled-coil bundle provides additional hexameric stability: the KaiCRS dodecamer is stable for extended periods of time in the presence of only ADP (Extended Data Fig. 3g,h), whereas for KaiCSE oligomers are not observed under these conditions21.

(a) The X-ray structure of KaiCRS-Δcoil was solved in the C2221 space group and contained three monomers in the asymmetric unit with ADP present in all active sites. The obtained electron density map allowed for model building up to Pro463, indicating that the truncation at position 490 results in enhanced flexibility beyond Pro463. Phosphorylation of Ser414 was observed in chain B (cyan) as shown by the electron density mFo-DFc polder map (green mesh, 3σ contour level). (b) Assembly analysis using the PISA software37 revealed a hexamer as the most probable quaternary structure (top view). (c) Structural comparison of the coiled-coil domain for unphosphorylated KaiCRS (dark and light green; X-ray structure) and the KaiCRS-S413E/S414E phosphomimetic mutant (dark and light blue; cryo-EM structure). (d) Overlay of interacting dimers of the structures in (c) using CII domain of chain A as reference (dark shades; bottom). Unphosphorylated KaiCRS (dark green) interacts with the opposite partner on the right (light green), whereas KaiCRS-S413E/S414E (dark blue) interacts with the partner on the left (light blue). The hydrophobic packing in the coiled-coil domain is mediated by only the Cβ atoms of alanine and arginine residues in unphosphorylated KaiCRS but involves the entire side chain of leucine and isoleucine residues in the phosphomimetic structure. (e) Allosteric network in the phosphomimetic state (blue) from the coil (light blue) propagating through the KaiCRS CII domain to the active site (dark blue) compared to the unphosphorylated state (dark green). (f) Auto-dephosphorylation of KaiCRS and KaiCRS-Δcoil over time in the presence of 4 mM ADP at 30 °C. The phosphorylated (P) and unphosphorylated (U) proteins were separated by Zn2+ Phos-tag™ SDS-PAGE.
Long-range allosteric network in KaiCRS
The change in phosphorylation state of KaiC has been well established to be the central feature for the circadian rhythm22,23. Interestingly, when comparing the unphosphorylated form of full-length KaiCRS (PDB 8dba) and its phosphomimetic mutant (S413E/S414E, PDB xxxx, Extended Data Fig. 4, and Extended Data Table 2) we observe two distinct coiled-coil interactions. Upon phosphorylation, the coiled-coil pairs swap partners by interacting with the other neighboring chain from the opposite hexamer resulting in a register shift that propagates ∼115 Å along the entire coiled-coil (Fig. 2c and Extended Data Fig. 5). In the phosphomimetic state, the register comprises bulkier hydrophobic residues thus resulting in a more stable interaction than for the dephosphorylated form (Fig. 2d and Extended Data Fig. 3g). Furthermore, the C-terminal residues of KaiCRS-S413E/S414E interact with the CII domain of the opposite hexamer, whereas the lack of electron density for the last 30 residues in the wild-type structure indicates more flexibility in the dephosphorylated state. Importantly, we discover that these conformational changes in the coiled-coil domain seem to be coupled through a long-range allosteric network to the phosphorylation sites. The rotameric states of residues Ser413, Ser414, Trp419, Val421, Tyr436, Leu438, Val449, and Arg450 move concertedly and point towards the nucleotide-binding site when the protein is phosphorylated or away in the absence of a phosphate group (Fig. 2e, Extended Data Fig. 5d). We hypothesize that the proximity of the nucleotide to the phosphorylated residue allows for a more efficient phosphoryl transfer and, therefore, determined experimentally the impact of the coiled-coil domain on the auto-dephosphorylation of KaiCRS. Indeed, we observe a noticeable effect: wild-type protein dephosphorylates comparatively quickly (kobs = 11.5 ± 0.8 h-1) in the presence of only ADP, whereas little dephosphorylation was observed for KaiCRS-Δcoil (Fig. 2f and Extended Data Fig. 3i) where the allosteric propagation is disrupted (Extended Data Fig. 5d). Consistent with this accelerated dephosphorylation due to the coiled-coil domain, our crystallographic data show a phosphate-group on Ser414 for KaiCRS-Δcoil but not for the wild-type protein (Fig. 2a and Extended Data Fig. 5d).
ATP-to-ADP ratio to reset the clock
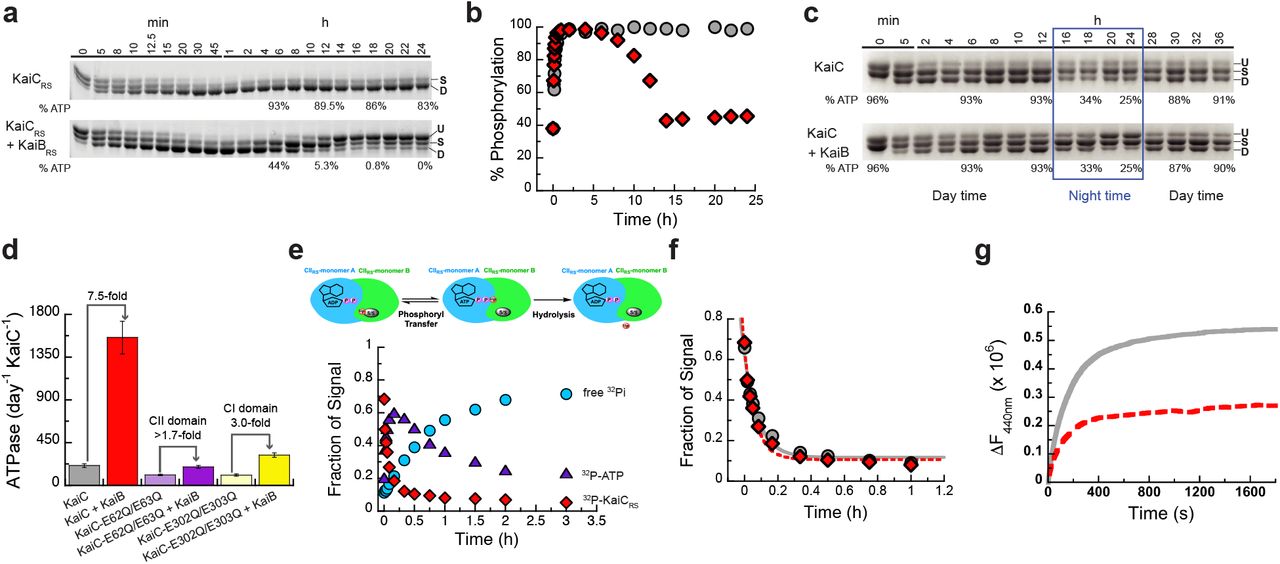
The surprising result here is that KaiCRS can auto-dephosphorylate on its own despite being constitutively active for phosphorylation due to its extended A-loop conformation. In the canonical kaiABC system, the interaction between KaiB and KaiC is required to provide a new binding interface which sequesters KaiA from its “activating” binding site, thereby promoting auto-dephosphorylation at the organism’s optimum temperature24-26. The obvious next questions are whether the KaiCRS system can oscillate and secondly if there is a regulatory role for KaiBRS in this process. Comparing the in vitro phosphorylation states of KaiCRS in the absence and presence of KaiBRS shows an initial, fast phosphorylation followed by an oscillatory-like pattern in the presence of KaiBRS (hereafter referred to as KaiBCRS), whereas KaiCRS alone remains phosphorylated (Fig. 3a,b). Interestingly, the ATP consumption during the reaction with KaiBRS is significantly higher than without (Fig. 3a) and we have seen earlier that KaiCRS will dephosphorylate completely in the presence of only ADP (cf. Fig. 2f). These results suggest that the phosphorylation state of KaiCRS and thus the observed oscillatory half-cycle (Fig. 3a,b), is likely related to a change in ATP-to-ADP ratio and we conjectured this could constitute the environmental cue to reset the timer. To test our hypothesis, an ATP-recycling system was added after complete dephosphorylation of KaiBCRS and, as predicted, KaiCRS was able to restart the cycle and phosphorylate again (Extended Data Fig. 6a). Naturally, in vivo the ATP-to-ADP ratio will not vary as drastically as in this in vitro experiment since nucleotide homeostasis is tightly regulated. To mimic the day- and night-period for Rhodobacter sphaeroides we repeated the experiments while keeping the ATP-to-ADP ratio constant (mostly ATP at daytime due to photosynthesis versus 25:75% ATP:ADP during nighttime, respectively)27. In the presence of high ATP (i.e., mimicking daytime) KaiCRS remains singly or doubly phosphorylated (Fig. 3c and Extended Data Fig. 6b) irrespective of KaiBRS, whereas a constant 25:75% ATP-to-ADP ratio (i.e., mimicking nighttime) results in a much higher fraction of dephosphorylated KaiCRS in the presence of KaiBRS (Fig. 3c). Moreover, when the ATP-to-ADP ratio is flipped to mimic the daytime, KaiCRS is able to phosphorylate again (cf. Fig. 3c; around the 28-hour mark). Our data support the notion that the phosphorylation behavior depends strongly on the ATP-to-ADP ratio and, more importantly, demonstrate that the physical binding of KaiBRS results in a higher level of KaiCRS dephosphorylation at nighttime.

