

Abstract
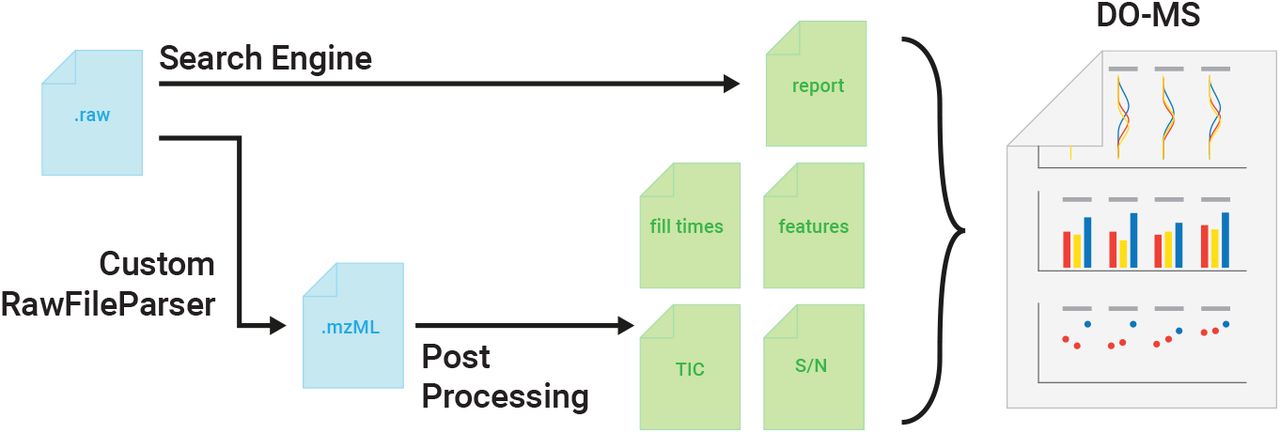
Mass-spectrometry (MS) enables specific and accurate quantification of proteins with ever increasing throughput and sensitivity. Maximizing this potential of MS requires optimizing data acquisition parameters and performing efficient quality control for large datasets. To facilitate these objectives, we extended the DO-MS app (do-ms.slavovlab.net) to optimize and evaluate results from data independent acquisition (DIA) MS. The extension works with both label free and multiplexed DIA (plexDIA) and supports optimizations particularly relevant for single-cell proteomics. We demonstrate multiple use cases, including optimization of duty cycle methods, peptide separation, number of survey scans per duty cycle, and quality control of single-cell plexDIA data. DO-MS allows for interactive data display and generation of extensive reports, including publication quality figures, that can be easily shared. The source code is available at: github.com/SlavovLab/DO-MS.
Competing Interest Statement
The authors have declared no competing interest.
Footnotes
∈ Data, code & protocols: do-ms.slavovlab.net





{kind=link}