

Abstract
Bacteriophages can adapt to new hosts by altering sequence motifs through recombination or convergent evolution. Where such motifs exist and what fitness advantage they confer remains largely unknown. We report a new method, Bacteriophage Library Informed Sequence Scoring (BLISS), to discover sequence motifs in metagenomic datasets governing phage activity. BLISS uses experimental deep mutational scanning data to create sequence profiles to enable deep mining of metagenomes for functional motifs which are otherwise invisible to searches. We experimentally tested 10,073 BLISS-derived sequence motifs for the receptor-binding protein of the T7 phage. The screen revealed hundreds of T7 variants with novel host specificity with functional motifs sourced from distant families besides other major phyla. Position, substitution and location preferences on T7 dictated different specificities. To demonstrate therapeutic utility, we engineered highly active T7 variants against urinary tract pathogens. BLISS is a powerful tool to unlock the functional potential encoded in phage metagenomes.
Introduction
Bacteriophages (or ‘phages’) constitute unparalleled biological variation in nature. The sequence diversity of phages is coming to light in the growing volume of viral metagenomes curated from the gut [1–4], oceans [5–8], lakes and soil microbiomes [9–12]. However, the functions encoded by many sequences remain largely unknown. For instance, it is estimated fecal viromes contain 108-109 virus-like particles per gram of feces, but only a fraction of phages can be identified using existing databases [13]. Over the decades, phage biologists have painstakingly characterized natural phage variants one at a time. This effort is very time and labor-intensive because traditional phage assays do not scale for evaluating many phages, leaving large swathes of phage sequence space unexplored. Developing high throughput approaches for functionally characterizing phage metagenomic sequences to understand sequence-function relationships is critical to close this daunting knowledge gap.
The general lack of sequence conservation among phages makes identifying functionally related metagenomic sequences difficult [6, 14]. Even amongst closely-related phages, genes that are under constant evolutionary pressure can have little sequence homology [15]. Routine sequence searches starting from a single query often fail to unearth homologs due to low sequence conservation. One such phage gene is the receptor-binding protein (RBP), a primary determinant of host range that mediates the interaction between phage and host receptors. The mutational adaptability of the RBP is intimately tied to the survival of the phage in the context of new hosts and environments. Since the RBP is under selective pressure to diversify, portions of this gene can be exchanged between phages, allowing phages to reuse optimal sequences as an evolutionary shortcut [15–17]. The minimal unit of such recombinogenic blocks is thought to cover the spectrum from full-length genes to partial domains to short sequence windows [15, 18]. For highly divergent genes, we reasoned that sequence similarity likely occurs over short sequence windows i.e., motifs, instead of full-length genes or domains. An evolutionary perspective strengthens this hypothesis. Convergent evolution may drive phages towards the same solution for binding to a particular receptor using a small motif in otherwise non-homologous sequences. However, the extent to which such motifs exist in metagenomic databases and if they can be harnessed to confer a fitness advantage to phages remains largely unknown.
In this study, we report a new method called BLISS (Bacteriophage Library Informed Sequence Scoring) to discover sequence motifs in metagenomic datasets. In contrast to other exclusively bioinformatics-driven motif discovery tools, BLISS uses experimental deep mutational scanning (DMS) data to quantitatively score the importance of every possible substitution within a defined sequence window, preserving functionally important mutations and curating every theoretical motif that can influence phage activity. This weighted substitution profile is used as a query to find all relevant sequence matches from metagenomic databases. Using BLISS, we identified metagenomic sequence motifs for the RBP of the T7 phage. Because they are crucial determinants of phage activity, RBPs have proven to be excellent targets for engineering phage activity [18–27]. However, the sequence space explored for the T7 phage RBP has been greatly limited by the small set of under 100 recognizable sequence homologs. BLISS identified 104-105 relevant motifs from diverse, non-homologous metagenomic phage sequences for the tip domain of the T7 RBP. Pooled screening of T7 phages engineered to contain BLISS motifs revealed hundreds of phages with novel host specificity. Surprisingly variants with motifs from the T7 family, Podoviridae, had the least activity, while variants with motifs sourced from Autographiviridae, Demerecviridae and Drexlerviridae were highly active. These motifs were well distributed in metagenomic space, indicating that deep mining of metagenomic datasets is a powerful tool for tuning host range. Position and substitution preference across different hosts showed key substitutions that drive activity and host range and revealed epistatic combinations of substitutions that are individually deleterious but show dramatic increases in activity when combined. To demonstrate the therapeutic utility of this method, we identified several BLISS variants that eliminate pathogenic Escherichia coli causing urinary tract infections (UTIs) that are insensitive to wildtype T7.
Results
BLISS identifies motifs from diverse phages
Three considerations guided the development of our method. First, relevant sequence motifs may be hidden in metagenomic proteins with little sequence homology to the original phage protein. A homology search using BLAST of the T7 RBP tip domain identified 83 unique sequences, an insignificant fraction of the potential matches in metagenomic sequences. Second, metagenomic motifs influencing phage activity may bear little resemblance to the wildtype motif. We thus needed to create a profile of query motifs instead of using just one sequence. Third, since motif activity depends on local protein context, we needed to determine where motifs needed to be inserted to best influence phage function.
We developed BLISS (Bacteriophage Library Informed Sequence Scoring), a bioinformatic motif-finding tool that uses experimental data from deep mutational scanning (DMS) to identify metagenomic motifs that influence phage activity (Figure 1A). BLISS is not constrained by overall sequence homology but can reach deep into metagenomic space for motifs from phages that are not closely related to the wildtype phage. Furthermore, by using DMS data to guide the search, BLISS evaluates every possible substitution at every position in candidate motifs, resulting in functionally relevant motifs that can significantly differ from the wildtype sequence. Finally, BLISS uses DMS data to precisely identify where motifs need to be inserted within the wildtype sequence to influence phage activity. We used BLISS to mine metagenomic motifs for the RBP tip domain of the T7 phage.

(A) Illustration of BLISS. Results from DMS of the T7 phage RBP tip domain (1) are used to seed scores for probable motifs (2), which are then mined in metagenomic databases (3). Motifs passing a hit threshold are substituted into phages in a phage library using ORACLE (4). Crystal structure (PDB: 4A0T) and secondary structure topology shown color coded with interior loops as red, β-sheets as yellow and exterior loops as blue. (B) Heat map showing seed score (red gradient) for substitutions in the tip domain. Substitutions are shown top to bottom while position (residue numbering based on PDB 4A0T), wildtype amino acid and secondary structure topology is shown left to right. (C) Number of 6-AA (light grey) or 10-AA (dark grey) motifs found passing hit threshold using BLISS and metagenomic phage structural proteins or non-structural proteins (left), using BLISS with metagenomic structural proteins after fully randomizing seed scores or randomizing seed scores at each position (middle, mean ± SD of triplicate randomizations), or number of hits using BLAST (right). (D) Number of non-unique variants sourced from different phage families derived from proteins annotated in NCBI. The family for T7 phage (Podoviridae) is marked with a yellow star.
BLISS first determines a ‘seed score’ for every possible substitution at every position using DMS data. (Figure 1B, Supplementary File 1). We computed seed scores using a prior DMS study from our group where we screened 1660 T7 RBP tip domain saturated single-amino acid substitution variants on multiple hosts [23]. The seed score is generated based on the performance of a substitution relative to other substitutions (including wildtype) at that position on various hosts (see Methods). Weighing both substitution performance and the difference in activity between hosts refines seed scores for contextual relevance for phage activity across different hosts. Furthermore, this approach retains the relevant position of each candidate substitution to determine where motifs need to be inserted to influence phage activity. We found that the seed scores are well distributed throughout the tip domain and favor positions known to influence phage activity such as exterior loops and outward-facing β-sheets (Figure 1B).