(a) SDS-PAGE gel of 3.5 μM KaiCRS and 4 mM ATP in the absence (upper gel) and presence (lower gel) of 3.5 μM KaiBRS at 35 °C with the percentage of ATP indicated at specific time points. U, S, and D represent unphosphorylated, singly, and doubly phosphorylated KaiCRS, respectively. (b) Phosphorylation (single + double) of KaiCRS during the reaction in the absence (gray circles) or presence (red diamonds) of KaiBRS demonstrates its role in the dephosphorylation of KaiCRS. (c) Phosphorylation-dephoshorylation cycle of 3.5 μM phosphorylated KaiCRS in the absence and presence of 3.5 μM KaiBRS in a constant ATP-to-ADP ratio of high ATP (4 mM) to mimic daytime and about 25% ATP to mimic the nighttime (exact percentage of ATP indicated at specific time points) at 30 °C. U, S, and D in both panels represent the unphosphorylated, singly phosphorylated (at Ser413 or Ser414), and doubly phosphorylated state of KaiCRS, respectively. These data show that the physical binding of KaiBRS is crucial to dephosphorylate KaiCRS at nighttime where ATP concentrations are still sufficiently high to otherwise keep KaiCRS in the phosphorylated state. (d) ATPase activity of wild-type KaiCRS in the absence (gray bar) and presence (red bar) of KaiBRS, KaiCRS-E62Q/E63Q in the absence (light purple bar) and presence (dark purple bar) of KaiBRS, and KaiCRS-E302Q/E303Q in the absence (light yellow bar) and presence (dark yellow bar) of KaiBRS at 30 °C. Bar graphs show mean ± S.D from three replicates. (e) Time-dependent auto-dephosphorylation of 32P-labeled KaiCRS bound with ADP in the presence of 20 μM KaiBRS and 4 mM ADP at 30 °C showing phosphorylated 32P-KaiCRS (red diamonds), 32P-ATP (purple triangles), and free 32Pi (cyan circles). The reaction products were separated by TLC. (f) The decay of phosphorylated 32P-KaiCRS bound with 4 mM ADP in the absence (gray circles) and presence (red diamonds) of KaiBRS at 30 °C is obtained from autoradiography quantification (see Extended Data Fig. 7). (g) The nucleotide exchange of 3.5 μM KaiCRS (gray trace) and 3.5 μM KaiCRS in complex with 30 μM KaiBRS (red dotted trace) in the presence of ATP with mant-ATP. The proteins were incubated at 30 °C for 24 h before the addition of mant-ATP. Representative traces are shown and the fitted parameters (mean ± S.D) were obtained from three replicate measurements.
Next, we investigated the accelerated ATPase activity in KaiCRS upon complex formation. The ATPase activity reported for KaiCSE is very low (∼15 ATP molecules day-1 molecule-1 KaiCSE) and was proposed as a reason for the “slowness”28. KaiCRS alone already shows a significantly faster ATPase rate that gets further enhanced by binding of KaiBRS (208 ± 19 and 1,557 ± 172 ATP molecules day-1 KaiCRS-1, respectively; left two bars in Fig. 3d and Extended Data Fig. 6c-g). Furthermore, KaiCRS exhibits no temperature compensation for its ATPase activity (Q10 ∼1.9; Extended Data Fig. 6c), a feature that is present in KaiCSE and proposed to be a prerequisite for self-sustained rhythms28. The deviation from unity for Q10 is consistent with our earlier observation that the KaiBCRS system is not a true circadian oscillator but rather an hourglass-timer (cf. Fig. 3b).
Regulatory role of KaiBRS
The mechanistic details of how KaiB binding in the CI domain allosterically affects the auto-dephosphorylation of KaiCRS in the CII domain remain unclear. Intuitively, there are three plausible scenarios to explain this, namely that KaiBRS binding (i) stimulates the phosphoryl-transfer from pSer back to ADP (Extended Data Fig. 7a), (ii) increases the hydrolysis rate of the active-site ATP (Extended Data Fig. 8a), or (iii) accelerates the nucleotide exchange in the CII domain (Extended Data Fig. 8e). To differentiate between these possibilities, we performed radioactivity experiments to follow nucleotide interconversion, measured ATPase activity for wild-type KaiCRS and mutant forms that are incapable of ATPase activity in the CI or CII domain, and quantified nucleotide-exchange rates by fluorescence using mant-ATP. First, we detected fast, transient 32P-ATP formation in our radioactivity experiments when starting from 32P-phosphorylated KaiCRS due to its ATP-synthase activity in the CII domain (Fig. 3e and Extended Data Fig. 7b-d). The observed phosphoryl-transfer rate is independent of KaiBRS (kobs = 12.0 ± 1.7 h-1 and 15.4 ± 1.7 h-1 in its absence and presence, respectively; Fig. 3f) and agrees well with the rates determined from our gel electrophoresis experiments (11.0 ± 0.8 h-1 and 11.5 ± 0.8 h-1 with/without KaiBRS, respectively; Extended Data Fig. 7e,f). Our experimental data confirm that KaiCRS undergoes dephosphorylation via an ATP-synthase mechanism similarly to what was observed for KaiCSE29; KaiB does not expedite the actual phosphoryl-transfer reaction, which is never the rate-limiting step. Since we were unable to stabilize the first phosphorylation site (Ser414) in the presence of ADP, the rates reported here correspond exclusively to dephosphorylation of Ser413. Secondly, to deconvolute the contributions of the CI and CII domains to the observed ATPase activity, we measured ADP production by KaiCRS mutants that abolish hydrolysis in either the CI (KaiCRS-E62Q/E63Q) or CII (KaiCRS-E302Q/E303Q) domain. For wild-type KaiCRS, the binding of KaiBRS results in a 7.5-fold increase in ATPase activity, and we show that both domains are affected and contribute additively (3-fold for CI and at least 1.7-fold for CII, respectively) to the overall effect (Fig. 3d and Extended Data Fig. 8b-d). Of note, the fold increase in the CII domain represents a lower limit since the KaiCRS-E62Q/E63Q mutations interfere with KaiBRS binding as reported before for KaiCSE30. Thirdly, our measurements of the nucleotide exchange rate show that it is also unaffected by KaiBRS binding (19.8 ± 1.8 h-1 and 18.0 ± 1.5 h-1 with/without, respectively; Fig. 3g); since there is no Trp residue near the nucleotide binding site in the CI domain, only the exchange rate in the CII domain could be determined. Interestingly, the change in fluorescence amplitude is much smaller in the presence of KaiBRS, demonstrating that even though the binding of KaiBRS does not accelerate nucleotide exchange, it appears to induce a conformational rearrangement in the CII domain especially at higher temperature (Fig. 3g and Extended Data Fig. 8f-h).
