

ABSTRACT
Enhancers function with a basal promoter to control the transcription of target genes. Enhancer regulatory activity is often studied using reporter-based transgene assays. However, unmatched results have been reported when selected enhancers are silenced in situ. In this study, using genomic deletion analysis in mice, we investigated the roles of two previously identified enhancers and the promoter of the Rho gene that codes for the visual pigment rhodopsin. The Rho gene is robustly expressed by rod photoreceptors of the retina, and essential for the subcellular structure and visual function of rod photoreceptors. Mutations in RHO cause severe vision loss in humans. We found that each Rho regulatory region can independently mediate local epigenomic changes, but only the promoter is absolutely required for establishing active Rho chromatin configuration and transcription and maintaining the cell integrity and function of rod photoreceptors. To our surprise, two Rho enhancers that enable strong promoter activation in reporter assays are largely dispensable for Rho expression in vivo. Only small and age-dependent impact is detectable when both enhancers are deleted. Our results demonstrate context-dependent roles of enhancers and highlight the importance of studying functions of cis-regulatory regions in the native genomic context.
INTRODUCTION
Transcriptional regulation is a tenet of cellular development and homeostasis. Each cell type uses a unique set of regulatory sequences, namely promoters and enhancers, to enact expression of specific genes. A gene’s promoter is the DNA region immediately upstream of the transcription start site (TSS). Enhancers are often found distally upstream or downstream of a gene’s coding region and modulate the promoter activity. The cooperative regulation by promoter and enhancers is important for directing gene transcription within a specific cell type(s) at a precise timing and an appropriate level. In particular, promoters and enhancers contain accessible regions of sequence-specific binding sites for cell-type specific and general transcription factors (TFs). Base-content analysis indicates the differences between the two classes: promoters display high-GC content and contain the requisite TATA box, initiator sequences and binding sites for a variety of TFs; enhancers are often AT-rich and contain binding sites for cell-type specific TFs1-4. Both classes can be defined by the presence of active histone modifications; specifically, promoters with H4K4me3 and H3K27Ac marks5,6, and enhancers with H3K27Ac and H3K4me1 marks7. Studies have further classified the regulatory units based on the landscape and density of the enrichment of these histone modifications. Super enhancers and broad H3K4me3 domains8-13 are hence defined as genomic regions regulating the specific gene expression that is imperative to a cell’s state and function.
Rod and cone photoreceptors are the cell types of the retina, and mediate the conversion of light photons into electrical signals in a process called phototransduction. In mouse and human retinas, rod photoreceptors reside within the outer nuclear layer (ONL) and comprise 70% of all retinal cells14-16.
Rod photoreceptors make synaptic connections at the outer plexiform layer (OPL) with horizontal neurons and bipolar cells that reside in the inner nuclear layer (INL) to transfer light-evoked signals further to the brain via ganglion cell axons that comprise the optic nerve. The first step of phototransduction is accomplished within a unique cellular structure called the outer segment (OS) which consists of a stack of membrane discs packed with the light-sensing visual pigment, Rhodopsin17-19.
Rhodopsin is a G-protein-coupled receptor consisting of an opsin apo-protein and its chromophore 11-cis-retinal that accounts for the absorption properties. Rhodopsin is also required for OS structural integrity20,21. Mutations in the human Rhodopsin gene (RHO) cause blinding diseases, such as retinitis pigmentosa (RP) and night blindness 22-25. The Rho gene is tightly regulated and highly transcribed, with its mRNA representing the most abundant transcript in rod photoreceptors 26-28. Irregular Rho expression, either at lower or higher than normal levels, can lead to photoreceptor degeneration in animal models 29-31. Mechanisms of the precise regulation on Rho expression have been extensively investigated. Initial studies identified the Rho proximal promoter region (PPR), a conserved ∼200bp region immediately upstream of Rho TSS, which was sufficient to drive the expression of the LacZ reporter in mouse photoreceptors32,33. Addition of a conserved genomic region, the Rho enhancer region (RER) lying 2kb upstream of PPR, greatly increased PPR-mediated LacZ expression 32,33.
Individual cis-regulatory elements (CREs) within PPR are detailed as sequence motifs bound by photoreceptor-specific TFs, including Cone-Rod Homeobox (CRX) 34,35 and Neuroretina Leucine Zipper (NRL)36-38. ChIP-seq targetome analysis has discovered that CRX and NRL cooperatively bind not only to PPR and RER, but also to a region 4 kb upstream of Rho TSS, which is designated as CRX-Bound Region 1 (CBR)39,40. Both CBR and RER could further activate a PPR-driven reporter by 8-10 fold in retinal explant reporter assays39. CRX/NRL synergistically activated PPR activity in transient HEK293 cell transfection assays, and loss of CRX or NRL in mice caused reduction in Rho expression and defects to rod photoreceptor development and maintenance36,41,42. In Crx-knockout mice, photoreceptor cells failed to gain appropriate active chromatin configuration at cis-regulatory regions of CRX-dependent genes, including PPR, RER and CBR of the Rho locus43, implicating the role of these regions in epigenomic remodeling of Rho expression.
In vivo chromosome conformation capture assays (3C) have illustrated CRX/NRL-dependent intrachromasomal loop interactions between RER and Rho PPR/gene body44. These findings collectively suggest that CBR and RER are Rho enhancers, which potentially act by mediating local epigenomic changes to increase PPR activity. However, it is unknown if such enhancer-promoter interactions are necessary for Rho expression in vivo. More specifically, it remains to be determined if these two previously identified Rho enhancers are essential for the epigenomic modulation and transcriptional activation of the Rho gene, and what degree of regulation each contributes.