After compiling a list of seeded motifs, the next step of BLISS is mining metagenomic datasets for matching sequences. We searched for motifs in publicly available NCBI and IMG/VR databases, representing some of the largest databases for metagenomic phage sequences [28, 29]. Seed scores were used to mine 6-mer and 10-mer protein motifs from ∼60,000 unique metagenomic phage structural proteins. Since productive motifs are likely to be found in a greater abundance, only motifs meeting a minimum threshold of abundance were selected. This yielded 15,565 6-mer and 3,549 10-mer protein motifs (Figure 1C and Figure S1).
We then used two approaches to evaluate if BLISS identified motifs relevant to receptor recognition. First, we repeated BLISS using a comparably sized set of metagenomic non-structural proteins such as polymerases, ligases, and terminases, reasoning fewer motifs for receptor recognition should be found in non-structural proteins. Indeed, we mined ten-fold fewer motifs in non-structural proteins (Figure 1C). Second, we evaluated the importance of the DMS data by searching with randomized seed scores. Randomizing seed scores should result in fewer hits because scores lose relevance to phage activity, resulting in poorly seeded motifs that should be found less frequently than correctly seeded motifs. Randomizing either position or identity of the substitution scores (Supplementary File 1) produced ten-fold fewer motifs than correctly seeded scores (Figure 1C).
We next assessed if our approach sampled motifs broadly from different phage families or biased toward Podoviridae phages closely related to T7 phages. We examined protein taxonomy from 28,989 source proteins from the NCBI database that informed 84,650 source motifs. Motifs were derived from diverse phages reflecting the overall taxonomic distribution of the database, predominantly from the families Siphoviridae and Myoviridae, without restriction to closely related phages (Figure 1D). Overall, these results showed that recombinogenic motifs are likely distributed across diverse phage families. Identifying these distant sequence motifs requires systematic experimental calibration of residue substitutions to guide the search.
Library selection reveals family and location preferences for active phage variants
To experimentally test BLISS motifs, we synthesized 7,735 6-mers and 2,338 10-mers as chip oligonucleotides with motifs inserted at the appropriate location in the tip domain. The variant library was inserted into the T7 phage genome using a recently developed method in our laboratory, ORACLE (Optimized Recombination, Accumulation, and Library Expression) [23]. ORACLE is a high-throughput precision phage genome engineering tool designed to create a large, unbiased library of phage variants of defined sequence composition. Briefly, ORACLE uses Cre recombinase to insert variant sequences at a defined location on the phage genome, CRISPR-Cas9 to eliminate unrecombined phages, and a series of helper plasmids to prevent library bias. Notably, variant libraries created using ORACLE are not required to be active on the propagating host. This allowed us to create a large motif library without suffering a loss of diversity (see Methods). Variants in the final phage library contained between 2 and 10 substitutions with an average of 6.1 substitutions per sequence (see Figure S2A and Supplementary File 2).
We evaluated the activity of phage variants by selecting the library on a panel of five E. coli hosts. Three hosts, B strain derivative BL21, K-12 derivative BW25113, and DH10B derivative 10G, are susceptible to wildtype T7. Two hosts, E. coli deletion strains BW25113ΔrfaG and BW25113ΔrfaD, are resistant to wildtype T7 as they lack the host genes required for the biosynthesis of the full-length lipopolysaccharides (LPS) receptor. As a result, the activity of T7 phage on the resistant strains is lower than the permissive strains by ∼2-5 orders of magnitude [23,30–32]. The library was selected on each host for approximately two replication cycles. Each variant was assigned a functional score, FN, based on the ratio of the abundance of the variant before and after selection normalized to wildtype (Figure 2A) (see Methods). FN scores were well correlated across biological triplicates (Figure S3).

(A) Library variants are scored by comparing variant abundance pre- and post-selection on different hosts. (B) Number of non-unique variants active on any host (bottom, light grey) and percent of active non-unique variants compared to the pre-selection library (top, dark grey) sourced from different phage families derived from proteins annotated in NCBI. The family for T7 bacteriophage (Podoviridae) is marked with a yellow star. (C-D) Heat map showing total count (C) pre-selection and (D) post-selection of specific substitutions listed top to bottom (log10, red gradient) with substituted residue number (based on PDB 4A0T) and secondary structure shown left to right. Wildtype amino acids are shown blank with a black dot in the upper left. Total number of substitutions are shown at the top for every position as a stacked bar graph, with substitutions in 10-AA motifs shown dark grey and 6-AA motifs shown in light grey.
Following selection, approximately 6% (607 of 10,073) of phage variants showed activity on at least one host. The post-selection distribution of the number of mutations per sequence resembled the pre-selection distribution and averaged 4.7 mutations per sequence (Figure S2B), showing that active variants include distant motifs, not just close sequences matches to the wildtype motif. Contrary to our expectations, variants with motifs from T7 family Podoviridae had the least activity, with only 6.2% (47 of 754) active variants (Figure 2B). This was followed by Ackermannviridae, with 11.6% (16 of 138) active variants. Variants with motifs sourced from more distant phage families such as Autographiviridae, Demerecviridae and Drexlerviridae represented a smaller, but highly active portion of the library with 73.8% (31 of 42), 64.3% (9 of 14), and 73.3% (11 of 15) of variants active on any host. Mining deeper into metagenomic space from distantly related phages was thus important for creating active phage variants.
We then compared the composition and position of the motifs on the tip domain between the pre-selection input library and post-selection active variants. Most of the variants were concentrated in the exterior loop and proximal β-sheets (BC-C, DE-E, F-FG and H-HI) previously identified as functionally relevant [23]. We found a complete reversal in the regions with most motifs in pre- and post-selection populations. In the pre-selection library, approximately 79% of the motifs were concentrated in the exterior loops DE and proximal β-sheet E and 13% in the BC-C region. However, in the post-selection library, approximately 45% are located in BC-C and 16% in DE-E (Figure 2C and 2D, Figure S2C and S2D). One possible reason for the greater preference for BC-C is that this region faces away from possible interactions with other monomers of the trimer RBP and may thus be more tolerant to substitutions [33].
Several important insights emerged from the screen. While only 6% of variants with a sequence motif were active, this constitutes a substantial fraction given the importance of the T7 RBP to phage activity. Our prior screen of single amino acid mutations produced less than 40% functional phage variants on E. coli 10G. In comparison, the phage variants with the motif library carry approximately 5 mutations per sequence post selection, and BLISS promotes identification of motifs that have mutations expected to be impactful to phage fitness. This suggests that BLISS unearthed sequence motifs that are non-disruptive to the RBP and confer a fitness benefit to the phage. The fraction of functional motifs may also increase as more E. coli strains and close phylogenetic neighbors are tested. The drastic change in the locational preferences of the motifs pre- and post-selection shows that both the composition of the sequence motif and where it is located is important for function.
Hierarchical Clustering Reveals Patterns in Motif Selection
To understand how motif preferences governed phage activity and specificity, we classified phage variants based on their activity on the five hosts using hierarchical clustering using the log2 FN score of each phage variant on each bacterial hosts as input. To accurately interpret our dataset, we used agglomerative hierarchical clustering with Spearman distance and Ward’s clustering. This unsupervised correlation-based approach is not sensitive to expected outliers but can still identify smaller clusters, making it ideal for granular classification of host specificities. This clustering approach gave a suitable cophenetic score of 0.91, indicating the dendrogram faithfully retained the pairwise distance between variant scores and that the clustering approach was valid (see S4A). Variants with similar activity profiles were grouped into 21 clusters based on a measurement of gap statistic (C1-C21, Figure 3A and S4B) and motifs in each cluster were sourced from a variety of phage families (Figure S4C). We found the calculated 21 clusters effectively separated variants based on similar functional profiles, and other clustering thresholds gave comparable functional profiles (Figure S4D-E).