Structure of KaiBCRS complex
To elucidate the structural underpinning of the faster ATPase activity upon KaiBRS binding, we solved the cryo-EM structures of KaiCRS alone (PDB xxxx) and in complex with KaiBRS (PDB xxxx) (Extended Data Table 2). Twelve KaiBRS molecules (monomeric in solution, Extended Data Fig. 9a) bind to the CI domain of the KaiCRS-S413E/S414E dodecamer (Fig. 4a-c and Extended Data Fig. 9b). The bound state of KaiBRS adopts the same “fold-switch” conformation as observed for KaiBTE25 and suggests that this is the canonical binding-competent state (Fig. 4b). Upon binding of KaiBRS, the CI-CI interfaces loosen up (Fig. 4c), which allows for the formation of a tunnel that connects bulk solvent to the position of the hydrolytic water in the active sites (Fig. 4d and Extended Data Fig. 9c). There are other lines of evidence for the weakened interactions within the CI domains. First, KaiBRS binding to either KaiCRS-CI domain (Extended Data Fig. 10a) or KaiCRS-Δcoil (i.e., missing the C-terminal extensions; Extended Data Fig. 10b) results in disassembly of the hexameric KaiCRS structure into its monomers. In contrast, full-length KaiCRS maintains its oligomeric state upon binding of KaiBRS likely due to the stabilization provided by the coiled-coil interaction. Secondly, a decrease in melting temperature (TM) of KaiCRS is observed with increasing KaiBRS concentration (Extended Data Fig. 10c). There is no interaction between neighboring KaiBRS molecules within the complex (Extended Data Fig. 9b), suggesting a non-cooperative assembly of KaiBRS to KaiCRS contrary to what is observed for the KaiBCSE/TE complex31,32.

Cryo-EM structure of KaiCRS-S413E/S414E (yellow) in complex with KaiBRS (blue) (PDB xxxx). (b) Superposition of KaiCRS-S413E/S414E (yellow) bound to KaiBRS (blue) (PDB xxxx) and KaiCTE-S413E (dark gray) bound to fsKaiBTE (light gray) (PDB 5jwq26). (c, d) Binding of KaiBRS (blue) creates a tunnel (gray mesh) that enables water to reach the catalytic position (red sphere) for ATP hydrolysis in the CI domain. (e) Binding of wild-type and mutants forms of KaiCRS to His-tagged KaiBRS in the presence of ADP or ATP/recycling system at 25 °C. Bar graphs show mean ± S.D. from three replicates. (f) Fluorescence anisotropy of unlabeled KaiBRS competitively displacing KaiBRS-6IAF (where 6IAF is the fluorophore) from unphosphorylated KaiCRS. in the presence of ADP (dark green circles) and phosphorylated KaiCRS in the presence of the ATP/recycling system (light green triangles) at 30 °C. The average anisotropy and standard error were calculated from ten replicate measurements. (g) Schematic diagram of the uncovered mechanism of KaiCRS regulated by coiled-coil interaction and KaiBRS in the CI and CII domain.
Furthermore, we noticed that KaiB-bound structures in phosphomimetic variants of KaiCRS (Fig. 4c,d) and KaiCSE26 have ADP bound in their CI domain, demonstrating that the post-hydrolysis state is also the binding-competent state for KaiBRS. To test this hypothesis, a His-tagged KaiBRS protein was used in pull-down assays to detect its physical interaction with wild-type and mutant forms of KaiCRS bound with either ADP or ATP. Nearly all KaiBRS is complexed to ADP-bound KaiCRS, whereas less than 30% co-elutes in the ATP-bound form regardless of the phosphorylation state (Fig. 4e and Extended Data Fig. 10d,e). The complex formation depends inversely on the ATP-to-ADP ratio (Extended Data Fig. 10f). We performed fluorescence anisotropy competition experiments for a more quantitative description of the binding interaction between KaiCRS and KaiBRS: very similar KD values were obtained for unphosphorylated, wild-type KaiCRS (Fig. 4f) and its phosphomimetic form (Extended Data Fig. 10g) bound with ADP (0.42 ± 0.03 μM and 0.79 ± 0.06 μM, respectively). No measurable binding curves were obtained for ATP-bound phosphorylated wild-type KaiCRS (Fig. 4f) or KaiCRS-S413E/S414E (Extended Data Fig. 10g) with ATP-recycling system, likely due to the small fraction of complex present. Our data show that the post-hydrolysis state in the CI domain is key for KaiBRS binding, whereas the phosphorylation state of KaiCRS has only a marginal effect.
In summary, we unequivocally demonstrate that binding of KaiBRS at the CI domain in the post-hydrolysis state facilitates the hydrolysis of transiently formed ATP after dephosphorylation of KaiCRS in the CII domain (Fig. 4g). Our fluorescence experiments (Fig. 3g and Extended Data Fig. 8f) detect a conformational change in the CII domain upon KaiBRS binding, but we do not observe major structural changes in the cryo-EM structures. Based on the temperature dependence of the fluorescence amplitudes (Extended Data Fig. 8f) we conjecture that the inability to detect conformational differences is likely because of the low temperature. Since the CII domain prefers to bind ATP over ADP (Extended Data Fig. 10h), ATP hydrolysis in the CII domain stimulated by KaiBRS is particularly important to keep KaiCRS in its dephosphorylated state at nighttime, where the exogenous ATP-to-ADP ratio remains sufficiently high to otherwise result in ATP-binding in the CII active site (cf. Fig. 3c and Extended Data Fig. 6b).
Discussion
The KaiBCRS system studied here represents a primordial, hourglass timekeeping machinery and its mechanism provides insight into more evolved circadian oscillators like KaiABC. The dodecameric KaiCRS shows constitutive kinase-activity due to its extended C-terminal tail that forms a coiled-coil bundle with the opposing hexamer and elicits a conformation akin to the exposed A-loop conformation in KaiACSE, and auto-phosphorylation occurs within half an hour. In the KaiABCSE system, the transition from unphosphorylated to doubly phosphorylated KaiC takes place over about twelve hours and the fine-tuning of this first half of the circadian rhythm is accomplished by the emergence of KaiASE during evolution. The second clock protein, KaiB, binds at the CI domain with the same “fold-switched” state in both systems. The interaction is controlled by the phosphorylation state in the KaiABCSE system, and its sole function is to sequester KaiASE from the “activating” binding site, whereas KaiB binding directly accelerates the ATPase activity in the KaiBCRS system regardless of the phosphorylation state. The KaiBCRS system requires an environmental switch in ATP-to-ADP concentration to reset the clock and thus follows the day-night schedule when nucleotide concentrations inherently fluctuate in the organism. By contrast, the self-sustained oscillator KaiABCSE remains functional over a wide range of nucleotide concentrations and responds to changes in the ATP-to-ADP ratio by changing its phosphorylation period and amplitude to remain entrained with the day and night cycle33.