In order to fill this knowledge gap, we aimed to dissect and determine in vivo functional roles of the Rho enhancers (CBR and RER) and promoter (PPR) using a loss-of-function approach in mice. By delivering CRISPR/Cas9 and specific guide RNAs (gRNAs) to C57BL/6J mouse embryos, we generated mouse lines carrying <500bp deletions at each genomic region individually and a line carrying a paired deletion of both CBR and RER. We performed gene expression analyses and cellular phenotype characterization for each mouse line at various ages. Our results indicate that, as expected, the Rho promoter PPR is absolutely required for Rho expression, rod photoreceptor integrity and survival. However, to our surprise, the two enhancers (CBR and RER) collectively play only minor roles in Rho expression, which is only applicable in developing and aging retinas. CBR/RER deficiency did not largely impact rod development and maintenance up to one year of age. Together, our results suggest that Rho proximal promoter is necessary and sufficient for Rho expression in rod photoreceptors, while Rho enhancers are largely dispensable. Our findings highlight the importance of investigating the function of cis-regulatory regions in the native genomic context.
RESULTS
Generation of Rhodopsin (Rho) promoter and enhancer knockout mice
In order to examine the necessity of the Rho promoter and enhancers in Rho transcription, 4 mouse lines carrying a knockout of each genomic region were generated using CRISPR-Cas9/gRNA mediated excision. Based on the locations of CRX-ChIP-seq peaks 39, a pair of gRNAs targeting each peak at Rho promoter and two enhancer regions were introduced together with Cas9 enzyme into C57BL/6J mouse embryos to generate a 200-400bp deletion at each region. A mouse line lacking both enhancers (CBR-/-RER-/-) on the same allele was made using a sequential targeting approach, i.e. RER deletion on the CBR-/- allele by introducing RER gRNAs and Cas9 enzymes to the CBR-/- embryos. PCR combined with sanger sequencing confirmed the absence of each region with the indicated length of deletion: 217 for PPR-/- (retaining the TATA box and transcription start site), 234bp for RER-/- and 491bp for CBR-/- (Figure 1A). Mouse lines homozygous for each deletion appeared healthy and reproductive.

(A) Drawing of the Rho cis-regulatory regions (as black boxes) identified by previous studies and the specific knockouts involved in this study. (B) Browser tracks (mm10) of average ATAC-seq reads from duplicate retinal samples of the indicated genotypes at P14. Names of Rho CREs and peak calls are shown at the top of tracks. Scale bar represents 5kb. (C) Bar plot of average ATAC-seq signals relative to the WT control. One-way ANOVA test is performed. n=2 for each genotype. CBR, CRX-bound region 1. RER, Rhodopsin enhancer region. PPR, Rhodopsin proximal promoter region. INT, Rhodopsin Intragenic region. 3’ UTR, 3’ untranslated region. Asterisks (*, **, ***, ****) denote p ≤ 0.05, p ≤ 0.01, p ≤ 0.001, p ≤ 0.0001, respectively.
Deletion of Rho promoter, but not enhancers, alters local chromatin accessibility
Since the chromatin of Rho cis-regulatory regions and gene body are activated during postnatal rod photoreceptor differentiation45-47, we tested if removal of Rho promoter or enhancers altered the chromatin accessibility of the Rho locus at postnatal day 14 (P14) using Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq). In wildtype (WT) retina, rod photoreceptor specification is completed by P14 and the Rho gene is fully expressed; this timing is consistent with epigenetic remodeling of the Rho locus measured by ATAC-seq and DNAse I hypersensitivity profiling47-49. Duplicate ATAC-seq experiments on P14 whole retinas of WT and each of the four knockout mouse lines, PPR-/-, CBR-/-, RER-/-, and CBR-/-RER-/- yielded highly reproducible results. Consistent with the published results, WT samples displayed five significant open chromatin regions within the roughly 12kb region spanning the Rho locus (Figure 1B, WT), including the peaks at CBR, RER, PPR, and two intragenic regions (INT and 3’UTR). The PPR-/- retina lost ATAC-seq signal not only at Rho promoter as expected, but also at INT and 3’UTR (Figure 1B & 1C PPR-/-). Furthermore, PPR removal also caused a significant reduction of ATAC-seq signal upstream of the promoter, though the reduction was more profound at the proximal RER than at the more distal CBR (Figure 1B & 1C). These results suggest that PPR plays an essential role in achieving appropriate open chromatin configuration within Rho gene body, and is necessary for normal DNA accessibility at upstream enhancers RER and CBR. This role of PPR is specific to the Rho locus, as no significant DNA accessibility changes were detected at other loci in PPR-/- retina (Supplemental Figure 1A, Supplemental Figure 2A). Next, we analyzed changes in ATAC-seq signal in retinas of enhancer knockout lines. Interestingly, the removal of RER yielded no significant effect on any other regions, and the removal of CBR only affected the accessibility at the 3’UTR (Figure 1B & 1C). Thus, neither CBR nor RER is required to establish the normal open chromatin configuration of the Rho locus during photoreceptor differentiation. Surprisingly, the removal of both RER and CBR did not produce an additive effect, but showed a similar impact as CBR single knockout (Figure 1B & 1C, CBR-/-RER-/-). Similar to the case of PPR-/-, global changes in chromatin accessibility were not observed in single- or double-enhancer knockout samples (Supplemental Figure 1B-D, Supplemental Figure 2A). Overall, these results suggest that Rho enhancers have limited functions in establishing Rho chromatin accessibility, but PPR plays an essential and enhancer-independent role.