(A) Hierarchical clustering of normalized functional scores (log2 FN, blue to red gradient, wildtype FN = 0) for active phage variants on five E. coli hosts (listed left). FN clustered is the average of three biological replicates. (B) Heat map showing substitution frequency (red gradient) at every position in the tip domain for each cluster. Clusters are organized top to bottom, while position, wildtype amino acid and secondary structure topology is shown left to right. (C-E) Frequency plots comparing substitution frequency for (C) clusters 8 and 18 at positions 478 through 483, (D) clusters 9 and 4 at positions 498 through 503, and (E) clusters 9 and 8 at positions 516 through 521. The wildtype sequence and position is shown top and amino acids are colored based on similarity of physicochemical properties.
Each of the 21 clusters contained variants with similar functional profiles. For instance, C9 contained phage variants with high specificity that were active on BW25113 but not the remaining four hosts. In contrast C10 contained phage variants with broadened specificity. These variants were active on BW25113, BL21 and 10G, but not on the remaining two hosts. The largest cluster, C8, comprised of variants that were active on all strains with lower activity on E. coli BW25113. Other clusters specific to a single strain included C2, C18, and C20. Host range expanded to two hosts in C11 or C19 and three hosts in C10 and C16. These results revealed metagenomic sequence motifs can alter specificity, suggesting that sequence motifs could be an important driver of phage adaptation. Further, using metagenomic motifs allowed for customizing host specificity to different host combinations that were unachievable using only single substitutions [23].
We examined if locational preferences i.e., where motifs were incorporated on the tip domain, had a role in functional differentiation. We compared the location of enriched motifs for all the clusters along the tip domain and found locational preferences to be an important driver of host range (Fig. 3B). For instance, consider clusters C4 and C8. C8 variants have higher activity on BW25113 over BL21, whereas the activity of C4 variants is reversed. C8 variants almost completely eschew substitution in the DE-E region, with only 1.5% of variants (4 of 266) having a substitution in DE-E. In contrast, C4 has a stronger preference for the DE-E region with 45% (15 of 33) of variants located there compared to BC-C with 84% (223 of 266) variants. Other clusters, such as C10 and C11, also show location-specific trends. C11 contains variants active only on BL21 and BW25113, but not 10G, and has 31.3% (5 of 16) of variants in the DE-E region. In contrast, C10, which contains variants that can infect all three hosts, has no substitutions in this region. These results indicate that all regions of the RBP tip domain are not created equal and that the location of the motif plays a major role in governing phage activity.
We further explored sequence drivers of functional differentiation by examining the substitution profile for different clusters. C8 and C18 shared similar substitution frequency (C8 – 84%, C18 – 83%) between residues 478 to 483 in the BC-C region (Fig. 3B) but showed dramatic functional differentiation, with C8 variants active on all strains, whereas C18 variants were active only on BL21 (Figure 3A). The average change in physicochemical properties of motifs in either cluster were similar except motifs in C8 had significantly higher average isoelectric point (Figure S5A). There were two key sequence differences between C8 and C18 motifs (Figure 3C). At position 480, C8 variants largely contained lysine or arginine, whereas C18 variants contained serine. At position 482, C8 variants were largely aliphatic, whereas C18 variants carried a negatively charged aspartic or glutamic acid. Other clusters provided similarly intriguing comparisons. We examined substitutions between positions 498 and 503 in the DE-E region for variants in C9, which showed activity only on E. coli BW25113, and C4 to investigate what substitution(s) might drive specificity toward BW25113 in this region of the tip domain (Figure 3D). In addition to having a significantly lower isoelectric point (Figure S5B), variants in C9 tolerated glycine at positions 500 and 501, while variants in C4 only tolerated this substitution at positions 502 and 503. One possible hypothesis for this sequence preference is that local flexibility afforded by the glycine substitution may impact the relative conformation of the tip domain, preventing adequate receptor engagement for all strains except BW25113. Comparison between C8 and C9 between positions 516 and 521 of the F-FG region reveals further nuances for substitution preference (Figure 3E). Both C8 and C9 shared similar substitution frequency (C8 – 9%, C9 – 11%) in this region. Motifs in C8 have higher changes in volume and isoelectric point compared to motifs in C9 (Figure S5C). Our prior DMS screen showed that this region was largely intolerant to substitution except for key substitutions at G521 or A518 which enabled function on insensitive strains. Motifs in C8 are dominated by lysine and arginine substitutions at either G521 or A518, recapitulating DMS results. In addition, they also contain many substitutions that were not tolerated individually, notably proline substitutions, which likely change the local structure of the loop. In contrast, C9 contains no lysine or arginine substitutions at G521 but includes abundant proline substitutions in the region. We hypothesize the presence of enabling substitutions allows substantial changes to the local architecture of the loop to expand tolerable substitutions and alter the host range of the phage variants in each cluster.
These results show that both the location and the composition are key drivers of phage host specificity. BLISS created variants with unique combinations of substitutions and locational preferences that drove distinct patterns of host specificity. Many variants have substitutions that were poorly tolerated individually but in combination with other substitutions provided a novel path for altering phage activity. Substitution location was important for determining host specificity, with the area near the BC exterior loop and C β-strand overall more tolerant of substitution. In summary, metagenomic databases constitute a rich and as yet untapped resource for creating phages with novel specificity profiles.
BLISS reveals unique epistatic interactions in the tip domain
We examined if the motifs carried mutations that were individually deleterious but collectively beneficial due to favorable epistatic interactions. In other words, can the motifs provide a buffering genetic background for individually deleterious mutations. Indeed, we observed that certain mutations that were not tolerated individually in our prior DMS screen had been enriched in post-selection motifs. Due to the larger number of mutations introduced in each motif, this suggested specific epistasis may play an important role in the evolutionary sequence convergence of some functionally relevant motifs. Specific epistasis occurs when physical interactions between multiple mutations alter the biochemical function of a protein differently than the sum of each individual mutation. A point mutation in the tip domain that may be deleterious by itself could be highly functional in combination with other motif mutations. Understanding how local networks of residues interact provides insights into the sequence-function landscape of the RBP [34–37].
We examined epistatic activity for insensitive strains BW25113ΔrfaD and BW25113ΔrfaG. We chose to examine both insensitive strains because selection on these strains was most significant. We classified variants as epistatic if the sum activity of every individual mutation was worse than wildtype (sum log2 FN < 0) while the phage variant with the motif had increased activity compared to wildtype (log2 FN > 0). (Figure 4A and 4B, Supplementary File 2). We found 40 epistatic variants for BW25113ΔrfaD and 27 variants for BW25113ΔrfaG where every motif substitution was individually deleterious, but when combined together substantially increased activity. In fact, some epistatic motif variants considerably outperformed wildtype despite deleterious individual mutations. For example, a variant with motif QGVIHI replacing SGSAGG between positions S475 and G480 performed approximately 30 times better than wildtype without having any activity noted for any individual mutation.

(A-B) Comparison of variant activity (log2 FN) to the sum of activity of individual substitutions that comprised the variant motif on (A) BW25113ΔrfaG and (B) BW25113ΔrfaD. Shaded areas represent variants showing signs of epistasis (green), indicating substitutions in that motif show greater activity when combined than individually. Points are shown only if variant activity was greater than the limit of detection. (C-D) Frequency plots comparing substitution frequency for BW25113ΔrfaG and BW25113ΔrfaD at (C) positions 478 through 483 and (D) at positions 502 through 507 for variants showing strong epistasis. The wildtype sequence and position is shown top and amino acids are colored based on similarity of physicochemical properties.