The novel structural fold of KaiC utilizes the versatile coiled-coil architecture as part of a long-range allosteric network that regulates KaiCRS dephosphorylation. Nature uses conformational changes in coiled-coil domains for a variety of regulatory functions34, including the activity of the motor protein dynein in cellular transport of cargo along the actin filament12. A similar register shift, although in a coiled-coil interaction formed by only two helices is used in dynein motility. Given that this simple heptad repeat sequence emerged multiple times and is found throughout all kingdoms of life35 it is an example of convergent evolution.
Data availability
Structure factors and refined models are deposited in the Protein Data Bank (PDB) under accession codes 8dba (wild-type KaiCRS, x-ray), 8db3 (KaiCRS-Δcoil, x-ray), xxxx (KaiCRS-S413E/S414E, cryo-EM), and xxxx (KaiCRS-S413E/S414E:KaiBRS, cryo-EM), respectively. Cryo-EM maps are deposited in the Electron Microscopy Data Bank (EMDB) under accession codes xxxx (KaiCRS-S413E/S414E) and xxxx (KaiCRS-S413E/S414E:KaiBRS), respectively.
Author contributions
W.P., R.A.P.P., and D.K. conceived the project and designed experiments. W.P. performed and analyzed all biochemical data. W.P. and R.A.P.P. set up the crystal trays and R.A.P.P. collected and analyzed the X-ray crystallographic data. W.P. prepared the samples for the cryo-EM studies and collected negative stain images to screen for optimal sample conditions; T.G. collected and processed all cryo-EM data, and reconstructed the cryo-EM maps under supervision of N.G.; R.A.P.P. built and interpreted the structural models. W.P. and N.B. performed and analyzed experiments with radioactively labeled KaiC. M.H. built the KaiC phylogeny. W.P., R.O., and D.K. wrote the paper; and all authors commented on the manuscript and contributed to data interpretation.
Competing interests
D.K. is co-founder of Relay Therapeutics and MOMA Therapeutics. The remaining authors declare no competing interests.

(a) Phylogenetic tree of kaiChomologs, where kaiC genes that have an approximately 50 amino acids C-terminal extension are labeled in red Rhodobacter sphaeroides strain KD131 studied here and Synechococcus elongatus PCC 7942 (widely studied in the literature) are highlighted in green and pink, respectively. The accession code and organism are shown at the tip of the branches, the numbers at each node represent the aBayes bootstrap values38, and the legend for branch length is shown. (b) A sequence alignment of the CII domain of the kaiC subgroups annotated with its sequence similarity. Residue Glu490, the position where the stop codon was introduced in the truncated KaiCRS-Δcoil construct, is shown in red and marked with an arrow.

(a) 10% SDS-PAGE gel of 3.5 μM KaiCRS in the presence of 4 mM ATP and using an ATP-recycling system at 30 °C. U, S, and D represent unphosphorylated, singly, and doubly phosphorylated KaiCRS, respectively. (b) Densitometric analysis of auto-phosphorylation (single + double phosphorylation) from panel (a) over time yields a rate of 6.5 ± 1.0 h-1. (c) 6.5% SDS-PAGE gel of 3.5 μM KaiCSE in the presence of 1.2 μM KaiASE and 4 mM ATP at 30 °C. U and P represent unphosphorylated and phosphorylated KaiCSE, respectively. (d) Densitometric analysis of auto-phosphorylation of KaiCSE activated by KaiASE (panel (c)) shows a rate of 0.40 ± 0.02 h-1 and is substantially slower than for KaiCRS. The standard deviation for parameters in (b) and (d) were obtained from data fitting. (e) 10% SDS-PAGE gels for experiments with 3.5 μM KaiCRS in the presence of 4 mM ATP and using an ATP-recycling system between 20 and 35 °C show that the level of phosphorylation increases with temperature. U, S, and D represent unphosphorylated, singly, and doubly phosphorylated KaiCRS, respectively. (f) Bar graphs indicating the nucleotide exchange rate in the CII domain of KaiCRS incubated with 50 μM ATP in the presence of an ATP-recycling system, and then mixed with 250 μM mant-ATP. An increase in fluorescence intensity at 440 nm was recorded and the single-exponential time traces were fitted to obtain the exchange rate constants: 3.6 ± 0.8 h-1 (20 °C), 12.2 ± 1.0 h-1 (25 °C), 18.5 ± 1.5 h-1 (30 °C), and 25.2 ± 0.2 h-1 (35 °C). Experiments were performed in triplicate with error bars representing SD.