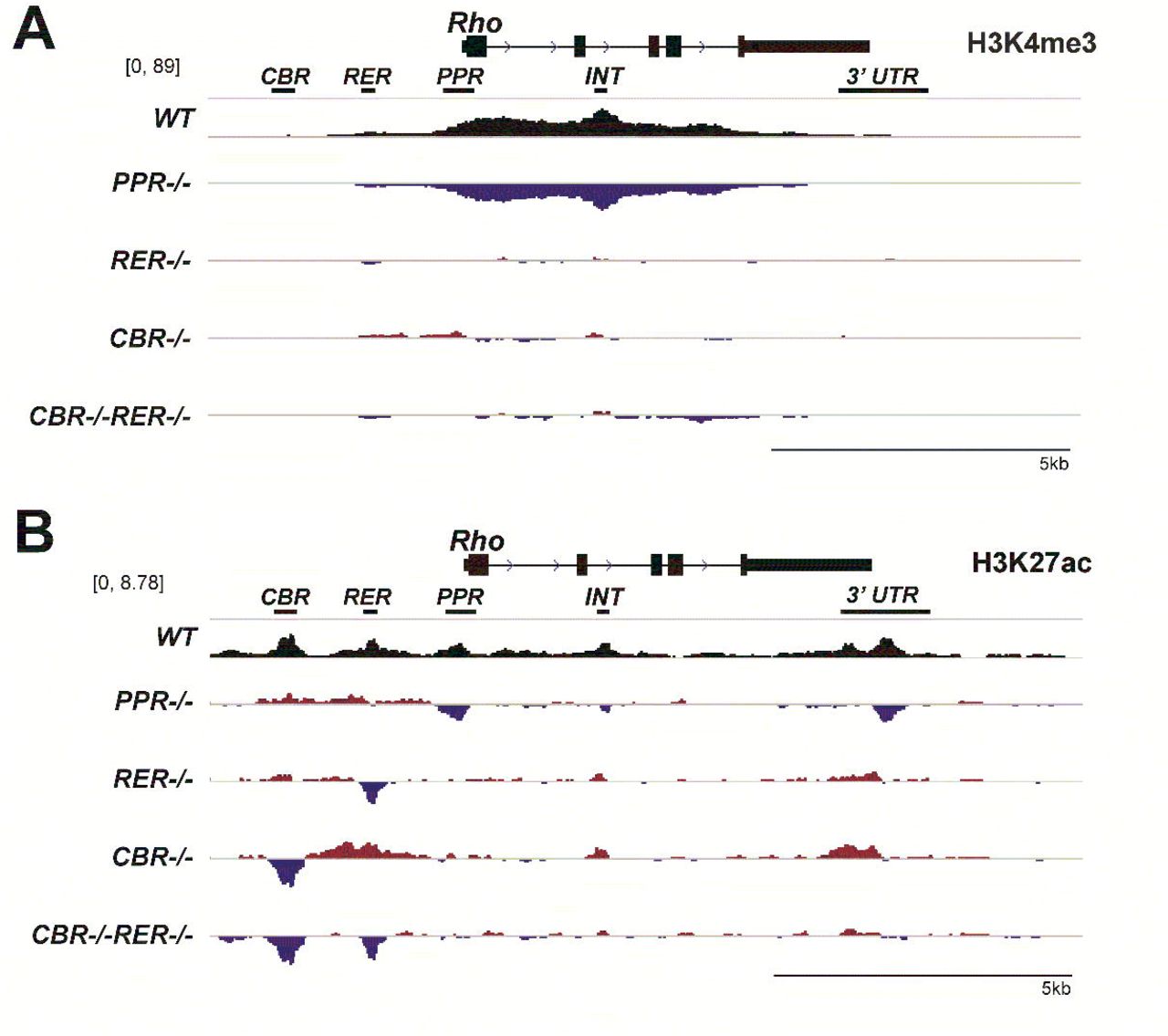
Histone modifications can also be used as a readout of the local chromatin and transcriptional activation state. Both H3K4me3 and H3K27ac are associated with transcriptionally active chromatin. H3K4me3 usually marks active gene promoters, while H3K27ac is enriched at active enhancers and promoters 5,9,50,51. Both marks are detected at the actively transcribed Rho locus in mature mouse retina, which is defined as an active epigenetic state established in a CRX-dependent manner during postnatal development 47,49. In order to assess changes in histone marks upon Rho promoter or enhancer knockout, Cleavage Under Targets and Tagmentation (CUT&Tag) analysis was performed to report the high-resolution chromatin profiling on P14 retinal samples. Two validated antibodies specific to these histone marks were used in CUT&Tag experiments. In WT retina, H3K4me3 peaks showed a broad domain encompassing the gene body, starting at the PPR and extending to the final exon of Rho gene (Figure 2A, WT), but the signal was absent at CBR and RER regions. Analysis of mutants showed strikingly different landscapes: PPR-/- retina lost H3K4me3 deposition at the Rho locus while all enhancer-knockout samples showed comparable H3K4me3 marks to the WT control (Figure 2A, PPR-/-). Inspection of the 50kb region surrounding Rho confirmed that no other H3K4me3 sites were affected in the mutants (Supplemental Figure 2B). The results of H3K4me3 CUT&Tag analysis suggest that Rho enhancers neither contribute to H3K4me3 deposition nor affect PPR activity, while PPR is required for recruiting H3K4me3 to the Rho locus.

Browser tracks display comparative analysis of H3K4me3 (A) and H3K27ac (B) peaks from retinal samples of the indicated genotypes at P14. Top track of each panel shows peaks in the WT control. The difference in H3K4me3 peaks ranges between -89 and 89. The difference in H3K27ac peaks ranges between -6.7 and 6.7. The following four tracks of each panel display increased (red) and decreased (blue) signals relative to the WT control in mutant retinas. Scale bar represents 5kb.