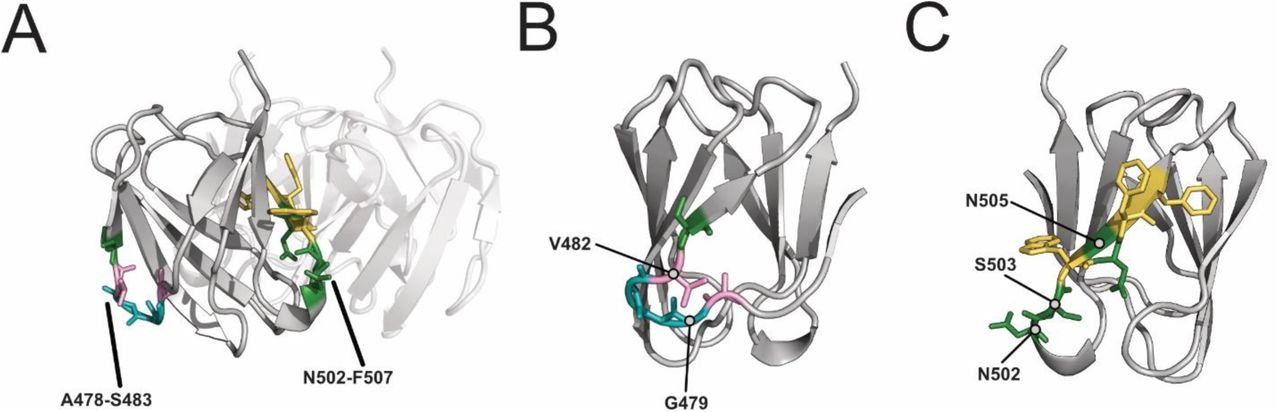
Most epistatic variants had substitutions between positions A478 and S483 in the BC exterior loop and C β-sheet (26 variants BW25113ΔrfaD,16 for BW25113ΔrfaG) or between positions N502 and F507 in the DE exterior loop and E β-sheet (11 variants BW25113ΔrfaD,6 for BW25113ΔrfaG). Different residue preferences drove activity between positions A478 and S483 (Figure 4C). This region faces outward and has fewer probable interactions with other monomers of the RBP (Supplementary Figure 6A and 6B). Epistatic variants frequently contained large, hydrophilic substitutions at G479 for BW25113ΔrfaD or V482 for BW25113ΔrfaG. These substitutions were not tolerated individually at these positions but increased activity in proximal positions [23]. We predict other substitutions changed the local structure of the domain, enabling a key binding event with these substitutions to increase phage activity.
Epistatic motifs with substitutions between positions N502 and F507 showed a similar substitution pattern for both hosts (Figure 4D). This region faces inward and likely has many interactions with other monomers (Supplementary Figure 6A and 6C). Epistatic motifs primarily had lysine and arginine substitutions at position N502 and replaced polar, uncharged residues or larger hydrophobic residues with small hydrophobic residues. We predict that the smaller hydrophobic substitutions stabilize interactions with other monomers, allowing the large hydrophilic substitutions to successfully interact with the host receptor. In summary, the role of epistasis in certain sequence motifs paints a fascinating picture. It shows that some metagenomic motifs carry the sequence background required to accommodate individually deleterious mutations without relying on the phage RBP to provide the buffering genetic background.
BLISS-generated motifs enhance activity on E. coli causing UTIs
An important test for the generalizability of BLISS was determining if we can generate phage variants with gain-of-function activity on new hosts outside of the DMS experiment used to seed the library. Since phage therapy is emerging as a promising solution to the antibiotic resistance crisis, we chose to evaluate gain-of-function on clinically relevant E. coli strains. For this purpose, we chose two isolates from patients with urinary tract infection (UTI), UTI33 and UTI46, which are insensitive to wildtype T7. We passaged the motif library on UTI33 and UTI46 on these strains, in a manner similar to the laboratory E. coli strains (Figure 2) and determined a normalized functional score, FN, for each motif variant (Figure S7A-B) [38]. The FN scores trended similarly for both strains, with several variants significantly outperforming wildtype (Figure 5A). Motifs enriched post-selection strongly favored the BC-C region (Figure S7C).

(A) Comparison of variant activity (log2 FN) on E. coli UTI46 and E. coli UTI33. (B) Efficiency of Plating (mean ± SD, biological triplicates) on E. coli UTI46 and E. coli UTI33 for wildtype T7 (WT), T7 variant with FGALKG replacing the wildtype region SGSAGG, and T7 variant contained the motif WCKENK replacing the wildtype region SAGGGV. E. coli 10G with a helper plasmid constitutively producing wildtype receptor binding protein is used as a reference host. Wildtype plaques are atypically small and hazy.
We isolated two high-scoring variants to determine if they could overcome the plaquing deficiency seen with wildtype T7. Both motifs are highly divergent from the wildtype sequence. The first variant contained motif FGALKG, replacing wildtype region SGSAGG from positions S475 to G480 (UTI33 log2 FN = 5.1±0.1, UTI46 log2 FN = 5.5±0.1). The second variant contained motif WCKENK, replacing wildtype region SAGGGV from positions S477 through V482 (UTI33 log2 FN = 3.6±0.3, UTI46 log2 FN = 3.7±0.1). Wildtype T7 has a 4-log total reduction in plaque efficiency on UTI46 and a 2-log total plaque deficiency on UTI33 (Figure 5B). Wildtype plaques are hazy with a diameter of ∼1-2 mm compared to ∼5-6 mm diameter on susceptible strains. The T7 variant with the motif FGALKG restored plaque morphology to wildtype but retained a ∼1 log deficiency on both strains, while the variant with the motif WCKENK completely restored plaque efficiency and plaque morphology on both strains (Figure 5B). BLISS was thus able to identify motifs that dramatically improve activity in clinically relevant strains outside the DMS panel used to seed the library. This result hints at the possibility that metagenomic motifs may be a path to broadening host specificity.
Discussion
In this study, we devised BLISS as a new approach to explore sequence-function relationships in diverse metagenomic databases and find metagenomic motifs that influence phage activity. Using BLISS, we found more than 104 6-mer and 10-mer motifs for the T7 RBP that significantly differed from the wildtype sequence. These motifs were sourced from disparate and distantly related structural metagenomic sequences, allowing us to overcome the problem of a lack of sequence conservation among phages to find functionally important motifs in diverse metagenomic databases. BLISS enables identification of otherwise invisible motifs that can be used to engineer activity and functional differentiation in phages and identify trends in sequence-function relationships in metagenomic proteins. By screening phage variants with these motifs, we revealed hundreds of T7 phage variants with novel host specificity and elucidated how motifs drove changes activity in the RBP.
Several key insights emerged from our results. First, leveraging DMS results was a successful strategy for correctly seeding scores to find motifs that broadly influenced phage activity and altered host specificity even if the motifs differ substantially in sequence identity [23]. This approach was able to generate phage variants that improved activity on UTI strains that had not been included in the host panel used for the DMS assay and created variants that had novel host specificity when compared to single substitutions. Second, many changes in phage activity and host range were dependent on curating motifs from distantly related phages. Motifs from Podoviridae, the family of T7 phage, performed notably poorly while motifs sourced from more distantly related phages drove activity and host specificity. This emphasized the importance of reaching deeply into metagenomic space to source motifs from non-homologous phages. Third, both the position and composition of motifs were important drivers of phage activity. The BC exterior loop and C β-strand were overall more tolerant of substitution while motifs inserted at other positions could drive host specificity. Novel combinations of substitutions appear to allow for substantial changes to the local architecture of the tip domain and revealed epistatic interactions that enable key residues to contact the host receptor. Unbiased libraries of phages with BLISS-identified motifs can reveal potent combinations of mutations that can tailor host range to specific hosts. Altogether these results improve our understanding of the sequence-function landscape of the T7 RBP and identify motifs that can drive highly specific changes in host activity.
Vast troves of phage metagenomic data are becoming increasingly available from diverse biomes. The enormous breadth of phage diversity makes querying these databases for relevant sequences a challenging prospect. BLISS is designed as a process that can tap into these complex datasets to identify relevant motifs that drive phage activity. This process is not limited to the RBP and can feasibly be used in any region with DMS results to seed and score potential motifs, including other structural proteins, lysins or holins. By using a DMS-seeded functional profile of possible motifs BLISS allows us to move beyond sequence similarity to deeply mine metagenomic databases for relevant sequence motifs. Through leveraging these metagenomic motifs we envision BLISS as a platform to transform our understanding of sequence-function relationships and engineer phages.