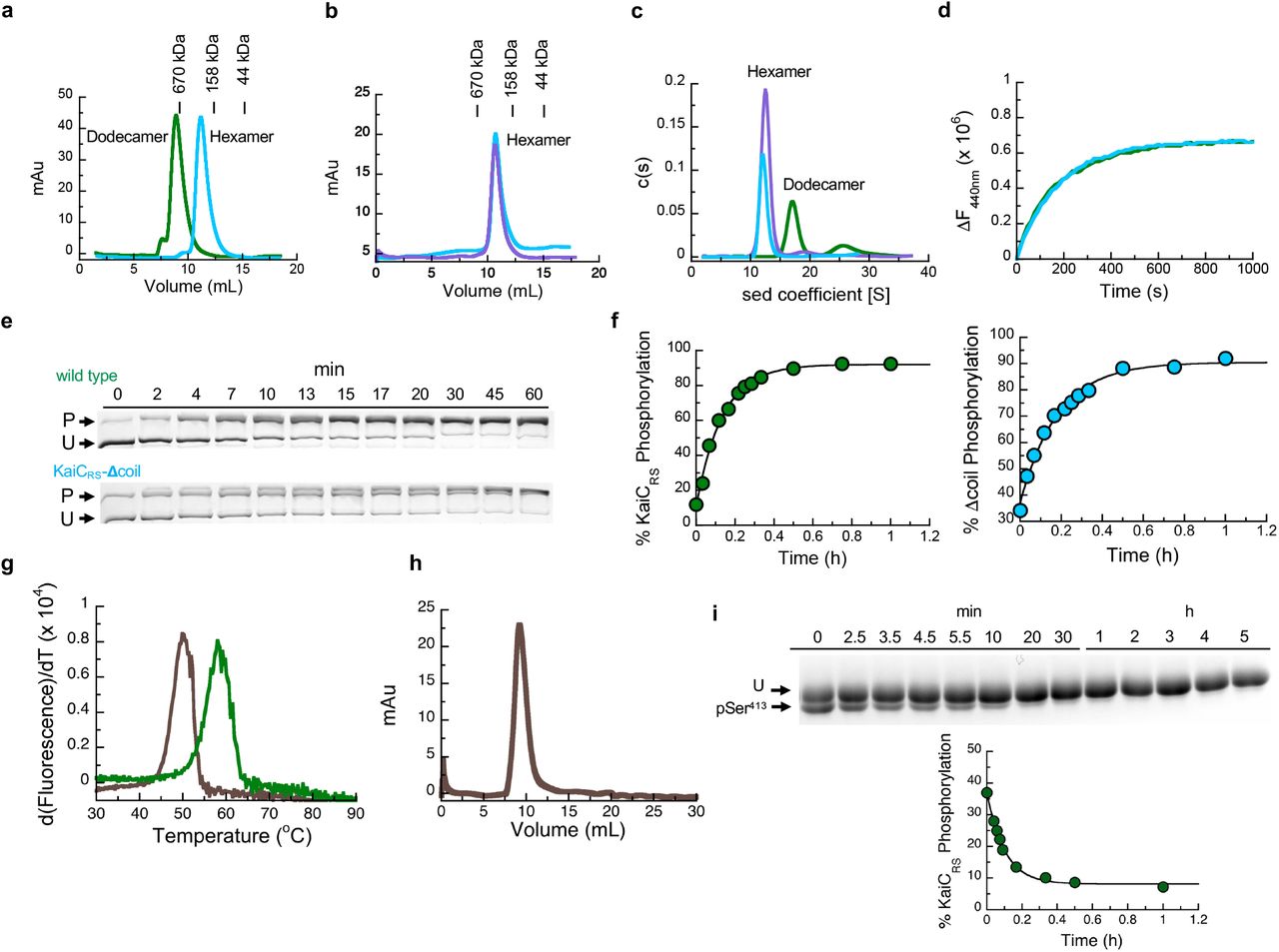
(a) Oligomerization analysis of KaiCRS (dodecamer, green line) and truncated KaiCRS-Δcoil (hexamer, cyan line) by analytical gel-filtration chromatography. The protein size markers are indicated at the top. (b) Comparison of the elution profiles of KaiCRS-Δcoil (cyan line) and KaiCSE (purple line) from size-exclusion chromatography shows a hexameric state for both KaiCRS-Δcoil and KaiCSE. (c) Oligomeric states of KaiCRS (dodecamer, green line), KaiCRS-Δcoil (hexamer, cyan line), and KaiCSE (hexamer, purple line) were also measured by analytical ultracentrifugation (sedimentation velocity at 30,000 rpm and 20 °C) and the results agree with the data shown in panels (d) and (e). The graph in panel (f) represents the sedimentation coefficient distribution [c(s)]. (d) The change in fluorescence at 440 nm (ΔF440nm) represents the nucleotide exchange between ATP and mant-ATP at 30 °C for KaiCRS (green trace, 18.0 ± 1.5 h-1) and KaiCRS-Δcoil (cyan trace, 19.1 ± 0.8 h-1). Representative traces are shown and the fitted parameters (mean ± S.D) were obtained from three replicate measurements. (e) Zn2+ Phos-tag™ SDS-PAGE gel shows the level of phosphorylation over time of KaiCRS (upper gel) and KaiCRS-Δcoil (lower gel) at 35 °C. P and U represent phosphorylated and unphosphorylated protein, respectively. (f) Phosphorylation level over time of KaiCRS (green circles, 7.4 ± 0.3 h-1) and KaiCRS-Δcoil (cyan circles, 5.5 ± 0.4 h-1) analyzed by densitometric analysis of Zn2+ Phos-tag™ SDS-PAGE gel in (b). (g) First derivative of thermal-stability curves measured for unphosphorylated KaiCRS bound with ADP (brown line) and phosphorylated KaiCRS bound with ATP (green line). The extracted temperatures of denaturation are 50 °C (unphosphorylated KaiCRS in the presence of 1 mM ADP) and 58 °C (phosphorylated KaiCRS in the presence of 1 mM ATP), respectively. (h) Dodecameric state of unphosphorylated KaiCRS (40 μM) bound with ADP measured by size-exclusion chromatography. (i) SDS-PAGE gel shows dephosphorylation of Ser413 over time at 30 °C in the presence of 4 mM ADP (U and pSer413 represent unphosphorylated and Ser413-phosphorylated KaiCRS, respectively) with the corresponding kinetics shown in the lower panel (confirmed by MS/MS) with a rate constant of 11.5 ± 0.8 h-1. This result suggests that the coiled-coil domain promotes KaiCRS dephosphorylation. The standard deviation for parameters in (f) and (i) were obtained from data fitting.

The workflow (see description in Methods section) demonstrates a typical image (scale bar: 60nm) and representative good class averages. The ab initio and final reconstructions are shown. Shown alongside the final reconstruction is the angular plot demonstrating the distribution of particle views and the Fourier shell correlation curve used for the global resolution estimation (a) KaiCRS-S413E/S414E alone and (b) KaiCRS-S413E/S414E:KaiBRS complex. To validate the final combined dodecamer structures, the data were reprocessed for the full dodecamer. The figure shows representative good class averages, the final reconstruction, angular distribution and Fourier shell correlation curve for the KaiCRS-S413E/S414E alone and (d) KaiCRS-S413E/S414E:KaiBRS complex dodecamers. (e) Comparison of the C1 and D6 reconstructions of KaiCRS-S413E/S414E alone and KaiCRS-S413E/S414E:KaiBRS, and Fourier shell correlation curves for the C1 reconstructions. The C1/D6 comparisons do not reveal discernable differences, suggesting that these complexes have D6 symmetry.
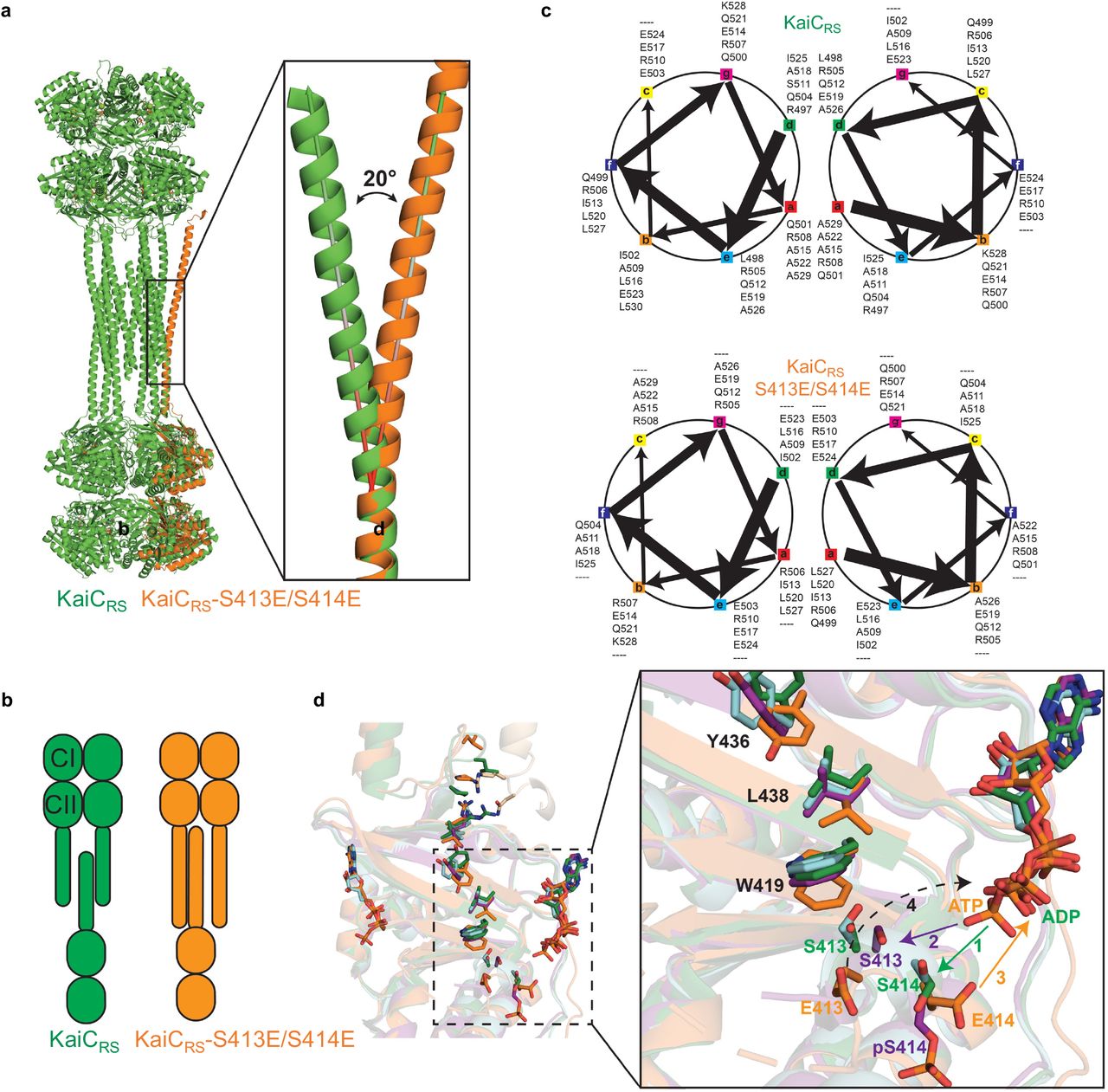
(a) Structural comparison between KaiCRS (green) and KaiCRS-S413E/S414E (orange, single-chain for clarity) reveals that the coiled-coil in the phosphomimetic structure points outwards, with an angle of about 20° relative to the KaiCRS coiled-coil. (b) The conformational change in the coiled-coil domain affects the dimer interface due to partner swaps with the opposite hexamer (see also Fig. 2). From an “outside perspective”: the C-terminal helix in KaiCRS interacts with the right chain from the opposite hexamer, whereas in KaiCRS-S413E/S414E the interactionis with the chain on the left. (c) Coiled-coil diagrams describe the heptad register shift that accompanies this structural rearrangement. (d) Based on the overlay of our structures, we propose the following model for