H3K27ac CUT&Tag analysis displayed 5 distinct peaks at Rho locus in WT retina at P14, including CBR, RER, PPR, INT, and 3’UTR, respectively (Figure 2B, WT). In PPR-/- retina, H3K27ac signal was missing or greatly reduced at PPR, INT and 3’UTR, but retained WT levels at CBR and RER, implying CBR and RER are capable of loading H3K27ac marks in the absence of PPR (Figure 2B, PPR-/-). In RER-/- retina, H3K27ac signal was similar to the WT control, besides a drop at the deleted RER region (Figure 2B, RER-/-). However, in CBR-/- retina, a compensatory increase in H3K27ac signal was observed at RER, INT and 3’UTR, as well as at a few regions outside the Rho locus (Figure 2B, Supplemental Figure 2C, CBR-/-), possibly suggesting coordination or compensation between enhancers. In CBR-/-RER-/- retina, no change in H3K27ac signal was found at PPR, suggesting that the epigenetic activation state of the Rho promoter is unaltered by CBR and RER knockouts (Figure 2B, CBR-/-RER-/-). Collectively, H3K27ac CUT&Tag results suggest that Rho promoter and enhancers can independently modify their histone tails to establish an active epigenetic environment and there is little cross-talk between these three regulatory regions in establishing the local epigenetic state. Altogether, these CUT&Tag data indicate that PPR is absolutely required for establishing the chromatin configuration and active histone marks at the accessible regions of the Rho locus, especially at the promoter and intragenic regions, but Rho enhancers are largely dispensable for this remodeling.
Rho enhancers make small and age-dependent contributions to Rho transcription levels
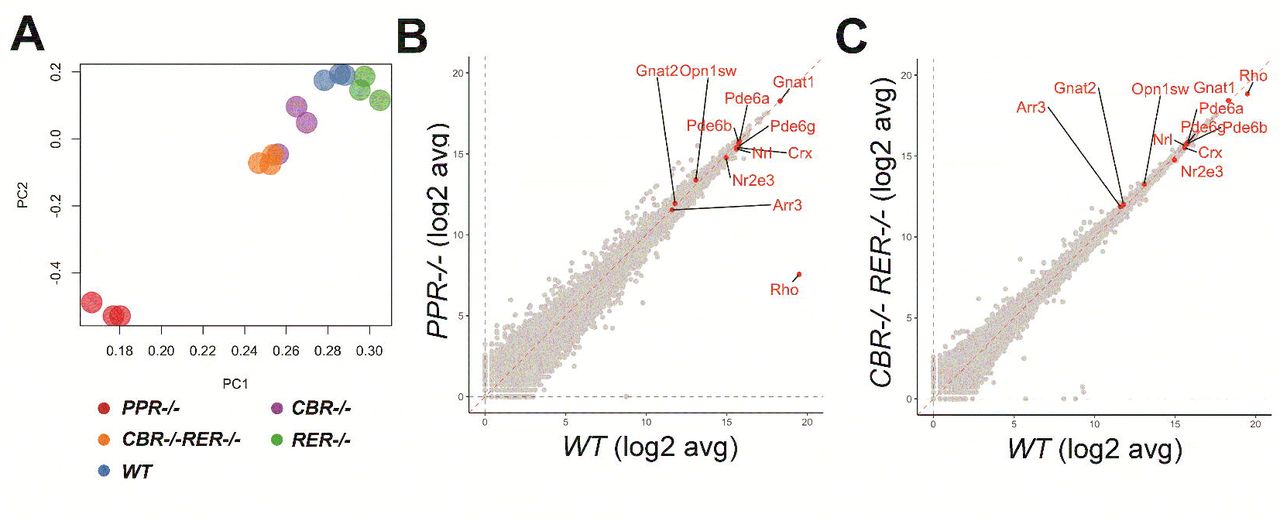
In order to analyze the effects of promoter/enhancer deletions on Rho expression, we performed next-generation RNA sequencing (RNA-seq) on P14 retinal samples of mutant and WT mice. Principal component analysis (PCA) showed that PPR samples were transcriptionally distinct from all other samples, evidenced by their distinct clustering (Figure 3A). CBR-/-RER-/- and CBR-/- samples were also separated from the WT controls (Figure 3A). Gene expression analysis showed that only Rho was differentially expressed in PPR-/- retina, and all other photoreceptor-enriched genes such as Crx, Nrl, Nr2e3, Gnat1, Pde6a, were not affected (Figure 3B). Analysis of other mutants showed similar profiles to the WT control, and Rho showed some degree of differential expression in CBR-/-RER-/- retina (Figure 3C). These results suggest that PPR plays a more dominant role in Rho transcription than either enhancer, and the combinatorial knockout of both enhancers causes only a minor decrease of Rho transcription.

(A) PCA plot shows clusters of WT and the indicated mutant samples. (B & C) Scatterplot displays expression distributions of selected genes in the indicated mutant relative to the WT level. n=3 for each genotype.
We further investigated the degree by which promoter and enhancer knockouts impacted Rho expression using qRT-PCR with retinal samples of specific allelic arrangements at various ages. P14 retinal samples were collected for qRT-PCR experiments from genotypes illustrated in Figure 4A. At P14, Rho expression was completely ablated in PPR-/- retina and roughly reduced by half in PPR+/- retina (Figure 4B, PPR-/-, PPR+/-), suggesting that no compensatory regulation or feedback from the intact allele when one PPR allele is lost. In contrast, RER-/- retina had comparable Rho expression to the WT control, while CBR-/- retina reduced Rho expression to roughly 70-80% of the WT level (Figure 4B, RER-/-, CBR-/-), suggesting that CBR, not RER, makes a small contribution to Rho expression at P14. CBR-/-RER-/- retina showed reduced Rho expression to approximately 70-80% of the WT level, similar to, or slightly lower than that of CBR-/- retina (Figure 4B, CBR-/-RER-/-). These results suggest that the regulatory capacity of Rho enhancers, specifically CBR, contributes 20-30% of Rho expression. When a copy of RER was present in Rho enhancer regions (CBR-/-RER+/-), Rho expression generally matched with those in CBR-/- and CBR-/-RER-/- retinas, collectively suggesting that RER is not required for Rho expression (Figure 4B, CBR-/-RER+/-). When a copy of CBR was present in Rho enhancer regions (CBR+/-RER-/-), Rho expression reached to an indistinguishable level as the WT control (Figure 4B, CBR+/-RER-/-), suggesting that at least one copy of CBR is needed to achieve maximum-level Rho expression in the presence of two PPR copies. Lastly, Rho expression in a retina heterozygous for PPR and Rho enhancers was comparable to that of PPR+/- retina, i.e. about 50% to the WT control (Figure 4B, CBR+/-RER+/-, PPR+/-). These results together indicate that PPR is absolutely required for Rho expression and the regulatory functions of Rho enhancers are detectable and secondary to PPR in retinal development.