Contributions
P.H.: Conceptualization, Data curation, Software, Formal analysis, Investigation, Visualization, Methodology, Writing - original draft, Writing - review and editing K.K.: Conceptualization, Data curation, Software, Formal analysis, Investigation, Methodology, Writing - review and editing
A.M.: Software
K.N.: Software
K.A.: Conceptualization, Resources, Supervision, Funding acquisition, Project administration, Writing - review and editing
S.R.: Conceptualization, Resources, Supervision, Funding acquisition, Project administration, Writing - review and editing

Total number of curated motifs (log10) found using different metagenomic hit cutoffs for 10-AA motifs (dark grey) and 6-AA motifs (light grey). The lowest cutoff indicated is 15 hits for 10-AA motifs and 50 hits for 6-AA motifs.

Count of substitutions for each phage variant in the phage library before (A) and after (B) selection for variants with activity on at least one host. Crystal structure (PDB: 4A0T) of tip domain shaded red based on the number of substitutions at that position in the library before selection (C) and after (D) selection for variants with activity on at least one host.

Correlation of FN scores between biological replicates of the phage variant library on (A) E. coli 10G, (B) BL21, (C) BW25113, (D) BW25113ΔrfaD and (E) BW25113ΔrfaG. R values and trendlines are displayed for all replicates as a black line.
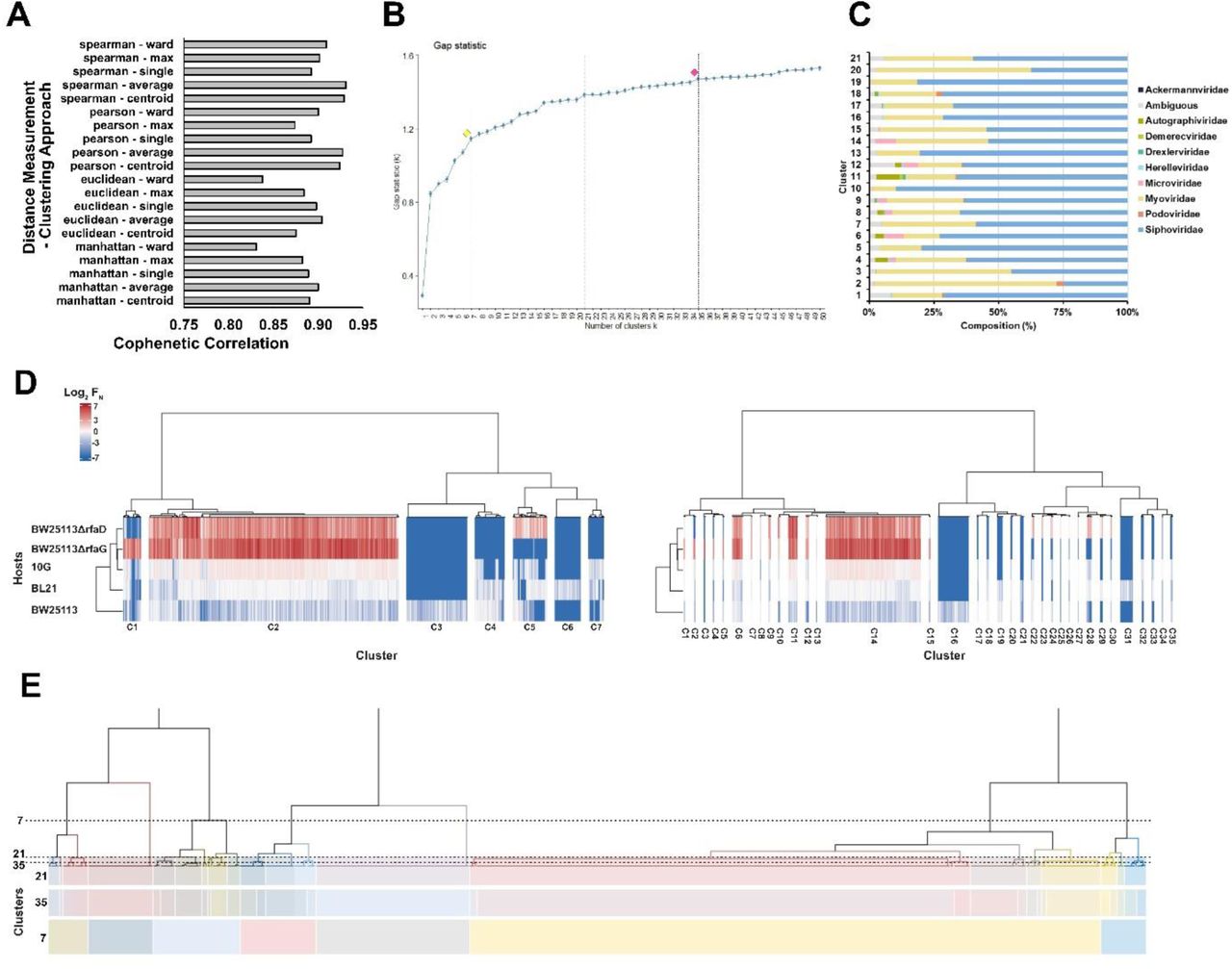
(A) Cophenetic correlation for different distant measurements and clustering approaches. (B) Gap statistic results for hierarchical clustering to determine cluster generation, with example clusters marked with dotted lines and alternative cutoffs of seven (yellow) and 35 (red) clusters indicated. (C) Percentage source protein taxonomy for clusters using 21 clusters. (D) Hierarchical clustering for 7 (left) and 35 (right) clusters using normalized functional scores (log2 FN, blue to red gradient, wildtype FN = 0) for active phage variants on five E. coli hosts (listed left). FN clustered is the average of three biological replicates. (E) Visual representation of cutoffs for 21, 35, and 7 cluster cutoffs on the dendrogram generated for clustering.
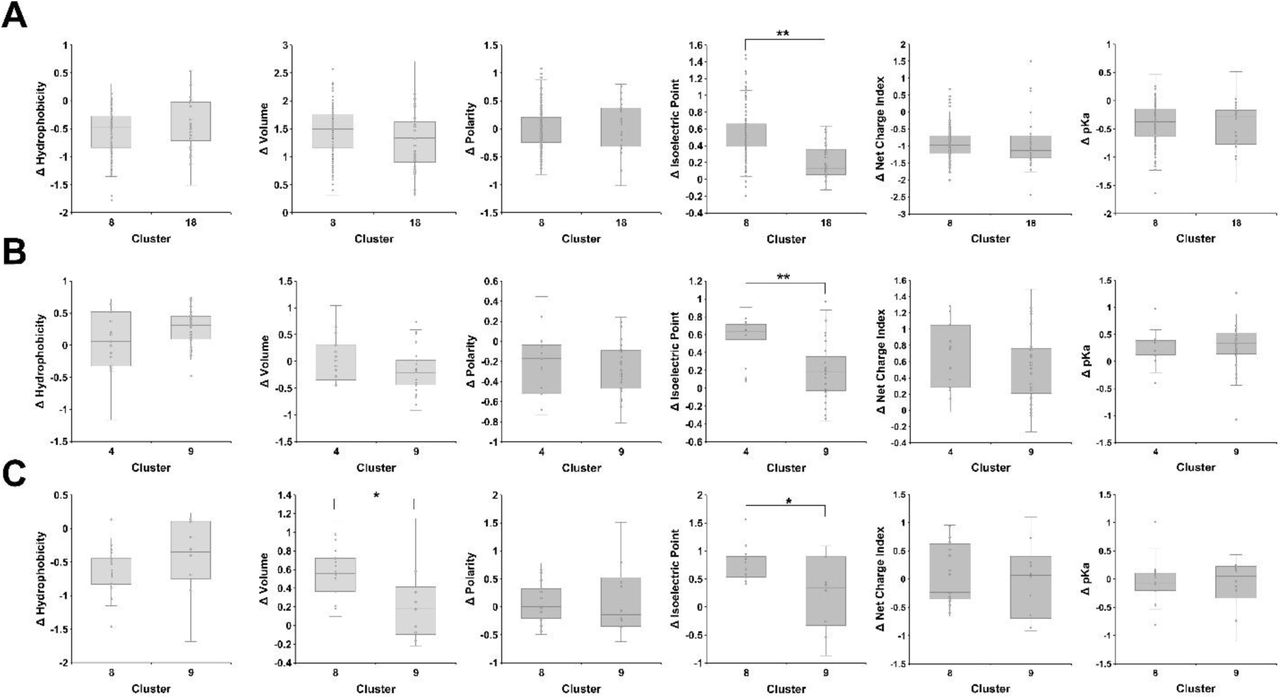
Physicochemical comparison of the change in hydrophobicity, volume, polarity, isoelectric point and pKa between motifs with substitutions in the specified region for (A) clusters 8 and 18 at positions 478 through 483, (B) clusters 9 and 4 at positions 498 through 503, and (C) clusters 9 and 8 at positions 516 through 521. The change in physicochemical properties is calculated as the sum of the change in property of all substitutions in the motif. (*) indicates significance of p >0.05, (**) indicates significance where p > 0.001 as calculated from Welch’s t-test.