the phosphorylation/ dephosphorylation events. First, the phosphorylation cycle starts with the transfer of the γ-phosphate of ATP to the hydroxyl group of Ser414 (1; green arrow) in unphosphorylated KaiCRS (green) or KaiCRS-Δcoil (cyan). Secondly, pSer414 of KaiCRS-Δcoil (purple, singly phosphorylated) moves away from the active site placing the hydroxyl group of Ser413 closer to the -phosphate of ATP for the second phosphorylation (2; purple arrow). Thirdly, the doubly phosphomimetic state (KaiCRS-S413E/S414E, orange) reveals that the phosphoryl group of pSer414 moves back towards the active site for dephosphorylation (3; orange arrow). Lastly, we hypothesize that the indole group of Trp419 “pushes” pSer413 into the active site for the second dephosphorylation event (dashed arrow), in agreement with the slower dephosphorylation rate observed in the KaiCRS-Δcoil construct (cf. Fig. 2d).

(a) 10% SDS-PAGE gel of 3.5 μM KaiCRS and 3.5 μM KaiBRS in the presence of 4 mM ATP and 10 mM 2-phosphoenolpyruvate at 30 °C shows that the auto-phosphorylation cycle restarts upon regeneration of ATP by the addition of 2 U/mL pyruvate kinase at the 24-hour time mark. (b) 10% SDS-PAGE gel of 3.5 μM KaiCRS without (upper panel) and with (lower panel) 3.5 μM KaiBRS in the presence of 4 mM ATP with an ATP-recycling system added from the beginning showing that under these conditions KaiB does not accelerate dephosphorylation. (c) Representative curves for ADP production of KaiCRS (3.5 μM) alone and (d) in the presence of KaiBRS (3.5 μM) in 4 mM ATP measured by HPLC. The data were analyzed as described in the Methods section and result in ATPase activities of 108 ± 10 day-1 KaiC-1 (with KaiBRS = 176 ± 29 day-1 KaiC-1) at 20 °C, 163 ± 16 day-1 KaiC-1 (with KaiBRS = 1052 ± 143 day-1 KaiC-1) at 25 °C, 208 ± 19 day-1 KaiC-1 (with KaiBRS = 1557 ± 172 day-1 KaiC-1) at 30 °C, and 300 ± 21 day-1 KaiC-1 (with KaiBRS = 2584 ± 245 day-1 KaiC-1) at 35 °C. The temperature coefficient, Q10, was calculated using the data obtained at 25 °C and 35 °C and yields a value of ∼ 1.9. The standard deviations of ATPase activity at each temperature were obtained from three replicate measurements. (e) The comparison of ADP production of KaiCRS in the absence (orange circles) and presence (orange diamonds) of KaiBRS at 30 °C indicate a 7.5-fold increase in ATPase activity for the complex. The binding of KaiBRS accelerates the ATPase activity of KaiCRS in both the CI and CII domains (see also Extended Data Fig. 8b, c). (f) The SDS-PAGE gel of KaiCRS (10 μg) and KaiBRS (10 μg) shows that both proteins were purified to homogeneity and the measured ATPase activity is, therefore, not due to impurities. (g) ADP production of KaiBRS in 4 mM ATP at 30 °C shows, as expected, no ATPase activity for KaiBRS alone and confirms the increase in ATPase activity shown in panel (d) is due to complex formation.
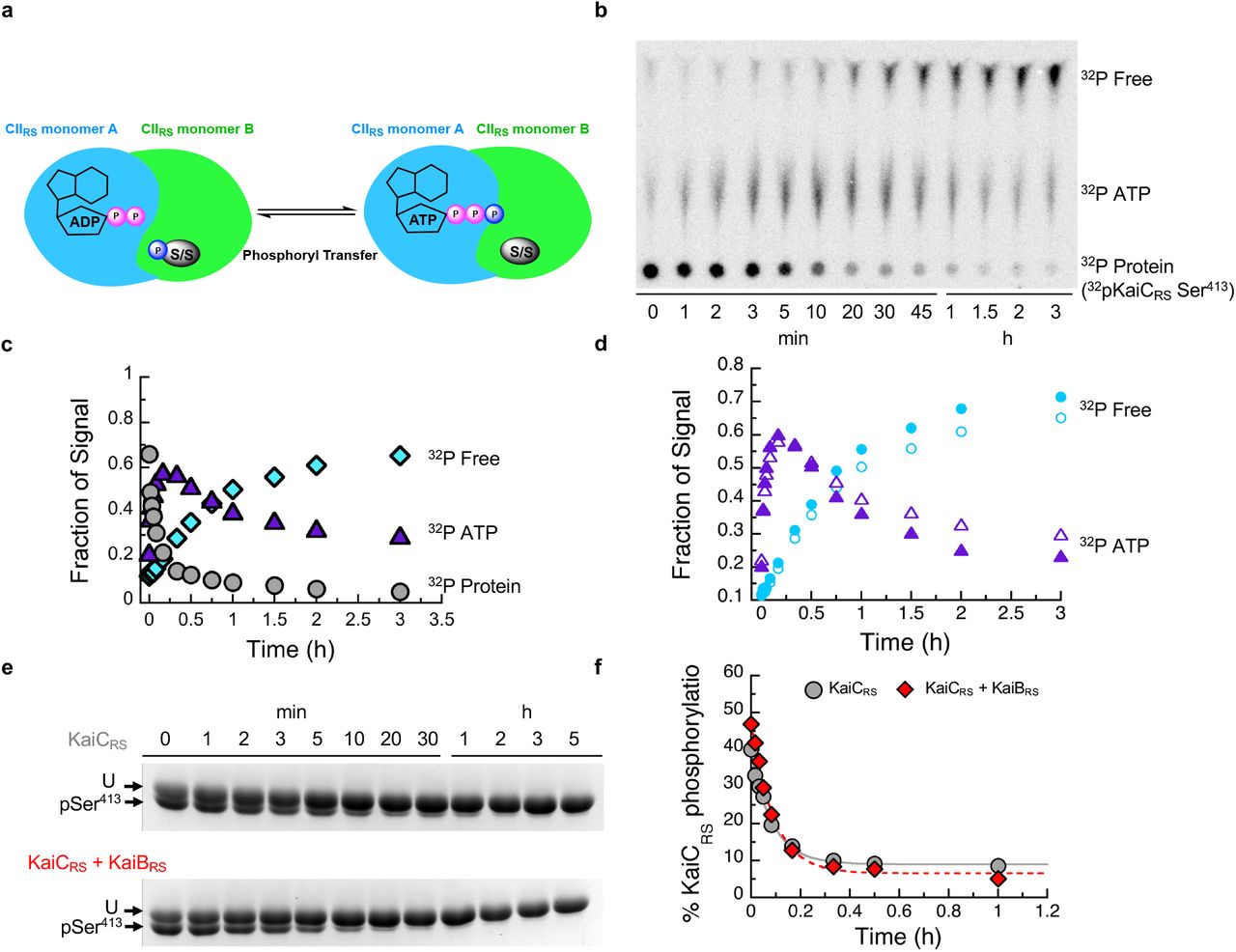
(a) Possible mechanisms how KaiBRS could accelerate KaiCRS dephosphorylation at nighttime. Binding of KaiBRS on the CIRS domain directly accelerates the phosphoryl transfer from pSer to bound ADP to generate transiently bound ATP. The cartoon represents the interface between two monomers in the CIIRS domain. (b) Autoradiograph of separation of 32P-KaiCRS at Ser413, transiently formed 32P-ATP, and free 32Pi via thin-layer chromatography (TLC) with 4 mM ADP at 30 °C, with the corresponding kinetics shown in (c) where gray circle, purple triangle, and cyan diamonds represent the relative concentrations of phosphorylated 32P-KaiCRS, 32P-ATP, and free 32Pi, respectively. (d) Comparison of transient 32P-ATP formation and decay in the absence (open triangle) and presence (solid triangle) of KaiBRS and free 32P formation in the absence (open circles) and presence of KaiBRS (solid circles). Faster decay of transient 32P-ATP together with higher free 32P production in the presence of KaiBRS indicated that KaiBRS accelerates hydrolysis in KaiCRS. (e) SDS-PAGE gel (10%) of dephosphorylation of phosphorylated 3.5 μM KaiCRS at Ser413 without (upper gel) and with (lower gel) 3.5 μM KaiBRS in the presence of 4 mM ADP at 30 °C. (f) Densitometric analysis of data in panel (e) shows the decay of total KaiCRS phosphorylation in the absence (gray circles) and presence (red diamonds) of KaiBRS and yields rates of 11.5 ± 0.8 h-1 and 11.0 ± 0.8 h-1, respectively. This result indicates that binding of KaiBRS does not accelerate the phosphoryl-transfer step in KaiCRS.