(A) Diagrams of WT and mutant samples measured in this study. (B) qPCR analysis of Rho mRNA expression in P14 WT (n=8) and mutant retinas (n=10 for CBR-/-; RER-/-; CBR-/-RER-/-; n=8 for CBR-/-RER+/-; CBR+/-RER-/-; n=6 for PPR-/-; PRR+/-; CBR+/-RER+/-, PPR+/-). (C) Relative Rho expression profiles of CBR-/- (□), RER-/- (Δ), and CBR-/-RER-/- (●) samples up to 1YO (n ≥ 6 for each age). Interpolation is performed with MATLAB and Wolfram Mathematica: dashed line represents the CBR-/- profile, dash-dotted line represents the RER-/- profile, solid black line represents the CBR-/-RER-/- profile. All results are plotted as relative expression to WT control. Asterisks (*, **, ***, ****) denote p ≤ 0.05, p ≤ 0.01, p ≤ 0.001, p ≤ 0.0001, respectively by one-way ANOVA with Tukey’s multiple comparisons. ns means not significant.
Furthermore, we tested if Rho enhancers functioned to maintain Rho expression in adult retina. qRT-PCR experiments were performed with enhancer-knockout retinas up to 1 year-old (1YO). Rho expression in RER-/- retina always remained indistinguishable from that in the WT control (Figure 4C, RER-/-). Rho expression in CBR-/- retina initially reached about 70-80% of the WT level at P14 but increased steadily to a comparable level after the completion of retinal development and throughout adulthood (Figure 4C, CBR-/-). Interestingly, although Rho expression in CBR-/-RER-/- retina was as low as 70-80% to the WT control at P14, the relative Rho expression profile became comparable from P21 to 4 month-old (MO). However, at 6MO, the relative Rho expression dropped back to 70-80% of WT expression and persisted to 1YO (Figure 4C, CBR-/-RER-/-). Overall, these results suggest that Rho enhancers possess age-dependent regulatory activity to control Rho expression during rod differentiation and aging, but this regulation is only responsible for 20-30% of the overall expression. Besides changes in Rho expression in the PPR-/-, PPR+/- and CBR-/-RER-/- mutants, other rod photoreceptor-related genes such as Crx and Gnat1 displayed comparable expression to the WT control at P14 (Supplemental Figure 4A) and 6MO (Supplemental Figure 4C). The immunohistochemistry (IHC) staining with anti-Rho and Gnat1 antibodies showed that both proteins were made in CBR-/-RER-/- and PPR+/- retinas and appeared at normal subcellular locations (Supplemental Figure 4B & 4D).
Redundant roles of Rho enhancers contribute to the maintenance of rod morphology and function in old adults
We next investigated if the decrease in Rho expression induced any phenotypic changes in Rho enhancer and promoter mutants. PPR-/- retina failed to elaborate OS during development and showed only one remaining layer of cells within ONL at 6MO by H&E staining of retinal cross-sections (Figure 5A, PPR-/-), while CBR-/-RER-/- and PPR+/- retinas maintained well-laminated structures (Figure 5A, CBR-/-RER-/-, PPR+/-). No mislocalized cells were observed in the mutants. OS length of CBR-/-RER-/- and PPR+/- retinas appeared shorter than that of the WT control (Figure 4B, OS Length). ONL thickness in the PPR+/- retina was reduced as compared to WT control (Figure 5B, ONL Thickness), while CBR-/-RER-/- retina had a comparable ONL thickness. Since rod photoreceptors represent the most abundant cell type within ONL, these morphological measurements suggest that CBR-/-RER-/- retina very likely has indistinguishable cell numbers of rod photoreceptors to the WT control without apparent cell death (data not shown). Therefore, the slightly shorter OS of CBR-/-RER-/- retina is associated with reduced Rho expression. On the other hand, the decreased ONL thickness of PPR+/- retina indicates loss of rod photoreceptors. Furthermore, visual function of mutant retinas was assayed by dark-adapted (rod response) electroretinography (ERG). As expected, PPR-/- mice had no ERG responses, while PPR+/- mice had decreased dark-adapted A-wave and B-wave amplitudes at high-light intensities as compared to the WT control (Figure 5C). CBR-/-RER-/- mice also showed lower A-wave and B-wave amplitudes at the high light intensities, but less severe than PPR+/- (Figure 5C). This is consistent with the reduction of Rho expression and OS length in the CBR-/-RER-/- retina that was less severe than observed in the PPR+/- retina. In addition, 6MO CBR-/- and RER-/- single mutants were indistinguishable from the WT control in retinal morphology (Supplemental Figure 5A), OS length (Supplemental Figure 5B) and ERG responses (Supplemental Figure 5C & 5D), echoing their comparable Rho expression as the WT control. Thus, CBR and RER act redundantly to constitute the Rho enhancer landscape for transcriptional regulation in adult retina, and their combined loss leads to measurable functional deficits.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Hematoxylin and Eosin (H&E) cross-section staining of 6MO WT and mutant retinas of the indicated genotypes. OS: outer segment; ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer. Scale bar represents 100 μm for all image panels. (B) OS thickness (left panel, in μm) and ONL thickness (right panel, in μm) in 6MO WT and mutant retinas of the indicated genotypes at various positions from the optic nerve head (ONH). Error bars represent mean (SD) (n ≥ 5). Dashed black line represents the comparison between WT and CBR-/-RER-/- samples. Solid black line represents the comparison between WT and PPR+/- samples. (C) Electroretinogram (ERG) analysis of 6MO WT and mutant mice of the indicated genotypes, showing amplitude changes of dark-adapted A-waves (left panel) and B-waves (right panel). Mean amplitudes (μV) are plotted against stimulus light intensity. Error bars represent SEM (n ≥ 7). All statistics is done by comparing to WT control. Asterisks (*, **, ***, ****) denote p ≤ 0.05, p ≤ 0.01, p ≤ 0.001, p ≤ 0.0001, respectively by two-way ANOVA with Tukey’s multiple comparisons. ns means not significant.