(A) Crystal structure of the tip domain with regions A478-S483 and N502-F507 in color on one monomer with the other monomers of the receptor binding protein shown transparent. Amino acids are colored based on similarity of physicochemical properties. (B-C) Rotated views of regions (B) A478-S483 and (C) N502-F507.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A-B) Correlation of FN scores between biological replicates of the phage variant library on (A) E. coli UTI33 and (B) UTI46. R values and trendlines are displayed for all replicates as a black line. (C) Substitution frequency (red gradient, total percentage of substitutions) for active variants on E. coli UTI33 (left) and UTI46 (right). Specific substitutions are listed top to bottom with substituted residue number (based on PDB 4A0T) and secondary structure shown left to right. Wildtype amino acids shown blank with black dot upper left.
List of Supplementary Files
Supplementary File 1. Seed scores for each substitution in the tip domain and randomized seed scores used to validate BLISS.
Supplementary File 2. List of phage variants and the wildtype region the motif replaced, the motif sequence and associated substitutions.
Supplementary File 3. Summary of deep sequencing results.
Methods
Microbes and Culture Conditions
T7 bacteriophage was obtained from ATCC (ATCC® BAA-1025-B2). T7 acceptor phages used in ORACLE were previously generated in our lab [23]. Escherichia coli BL21 is a lab stock, E. coli 10G is a highly competent DH10B derivative [39] originally obtained from Lucigen (60107-1). E. coli BW25113, BW25113ΔrfaD and BW25113ΔrfaG were obtained from Doug Weibel (University of Wisconsin, Madison) and are derived from the Keio collection [40]. UTI33 and UTI46 were obtained from Rod Welch (University of Wisconsin, Madison) and originates from a UTI collection [38].
All bacterial hosts are grown in and plated on Lb media (1% Tryptone, 0.5% Yeast Extract, 1% NaCl in dH2O, plates additionally contain 1.5% agar, while top agar contains only 0.5% agar) and Lb media was used for all experimentation and was used to recover host and phages after transformation. Kanamycin (50 μg/ml final concentration, marker for pHT7Helper1) and spectinomycin (115 μg/ml final concentration, marker for pHRec2, pHRec1-MLib and pHCas9 and derivatives) was added as needed. All incubations of bacterial cultures were performed at 37°C, with liquid cultures shaking at 200-250 rpm unless otherwise specified. Bacterial hosts were streaked on appropriate Lb plates and stored at 4°C.
T7 bacteriophage was propagated using E. coli BL21 after initial receipt from ATCC and then as described on various hosts in methods. All phage experiments were performing using Lb and culture conditions as described for bacterial hosts. Phages were stored in Lb at 4°C.
For long term storage all microbes were stored as liquid samples at -80°C in 10% glycerol, 90% relevant media.
SOC (2% tryptone, 0.5% yeast extract, 0.2% 5M NaCl, 0.25% 1M KCL, 1% 1M MgCl2, 1% 1M MgSO4, 2% 1M glucose in dH2O) was used to make competent cells.
General Cloning Methods
PCR was performed using KAPA HiFi (Roche KK2101) for all experiments. Golden Gate assembly was performed using New England Biosciences (NEB) Golden Gate Assembly Kit (BsaI-HFv2, E1601L). Restriction enzymes were purchased from NEB. DNA purification was performed using EZNA Cycle Pure Kits (Omega Bio-tek D6492-01) using the centrifugation protocol. Gibson assembly was performed according to the Gibson Assembly Protocol (NEB E5510) but Gibson Assembly Master Mix was made in lab (final concentration 100 mM Tris-HCL pH 7.5, 20 mM MgCl2, 0.2 mM dATP, 0.2 mM dCTP, 0.2 mM dGTP, 10 mM dTT, 5% PEG-8000, 1 mM NAD+, 4 U/ml T5 exonuclease, 4 U/μl Taq DNA Ligase, 25 U/mL Phusion polymerase). All cloning was performed according to manufacturer documentation except where noted in methods.
PCR reactions use 1 μl of ∼1 ng/μl plasmid or ∼0.1 ng/μl DNA fragment as template for relevant reactions. PCR reactions using phage as template use 1 μl of undiluted phage stock (genomic extraction was unnecessary) and have an extended 5 minute 95°C denaturation step.
DpnI digest was performed on all PCR that used plasmid as template. Digestion was performed directly on PCR product immediately before purification by combining 1.5 μl DpnI (30 units), 15 μl 10x CutSmart Buffer, 98 ul PCR product, and 35.5 ul dH2O, incubating at 37°C for 2 hours and 30 minutes then heat inactivating at 80C for 20 minutes.
Electroporation of plasmids was performed using a Bio-rad MicroPulser (165- 2100), Ec2 setting (2 mm cuvette, 2.5 kV, 1 pulse) using 50 μl competent cells and 100 ng of plasmid or 20 ul of golden gate reaction for transformation. Electroporated cells were immediately recovered with 950 μl Lb, then incubated at 37°C for 1 to 1.5 hours and plated or grown in relevant media.
E. coli 10G competent cells were made by adding 8 mL overnight 10G cells to 192 mL SOC (with antibiotics as necessary) and incubating at 21°C and 200 rpm until ∼OD600 of 0.4 as determined using an Agilent Cary 60 UV-Vis Spectrometer using manufacturer documentation (actual incubation time varies based on antibiotic, typically overnight). Cells are centrifuged at 4°C, 1000g for 20 minutes, the supernatant is discarded, and cells are resuspended in 50 mL 10% glycerol. Centrifugation and washing are repeated three times, then cells are centrifuged at 2000g for 20 minutes then resuspended in a final volume of ∼1 mL 10% glycerol and are aliquoted and stored at -80°C.