(a) Second possible mechanism to explain how KaiBRS accelerates KaiCRS dephosphorylation at nighttime: binding of KaiBRS to the CIRS domain could increase the hydrolysis rate in the CIIRS domain and, thereby, prevent the phosphoryl transfer back from transiently formed or external ATP back to serine residues. (b) ADP production of phosphorylated KaiCRS with catalytic mutations in the CI domain (KaiCRS-E62Q/E63Q, 3.5 μM) in the absence (dark green circles) and presence (light green circles) of 3.5 μM KaiBRS at 30 °C with 4 mM ATP was quantified using HPLC. From these data an ATPase activity in the CII domain of 112± 8 day-1 KaiC-1 and 195 ± 16 day-1 KaiC-1 in the presence of KaiBRS was determined. (c) ADP production measured by HPLC as in panel (b) of KaiCRS but with catalytic mutationsin the CII domain (KaiCRS-E302Q/E303Q, 3.5 μM) in the absence (dark pink circles) and presence (light pink diamonds) of 3.5 μM KaiBRS. The corresponding ATPase activities in the CI domain are 110 ± 12 day-1 KaiC-1 in the absence and 320± 22 day-1 KaiC-1 in the presence of KaiBRS. (d) ADP production of KaiCIRS-E62Q/E63Q (construct of only CI domain with catalytic mutations) in 4 mM ATP at 30 °C shows no ATPase activity indicating that Glu62 and Glu63 are the only two residues that are responsible for ATPase activity in CI domain of KaiCRS and confirms the ATPase activity shown in panel C is due to ATPase activity in CII domain of KaiCRS. (e) Third possible mechanism to explain how KaiBRS accelerates KaiCRS dephosphorylation at nighttime: binding of KaiBRS to the CIRS domain could promote faster nucleotide exchange in the CIIRS domain to displace transient ATP by ADP. (f) Time course of fluorescence intensity at 440 nm due to binding of mant-ATP to KaiCRS-S413E bound with ATP in the absence (solid blue trace) and presence (red dotted trace) of KaiBRS. KaiCRS-S413E (3.5 μM) was pre-incubated with 3.5 μM KaiBRS for 16 h at 20, 25, 30, and 35 °C in the presence of 50 μM ATP and an ATP-recycling system and then mixed with 250 μM mant-ATP. The observed exchange rates at each temperature are listed in the table (g). (h) Nucleotide exchange of KaiCIRS (i.e., only CIRS domain) cannot be measured since there is no tryptophan residue in close proximity of the nucleotide binding site. In summary, KaiBRS accelerates KaiCRS dephosphorylation by increasing the hydrolysis rate in the CI and CII domains and does not affect the nucleotide exchange rate. Representative traces are shown in (f) and (h) and the fitted parameters (g; mean ± S.D) were obtained from three replicate measurements.

(a) Size-exclusion chromatography of KaiBRS (blue) shows that it is monomeric in solution in contrast to KaiBSE (gray), which elutes as a tetramer. Molecular-weight standards are shown above the chromatogram. (b) Structural comparison of KaiBTE (gray, PDB 5jwq26) and KaiBRS (blue) when bound to their corresponding KaiC hexamers. The PISA software package37 determines that for the KaiBCTE complex the interface between the KaiBTE monomers is 255 Å2, whereas the average interface between KaiBRS monomersis only 45 Å2 in the KaiBCRS complex. (c) To understand how KaiBRS binding to KaiCRS-CI domain increases the hydrolysis rate, we investigated whether conformational changes modulated substrate access to the active site. The CAVER software39 was used to calculate tunnels (gray mesh) leading into the active site of the CI domain of KaiCRS-S413E/S414E alone (orange) and the KaiCRS-S413E/S414E:KaiBRS complex (yellow:blue) with varying probe radii. In both structures, the active site was occupied by ADP:Mg2+ (sticks and green sphere, respectively). The crystal structure of bovine F1-ATPase in complex with a transition-state analogue (PDB 1w0j, chain D)40 was used as a reference to determine the position of the catalytic water molecule in the active site (shown as a red sphere). The calculated tunnels connect bulk solvent to the catalytic water when KaiBRS is bound for probe radii larger than the default value of 0.9 Å, but never in its absence. These results suggest that KaiBRS facilitates the access of water into the active site of KaiCRS-CI via long-range conformational changes and thus enhances ATP hydrolysis.