Rod morphological and functional changes in mutant retinas were also assessed at early adult ages. At 6 weeks (6WK) and 3MO, OS length of the PPR+/- retina appeared shorter (Supplemental Figure 6A & 6B). This morphological defect in the PPR+/- retina echoed the lower amplitudes of dark-adapted ERG A-waves at the highest light intensity as compared to that of the WT control (Supplemental Figure 6E & 6F). OS length and the amplitudes of dark-adapted ERG A-wave of the CBR-/-RER-/- retina were comparable to WT controls (Supplemental Figure 6A, 6B, 6E & 6F). Importantly, ONL thickness of all mutants remained comparable to that of the WT control at 6WK and 3MO (Supplemental Figure 6C & 6D). Compared to the ONL reduction in 6MO PPR+/- retina, these results suggest that PPR+/- retinal degeneration occurs after 3MO. As a reference, OS and ONL measurements in heterozygous and homozygous Rho-knockout mice31 (Rho-/- and Rho+/-) showed similar results as PPR mutants: ONL thickness of PPR-/- retina degenerated dramatically after 6WK (Supplemental Figure 7A, PPR-/-). PPR+/- retina began to show ONL degeneration at 6MO but reduced OS length at 3MO, similar to that of Rho+/- retina (Supplemental Figure 7A & 7B, PPR+/- vs. Rho+/-).
DISCUSSION
Using CRISPR-Cas9/gRNA mediated excision, this study examined the specificity and strength of two Rho enhancers, namely CBR and RER, and of Rho promoter in regulating Rho expression. Previous reporter-based assays showed that both the RER and CBR were capable of enhancing Rho promoter activity by 8-10 fold in postnatal mouse retina33,39. However, this study shows that knockout of each or both enhancers surprisingly produce null or only moderate impact (20-30% loss) on endogenous Rho expression. Notably, RER, that was previously identified by a study with transgenic mice33 and known to interact with PPR by 3C assay44, fails to display any necessity for endogenous Rho expression in developmental and adult ages. Consistent with their minor roles, single Rho enhancer knockout also shows limited impact on rod photoreceptor identify, health and function. These results are in contrast to the essential function of the Locus Control Region (LCR) in its regulation of red/green (R/G) cone opsin expression52-54. The conserved LCR is absolutely required for mutually exclusive expression of red vs green opsin in the respective cone subtype in humans, mouse and zebrafish models. RER and R/G LCR share a conserved core region of similar TF binding motifs, including CRX binding sites33. Several models might explain the minimal relevance of Rho enhancers in situ, but high activity in reporter assays: firstly, the strength of a promoter might determine its cooperation with enhancers. Rho promoter (PPR) is highly robust and sufficient to drive cell type-specific expression of target genes both in episomal vectors39 and at ectopic genomic locations33. Future studies are needed to determine the relative strength of Rho promoter vs other photoreceptor gene promoters and how the promoter strength influences functional interactions with specific enhancers. Secondly, dynamic expression and precision might also be a determinant. Genes coding for cell fate-determining transcription factors with complex spatial and temporal expression patterns often harbor strong distal enhancers or enhancer clusters, such as those found in Pax655, Otx256-58, Vsx259, Atoh760 and Blimp161. Rho expression, despite being one of the highest expressed gene in the retina, follows a simple induction curve during rod photoreceptor development62, which may not require a prominent enhancer but a strong self-functioning promoter. Finally, results of reporter assays may have exaggerated Rho enhancer activities, as reporter gene constructs do not contain the correct genomic context, including native gene structure and epigenomic landscape. Overall, the findings of this study highlight the importance to validate functions of reporter-identified enhancers in a native genomic context.
This study shows age-dependent activities of Rho enhancers. During postnatal development, CBR clearly plays a dominant role, as it is responsible for 20-30% Rho expression, while RER’s activity was dispensable. Thus, CBR, but not RER, is required for appropriate Rho transcription during development. However, this minor contribution of CBR does not affect the development of rod subcellular structure and function. In adult retinas, deletion of Rho enhancers results in no detectable affects in young and mid ages, but after 6 MO, RER/CBR knockout causes a loss of 20-30% of Rho transcription. These suggest that CBR and RER play a redundant role in maintaining high-level Rho transcription in aged retinas. Similar “shadow” or redundant enhancers have been reported elsewhere, such as individual enhancers in β-globin LCR, Pax6 lens enhancers and Atoh7 distal enhancer 55,60,63,64. These enhancers mostly act during tissue/organ genesis and are required for regulating the expression timing of specific genes. Our discovery of redundant enhancers in aged retina provides a new insight into enhancers’ roles in aging and tissue-specific maintenance.
Consistently, CBR/RER knockout mutants affected Rho expression as well as morphological and functional integrity at 6MO. Since PPR+/- (50% Rho reduction) produced a more severe phenotype than CBR/RER knockout mutants (20-30% Rho reduction) in aged adults, our results confirm that rod homeostasis requires precise control of Rho expression. Aged rod photoreceptors with disrupted Rho enhancer activity may be more susceptible to genetic and/or environmental insults. Future studies are required to determine if Rho enhancer deficiency complicates (or modifies) defective phenotypes under disease/stress conditions, particularly in progressive rod photoreceptor degeneration.