Plasmid Cloning and Descriptions
pHT7Helper1 is used during optimized recombination and during accumulation in ORACLE to prevent library bias and depletion of variants that grow poorly on E. coli 10G. There are no changes to this plasmid from its initial construction as documented in our previous DMS assay, plasmid details are provided here for completeness [23]. This plasmid contains a pBR backbone, kanamycin resistance cassette, mCherry, and the T7 receptor binding protein gp17. Both mCherry and gp17 are under constitutive expression. Gp17 was combined with promoter apFAB47 [41] using SOE and the plasmid assembled by Gibson assembly. There is a single nucleotide deletion in the promoter that has no effect on plaque recovery for phages that require gp17 to plaque.
pHRec2 is used as template to generate the variant library containing metagenomic motifs, referred to as pHRec2-Motif and used during optimized recombination in ORACLE. This plasmid has been updated since its initial construction in our previous DMS assay, where it was called pHRec1. As before this plasmid contains an SC101 backbone, Cre recombinase, a spectinomycin resistance cassette, and the T7 tail fiber gp17 flanked by Cre lox66 sites with an m2 spacer, a 3’ pad region and lox71 sites with a wt spacer [42]. Cre recombinase is under constitutive expression. This plasmid was altered using sequential PCR and Gibson assembly to include a larger constant region and random, 20 bp barcode between the end of gp17 and the Cre-lox sites. The total region inserted was 105 bp. This barcode was added to facilitate smaller, rather less expensive NGS reads but was not used in this experiment.
pHCas9-3 is used during Accumulation in ORACLE. There are no changes to this plasmid from its initial construction as documented in our previous DMS assay, plasmid details are provided here for completeness [23]. This plasmid contains an SC101 backbone, a spectinomycin resistance cassette and cas9 cassette with a previously cloned gRNA targeting the T7 acceptor phage [43]. pHCas9 was created with Gibson assembly, while pHCas9-3 was assembled by phosphorylation and annealing gRNA oligos (100 uM forward and reverse oligo, 5 μl T4 Ligase buffer, 1 μl T4 PNK, to 50 μl dH2O, incubate at 37°C for 1 hour, 96C for 6 minutes then 0.1C/s temperature reduction to 23C), then Golden Gate cloning (1 μl annealed oligo, 75 ng pHCas9, 2 μl T4 DNA Ligase Buffer, 1 μl Golden Gate Enzyme Mix, dH20 to 20 ul. Incubation at 37°C for 1 hour then 60C for 5 minutes, followed by direct transformation of 1 ul, plated on Lb with spectinomycin). All plasmid backbones and gene fragments are lab stocks.
General Bacteria and Phage Methods
Bacterial concentrations were determined by serial dilution of bacterial culture (1:10 or 1:100 dilutions made to 1 mL in 1.5 microcentrifuge tubes in Lb) and subsequent plating and bead spreading of 100 μl of a countable dilution (targeting 50 colony forming units) on Lb plates. Plates were incubated overnight and counted the next morning. Typically, two to three dilution series were performed for each host to initially establish concentration at different OD600 and subsequent concentrations were confirmed with a single dilution series for later experiments.
Stationary phase cultures are created by growing bacteria overnight (totaling ∼20-30 hours of incubation) at 37°C. Cultures are briefly vortexed then used directly. Exponential phase culture consists of stationary culture diluted 1:20 in Lb then incubated at 37°C until an OD600 of ∼0.4-0.8 is reached (as determined using an Agilent Cary 60 UV-Vis Spectrometer using manufacturer documentation), typically taking 40 minutes to 1 hour and 20 minutes depending on the strain and antibiotic, after which cultures are briefly vortexed and used directly.
Phage lysate was purified by centrifuging phage lysate at 16g, then filtering supernatant through a 0.22 uM filter. Chloroform was not used.
To establish titer, phage samples were typically serially diluted (1:10 or 1:100 dilutions made to 1 mL in 1.5 microcentrifuge tubes) in Lb to a 10-8 dilution for preliminary titering by spot assay. Spot assays were performed by mixing 250 μl of relevant bacterial host in stationary phase with 3.5 mL of 0.5% top agar, briefly vortexing, then plating on Lb plates warmed to 37°C. After plates solidified (typically ∼5 minutes), 1.5 μl of each dilution of phage sample was spotted in series on the plate. Plates were incubated and checked after overnight incubation (∼20-30 hours) to establish a preliminary titer. After a preliminary titer was established, phage samples were serially diluted in triplicate for efficiency of plating (EOP) assays.
EOP assays were performed using whole plates instead of spot plates to avoid inaccurate interpretation of results due to spotting error [44]. To perform the whole plate EOP assay, 400 μl of bacterial host in exponential phase was mixed with between 5 to 50 μl of phages from a relevant dilution targeting 50 plaque forming units (PFUs) after overnight incubation. The phage and host mixture was briefly vortexed, briefly centrifuged, then added to 3.5 mL of 0.5% top agar, which was again briefly vortexed and immediately plated on Lb plates warmed to 37°C. After plates solidified (typically ∼5 minutes), plates were inverted and incubated overnight. PFUs were typically counted after overnight incubation (∼20-30 hours) and the total overnight PFU count used to establish titer of the phage sample. PFU totals between 10 and 300 PFU were typically considered acceptable, otherwise plating was repeated for the same dilution series. This was repeated at least in triplicate for each phage sample on each relevant host to establish phage titer.
EOP was determined using E. coli 10G with pHT7Helper1 as a reference host. EOP values were generated for each of the three dilutions by taking the phage titer on the test host divided by the phage titer on the reference host, and this value was subsequently log10 transformed. Values are reported as mean ± SD.
MOI was calculated by dividing phage titer by bacterial concentration. MOI for the T7 variant library after the variant gene is expressed was estimated by titering on 10G with pHT7Helper1.
Position Seed Scoring
Data from our previous DMS assay of the T7 RBP tip domain was used to seed scores for each possible substitution [23]. The maximum FN of each biological replicate for each substitution from susceptible strains 10G, BL21, and BW25113 in our DMS assay was compressed where if FN < 1 compressed FN (CFN) = FN and if FN > 1 then CFN = (FN – 1 / max(strain FN) -1) + 1. Scores were compressed to reduce the impact of variants that performed exceedingly well, as we wanted to weight scores higher for variants with functionally different scores across strains (i.e. it matters less if a variant does very well on one strain and comparable to wildtype on other strains, it matters more if it does poorly on one strain and comparable to wildtype or better than wildtype on another, as that is more functionally distinct.)
If the difference between CFN on all susceptible strains was less than 0.5, indicating the functional difference between strains was small, motif seed score MFF = CFN, else MFF = CFN + 0.5. This served to seed scores higher if there was a functional difference between susceptible strains. Finally max(⌊log(FN⌋) of each biological replicate for each substitution from resistant strains BW25113ΔrfaG and BW25113ΔrfaD was calculated and added to MFF. This further weighed substitutions higher if they had recovered function on resistant strains. These final values (see Supplementary File 1) were used to seed scores for every possible substitution to curate motifs.
Database construction
NCBI databases (release July 2019) were queried for the term “prokaryotic virus” and limited to sequences of at least 3 kb. The resulting sequences were manually curated by removing non-genomic sequences (e.g., containing the terms ‘open reading frame’ or ‘ORF’) and non-viral sequences. Additionally, the IMG/VR v2.1 (October 2018) database was downloaded and limited to sequences originating from human, wastewater, and animal environments according to IMG/VR metadata[29].
For each dataset, open reading frames were predicted using Prodigal v2.6.3 (-p meta) and proteins were dereplicated using CD-HIT v4.7 (-c 1)[45, 46]. To obtain proteins of interest, HMM profiles for 373 viral hallmark proteins were selected from VOGDB v94 (vogdb.org) according to viral hallmark proteins designated by VIBRANT v1.2.1[47]. The selected HMM profiles represented annotations containing the terms tail, capsid, spike, baseplate, sheath, lysin, and holin. Hmmsearch v3.1b (-e 1e-5) was used to query the database proteins to the selected HMM profiles[48]. In total, 34682 and 26335 non-redundant proteins from NCBI and IMG/VR databases, respectively, were selected for final analysis. Source code is available at https://github.com/raman-lab/Bliss.
Motif search
A custom motif search tool (motif_finder_tool.py) was used to query each of the protein databases separately for 6-mer motifs (-n 6 -c 1 -e 1e-50 -t 0.8) and 10-mer motifs (-n 10 -c 1 -e 1e-5 -t 0.045). The motif search tool scans n-mer windows of input database amino acids against n-mer windows of input matrix scores. The scores per amino acid window are compared to a set cutoff and predetermined constants. The maximum score per amino acid window across all matrix windows is summed to generate an overall score per protein. The overall score and e-value, calculated based on the number of windows searched, are compared to set thresholds. Any protein passing these thresholds is considered as a candidate protein that is similar to the motifs found within the original input matrix. All amino acid windows used in the generation of the overall score for that protein are extracted. To limit the results to a practical search space, 6-mer and 10-mer motifs were filtered to those that were extracted at least 50 and 15 times, respectively, by the motif search tool across all proteins.