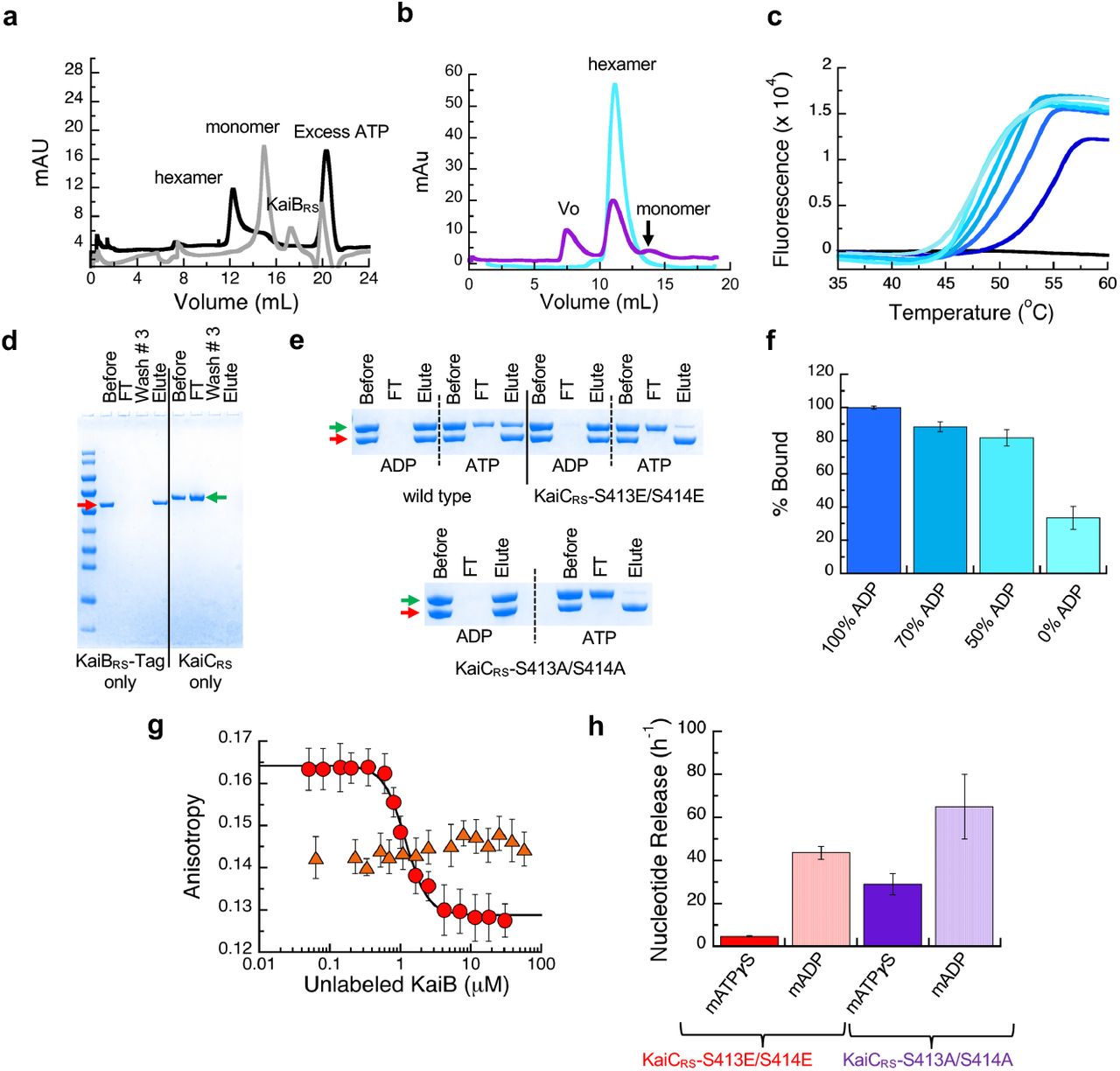
(a) Size-exclusion chromatography of 50 μM KaiCRS-CI (CIRS domain) in the absence (black line, hexamer) and presence (gray line, monomer) of 50 μM KaiBRS in 1 mM ATP buffer. (b) Size-exclusion chromatography of 50 μM KaiCRS-Δcoilin the presence of 50 μM KaiBRS (purple). The reference sample (50 μM KaiCRS-Δcoil) is a hexamer in solution (cyan) and after the addition of 50 μM KaiBRS the mixture was incubated at 30 °C for 3.5 h (purple) before running the samples again ona Superdex-200 10/300 GL column at 4 °C. These data show that binding of KaiBRS results in (i) disassembly of the hexameric KaiCRS-Δcoil structure into its monomers and (ii) aggregation as detected by the elution in the void volume of the column (v0). (c) Thermal denaturation profiles for KaiCRS-S413E/S414E in the presence of 1 mM ADP are shown from dark to light blue for increasing concentrations of KaiBRS (between 0 – 4 μM). The black line represents KaiBRS alone (15 μM), which shows no fluorescence signal as it does not bind to SYPRO Orange due to a lack of a hydrophobic core. The Tm decreases upon the addition of KaiBRS, indicating that binding of KaiBRS destabilizes the KaiCRS dodecamer. Likely due to loosening up of interface and the KaiCRS structure, thereby allowing for the formation of a tunnel that connects bulk solvent to the position of the hydrolytic water in the active site (see Extended Data Fig. 9). (d) SDS-PAGE analysis showing the control experiment for pull-down assay. The first four lanes after the molecular weight marker are KaiBRS-Tag samples (red arrow) and show that KaiBRS-Tag binds tightly to the column. The last four lanes are control pull-down assay experiments for KaiCRS (green arrow) and show that KaiCRS alone is unable to bind to the column. The lanes represent the initial sample used in pull-down assay (Before), flow-through after loading sample onto the column (FT), flow-through after washing the column three times with the binding buffer (Wash #3), and sample after elution with imidazole (Elute). (e) SDS-PAGE analysis of pull-down assay to measure the complex formation between KaiBRS-Tag and wild-type KaiCRS, KaiCRS-S413E/S414E, or KaiCRS-S413A/S414A in the presence of 4 mM ADP or ATP (with an ATP-recycling system). (f) Percentage of wild-type KaiCRS bound to KaiBRS-Tag protein for different ATP-to-ADP ratios (4 mM total nucleotide concentration) at 25 °C as measured from pull-down assays. (g) Fluorescence anisotropy at 30 °C of unlabeled KaiBRS competitively replacing the fluorophore-labeled KaiBRS (KaiBRS-6IAF) from KaiCRS-S413E/S414E in the presence of 4 mM ADP (red circles, KD value of 0.79 ± 0.06 μM) and 4 mM ATP with an ATP-recycling system (orange triangles). In the latter experiment no change in anisotropy is observed, indicating that only a small fraction of KaiBRS-6IAF is bound under these conditions. The average anisotropy and standard error were calculated from ten replicate measurements. (h) The mant-ATPγS or mant-ADP release is shown as bar graphs with observed rates of 4.8 ± 0.2 h-1 and 43.6 ± 3.0 h-1 for mant-ATPγS and mant-ADP releasing from KaiCRS-S413E/S414E, respectively, and 21.0 ± 3.0 h-1 and 65 ± 15 h-1 for mant-ATPγS and mant-ADP releasing from KaiCRS-S413A/S414A, respectively. The result shows that CII domain of KaiCRS prefers binding of ATP over ADP. Experiments in panels f and h were performed in triplicate with error bars representing SD.
Acknowledgements
D.K. and N.G. are supported by the Howard Hughes Medical Institute (HHMI). We would like to thank Mike Rigney for assistance with negative-stain data collection at the Brandeis University Electron Microscopy Facility, and Zhiheng Yu and the staff of the Janelia Research Campus cryo-EM facility for advice and assistance with data collection. The Berkeley Center for Structural Biology is supported in part by HHMI. Beamline 8.2.1 of the Advanced Light Source, a U.S. DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169-01. Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Core Facility, RRID:SCR_019063.
Footnotes
Remove the line number that overlap with the figure legend





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}