Typical “super enhancer” characteristics were detected in wildtype retina within 5 kb of the Rho upstream regulatory sequence. Changes of these characteristics in mutants, in general, agreed with the impact of each region on Rho transcription. In particular, loss of ATAC signals at Rho gene body was only seen in PPR-/-, but not in enhancer knockout mutants, highlighting the interdependency and necessity of PPR in epigenomic modulation during Rho transcription activation. Deleting CBR and/or RER enhancers had limited impact on the epigenomic landscape of the Rho locus, despite the 20-30% contribution to transcription, suggesting that the two enhancers do not act in a “trans” manner on DNA accessibility or chromatin configuration. Most likely, they promote RNA polymerase II activity at the promoter and gene body via CRX/NRL-dependent looping interactions44. A similar interactive pattern of predominant promoter versus subsidiary enhancers has been previously reported65. Although CBR and RER deletions are insufficient to elicit large changes in Rho expression and DNA accessibility of Rho PPR, this study cannot exclude the existence or importance of more distal Rho-specific CREs in regulating chromatin dynamics at the Rho PPR region.
The mouse Rho gene shares about 95% sequence homology with the human gene33 (Mouse Genome Informatics). Importantly, Rho enhancers at the Rho locus also share remarkably conserved topography between the two species in terms of CRE number and relative locations33,39. The profile of transcription factors bound to Rho enhancers and promoter is also conserved39,66. While numerous non-synonymous and nonsense variants within the Rho coding region have been found to cause human retinal disease, no regulatory variants have yet been discovered. This study suggests that future efforts should be primarily focused on the proximal promoter region, particularly at critical TF binding motifs. Distal regulatory variants within the RER and CBR are unlikely to cause pathogenic transcriptional deficits for severe developmental defects. These variants however could be genetic modifiers of other pathogenic mechanisms or independently yield minor but detectable effects on visual function and alter long-term photoreceptor health.
In conclusion, this study has described unexpectedly minor roles of two previously identified Rho enhancers on in vivo Rho transcription, and their redundant activity in rod functional maintenance in aging retinas. These findings contribute to our understanding of enhancer functions and mechanism of action in vivo, and highlight the importance of dissecting individual enhancer functions and interactions in the native genomic context at various ages.
MATERIALS AND METHODS
Animals
All mice in this study were on the C57BL/6J background. Both male and female mice were used in experiments. All animal procedures were conducted according to the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, and were approved by the Washington University in St. Louis Institutional Animal Care and Use Committee.
Generation of mutant mice
CRISPR/Cas9 gene editing was performed using two single-guide RNA (sgRNAs) flanking each element of CBR, RER, PPR in order to delete these elements (see Figure 1). The gRNA sequences are: For CBR, 5’:TAGCTCCGTTTCCACATTGA and 3’:TCAAGATACACTGTCCCCAC. For RER, 5’: GCTTCATCGTGGTCTCCGCG and 3’: TCCATGCAGGTGTCTTGTTT. For PPR, 5’: GAAGTGAATTTAGGGCCCAA and 3’: GCGGATGCTGAATCAGCCTC. Fertilized mouse oocytes injected with sgRNAs were allowed to proceed to birth to generate mouse lines. In order to generate the mutant carrying deletions of both CBR and RER on the same chromosome, a mouse line homozygous for CBR-/- was used to prepare fertilized oocytes that were subsequently injected with sgRNAs for RER deletion. F0 mutants were genotyped by DNA sequencing of PCR products overlapping the mutations.
Histology and immunohistochemistry
Eyes were enucleated at tested ages and fixed in 4% paraformaldehyde at 4°C overnight for paraffin embedded sections. Each retinal cross-section was cut 5 microns thick on a microtome. Hematoxylin and Eosin (H&E) staining was used to examine retinal morphology.
For IHC staining, sections firstly went through antigen retrieval with citrate buffer, and blocked with a blocking buffer of 5% donkey serum, 1% BSA, 0.1% Triton-x-100 in 1X PBS (pH-7.4) for 2 hours. Sections were then incubated with primary antibodies at 4°C overnight, then washed, followed by 2-hour incubation of specific secondary antibodies. Primary antibody to Rho (MilliporeSigma, O4886) or Gnat1 (MilliporeSigma, GT40562) and secondary antibodies were applied with optimal dilution ratios.
All slides were mounted with hard-set mounting medium with DAPI (Vectashield, Vector Laboratories, Inc., CA).
Electroretinogram
ERGs were performed on adult mice at different ages using UTAS-E3000 Visual Electrodiagnostic System (LKC Technologies Inc., MD). Mice were dark-adapted overnight prior to the tests. The experimental procedures were previously established in our lab. ERG responses of biological replicates were recorded, averaged and analyzed using Graphpad Prism 8 (GraphPad Software, CA). The mean peak amplitudes of dark-adapted A and B waves and light-adapted B waves were plotted against log values of light intensities (cd*s/m2). The statistical analysis was done by two-way ANOVA with multiple pairwise comparisons (Tukey’s).
Quantitative PCR
Each RNA sample was extracted from 2 retinae of a mouse using the NucleoSpin RNA Plus kit (Macherey-Nagel, PA). RNA concentrations were measured using a NanoDrop One spectrophotometer (ThermoFisher Scientific). 1 μg of RNA was used to synthesize cDNA with First Strand cDNA Synthesis kit (Roche, IN). The reaction master mix contained EvaGreen polymerase (Bio-Rad Laboratories, CA), 1 μM primer mix, and diluted cDNA samples. Samples were run using a two-step 40-cycle protocol on a Bio-Rad CFX96 Thermal Cycler (Bio-Rad Laboratories, CA). At least 3 technical triplicates were run for each gene. Primers (5’ to 3’) used in this study were Rho (F: GCTTCCCTACGCCAGTGTG, R: CAGTGGATTCTTGCCGCAG), Crx (F: GTCCCATACTCAAGTGCCC, R: TGCTGTTTCTGCTGCTGTCG), Gnat1 (F: ACGATGGACCTAACACTTACGAGG, R: TGGAAAGGACGGTATTTGAGG). Data were analysed with QBase software (Biogazelle, Belgium). The statistical analysis was done by Student’s t-test with p < 0.05, CI:95%.
RNA-seq data generation and analysis
RNA was extracted from retinal samples using the NucleoSpin® RNA kit (Macherey-Nagel) using the manufacturer’s protocol. Total RNA integrity was determined using Agilent Bioanalyzer or 4200 Tapestation. Library preparation and sequencing experiments were performed at Genome Technology Access Center, Washington University in St. Louis. A brief description of the experimental procedures was outline as follows. Library preparation was performed with 5 to 10ug of total RNA with a RIN score greater than 8.0. Ribosomal RNA was removed by poly-A selection using Oligo-dT beads (mRNA Direct kit, Life Technologies). mRNA was then fragmented in reverse transcriptase buffer and heating to 94 degrees for 8 minutes. mRNA was reverse transcribed to yield cDNA using SuperScript III RT enzyme (Life Technologies, per manufacturer’s instructions) and random hexamers. A second strand reaction was performed to yield ds-cDNA. cDNA was blunt ended, had an A base added to the 3’ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12-15 cycles using primers incorporating unique dual index tags. Fragments were sequenced on an Illumina NovaSeq-6000 using paired end reads extending 150 bases. Reads were trimmed using Trim-Galore! (v0.6.7) a wrapper for Cutadapt (v3.4) and FastQC (v0.11.9). Trimmed reads were mapped to mm10 using STAR (v2.7.0)67. Mapped reads were cleaned using Samtools (v1.9.4), gene counts generated by HTseq (v0.11.2)68, and visualizations generated in R (v3.6.1).
ATAC-seq data generation
ATAC-seq libraries were generated as published in Ruzycki et al49, a slightly modified protocol to that published by Buenrostro et al69. Briefly, retinas were dissected from P14 mice of each genotype and washed in PBS. Retinal cells were dissociated using 2% collagenase in TESCA buffer for 13 minutes at 37C. DNase I (0.5 Units; Roche, Basel, Switzerland) was added for the final 3 min to minimize clumping. DMEM+10% FBS was added to stop the reaction. Nuclei were stained with SYBR Gold (Thermo Fisher Scientific) diluted 1:100 in trypan blue and counted using a fluorescent microscope. 50,000 cells were resuspended in TD buffer for a 1hr incubation at 37C with TDE1 (Nexetra DNA Library Prep Kit; Illumina, San Diego, CA). Remaining steps of library prep were performed as published by Buenrostro et al. Final libraries were pooled and sequenced using the Illumina 2500.
CUT&Tag data generation
Retinal cells were dissociated and counted in same manner as described above for ATAC-seq. After quantification, cells were diluted, aliquoted and cryopreserved by adding DMSO to a final volumne of 10% and slowly freezing to –80C in a Mr. Frosty container. CUT&Tag reactions were performed using Epicypher reagents (protocol v1.5; Epicypher, USA). Briefly, cells were thawed on ice and 100,000 bound to ConA beads for each sample. Primary antibody incubation was left at 4C overnight on a nutator. 0.5ug of antibody was used for each reaction; H3K4me3 (MABE647; EMD Millipore Corporation, USA) and H3K27Ac (ab177178; Abcam Inc, USA). 0.5ug secondary antibody (CUTANA anti-rabbit secondary antibody; 13-0047; Epicypher) was incubated for 0.5hr at RT, and Tn5 binding and trnasposition done exactly as specified in CUTANA protocol. After transposition, custom 10bp unique-dual-indexed primers were used to amplify transposed fragments (P7-CAAGCAGAAGACGGCATACGAGAT-10bpBC-GTCTCGTGGGCTCGGAGATGTG, P5-AATGATACGGCGACCACCGAGATCTACAC-10bpBC-TCGTCGGCAGCGTCAGATGTGTAT) for 20 cycles. Libraries were cleaned and size-selected using Ampure XP beads. Libraries were pooled and sequenced using the Illumina NovaSeq 6000.
ATAC-seq and CUT&Tag data analysis
Reads were trimmed using Trim-Galore! (v0.6.7) a wrapper for Cutadapt (v3.4) and FastQC (v0.11.9). Trimmed reads were mapped to the mm10 genome build using bowtie2 (v2.4.1)70,71. Mapped reads were cleaned using Samtools (v1.9.4), Picard (v2.25.7), and Bedtools (v2.27.1). For visualization, reads were converted to bigwig format and compared using Deeptools (v3.5.1). ATAC-seq peak calling was accomplished using MACS2 (v2.2.7.1).
Data access
All raw and processed data has been uploaded to the NCBI SRA database.
ETHICS STATEMENT
All animal procedures were conducted according to the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, and were approved by the Washington University in St. Louis Institutional Animal Care and Use Committee.
AUTHOR CONTRIBUTIONS
SC conceived of the study, CS, PR and SC designed the experiments, CS and PR performed experiments and analyzed the results. CS, PR and SC wrote the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACKNOWLEDGMENTS
We thank Courtney Linne, Mingyan Yang, Guangyi Ling, Belinda Dana and Dr. Xiaodong Zhang for technical assistance; We also thank Susan Penrose and Mike Casey with the Molecular Genetics Service Core for generating Rho promoter/enhancer knockout mice. This work was supported by NIH grants EY012543 (to SC) and EY002687 (to WU-DOVS), and unrestricted funds from Research to Prevent Blindness (to WU-DOVS).
REFERENCES