Motif Variant Plasmid Library Preparation
To create the metagenomic motif variant plasmid library, oligos were first designed and ordered from Agilent as a SurePrint Oligonucleotide Library (OLS 230mers, Barcode 33049511001 OLS, internal freezer stock #2). Every oligo contained a single motif that replaced the wildtype region in the tip domain. We used the most frequently found codon for each amino acid in the gp17 tail fiber to define the codon for each substitution. Oligos contained BsaI sites at each end to facilitate Golden Gate cloning. Two pools of oligos were used for this library. Oligo pools were amplified by PCR using 0.005 pmol total oligo pool as template (∼0.5 ng total DNA) and 15 total cycles to prevent PCR bias, then pools were purified. pHRec2 was used as template in a PCR reaction to create two backbones for each of the three pools, which was then DpnI digested and purified. Backbones are not otherwise pretreated before Golden Gate Assembly. Golden gate assembly was performed using ∼300 ng of relevant pool backbone and a 1x molar ratio for oligos (∼20 ng) and a small ∼100 bp fragment containing constant regions and a 20 bp randomized barcode (∼15 ng), combined with 2 μl 10x T4 DNA ligase buffer, 1 μl NEB Golden Gate Enzyme mix and dH2O to 20 ul. For the 3’ pool this is a 4 part assembly, for the 5’ pool this is a 3 part assembly. These reactions were cycled from 37°C to 16°C over 5 minutes, 60x, then held at 60°C for 5 minutes to complete Golden Gate assembly and held at 4°C overnight. The following day the reaction was held at 60°C for 5 minutes before dialysis in accordance with NEB guidance. Membrane drop dialysis was then performed on each library pool for 90 minutes to enhance transformation efficiency. The entire 20 μl reaction was then transformed into 50 μl competent E. coli 10G cells. Drop plates were made at this point (spotting 2.5 μl of dilutions of each library on Lb plates with spectinomycin) and total actual transformed cells were estimated at ∼5×105 CFU/mL for the 5’ pool at ∼1×106 for the 3’ pool. Each 1 mL pool was added to 4 mL Lb with spectinomycin and incubated overnight, then plasmids were purified. Plasmids concentration was determined by nanodrop and pools were then combined at an equimolar ratio to create the final phage variant pool, denoted as pHRec2-Motif. pHRec2-Motif was transformed into E. coli 10G with pHT7Helper1 by transforming 50 ng of plasmid into 60 ul of E. coli 10G competent cells. Drop plates were made (spotting 2.5 μl of dilutions of each library onto Lb plates with spectinomycin and kanamycin) and total actual transformed cells were estimated at ∼6.7×105 CFU/mL. The 1 mL library was added to 4 mL Lb with spectinomycin and kanamycin and incubated overnight. This was repeated once to ensure >100 coverage of the entire library and all preparations were combined. This host, E. coli 10G with pHT7Helper1 and pHRec1-Motif, was the host used for Optimized Recombination during ORACLE.
Phage Library Preparation Using ORACLE
T7 Acceptor phages from our previous DMS study for the RBP tip domain were used for ORACLE [23]. Optimized Recombination was performed by adding T7 Acceptor phages (MOI ∼1) to 50 mL exponential phase 10G with pHT7Helper1 and pHRec2-Motif. Cultures were incubated until lysis and phages were purified, titer was estimated by spot plate at ∼5×107 variants/mL in a total phage population of ∼1×1010 PFU/mL. Accumulation was performed by adding ∼MOI of 0.2 of recombined phages to 50 mL of stationary phase E. coli 10G with pHT7Helper1 and pHCas9-3 resuspended in fresh Lb with kanamycin and spectinomycin. Cultures were incubated until lysis then phages were purified. Phage titer was estimated to be ∼3×109 PFU/mL on stationary E. coli 10G with pHT7Helper1 and ∼1.2×109 on stationary E. coli 10G. Library expression was performed by adding the accumulated library to 5 mL E. coli 10G (with no plasmid) at an MOI of ∼2. Cultures are incubated until lysis and phages were purified. This constitutes the final phage variant library with full expression of the variant gp17 tail fibers. This phage population is directly sequenced to establish the pre-selection library population.
Motif Library Selection
All motif library selection experiments were performed in the same way. The T7 variant library was added to 5 mL of exponential host at an MOI of ∼2×10-2 and the culture was allowed to fully lyse. This MOI was chosen to give an estimated 2 replication cycles for the phage library. The entire process was repeated in biological triplicate for each host. Phages were then purified and sequenced.
Deep Sequencing Preparation and Analysis
We used deep sequencing to evaluate phage populations. We first amplified the tip domain by two step PCR, or tailed amplicon sequencing, using KAPA HiFi. Primers for deep sequencing attach to constant regions adjacent to the tip domain, 3’ of the new barcode in pHT7Rec2. Constant regions are also installed in the fixed region of the acceptor phages for the similarly size amplicon so acceptor phages can also be detected. The first PCR reaction adds an internal barcode (used for technical replicates to assay PCR skew), a variable N region (to assist with nucleotide diversity during deep sequencing, this is essential for DMS libraries due to low nucleotide diversity at each position), and the universal Illumina adapter. Four forward and four reverse primers were used in each reaction, each with a variable N count (0, 2, 4, or 8). Primers were mixed at equimolar ratios and total primers used was per recommended primer concentration. PCR was performed using 13 total cycles in the first PCR reaction. The product of this reaction was used directly in the second PCR reaction, which adds an index and the Illumina ‘stem’. This PCR was run for 9 total PCR cycles. The product of this reaction was purified and was used directly for deep sequencing. Each phage population was sampled at least twice using separate internal barcodes, and no PCR reactions were pooled. Total PCR cycles overall for each sample was kept at 22x to avoid PCR skew. All phage samples were deep sequenced using an Illumina Miseq System, 2×250 read length using MiSeq Reagent Kit v2 or v2 Nano according to manufacturer documentation.
Paired-end Illumina sequencing reads were merged with PEAR using the default software parameters [49]. Phred quality scores (Q scores) were used to compute the total number of expected errors (E) for each merged read and reads exceeding an Emax of 1 were removed [50]. Wildtype, acceptor phages, and each variant were then counted in the deep sequencing output. We correlated read counts for each technical replicate to determine if there was any notable skew from PCR or deep sequencing. Technical PCR replicates correlated extremely well (R≥0.98 for all samples) indicating no relevant PCR skew and were aggregated for each biological replicate. Besides wildtype and acceptor counts, we included only sequences with known motifs in our analysis, greatly reducing the possibility of deep sequencing error resulting in an incorrect read count for a variant. With this in mind, and to avoid missing low abundance members after selection, we used a low read cutoff of 4. A pseudo-count was not utilized. Possible outliers were identified by data sorting for UTI strains. A summary of deep sequencing data can be found in Supplementary File 3.
To score enrichment for each variant we used a basic functional score (F), averaging results of the three biological replicates where  . To compare variant performance across hosts we normalized functional score (FN) to wildtype, where
. To compare variant performance across hosts we normalized functional score (FN) to wildtype, where  .
.
Other Statistical Analysis and Source Code
Hierarchical Clustering was performed using the Complex Heatmaps package in R [51]. Different distance values were computed using get_dist in R, clustering approaches were calculated using hclust in R, and cophenetic scores were calculated using cophenetic in R. See documentation on github for exact code used. Frequency plots in Figure 3 and Figure 4 were made using SequenceLogoVis (https://github.com/ISA-tools/SequenceLogoVis) [52]. P-values for EOP graphs compare plaque capability on the tested host to the reference host for the EOP graph. Counting motif variants was done in Pandas. All other calculations and plots were made in Excel. Relevant statistical details of experiments can be found in the corresponding figure legends or relevant methods section.
Acknowledgements
We thank Dr. Rodney Welch for UTI strains and Dr. Douglas Weibel for BW25113 deletion mutant strains. This work was supported by National Institute of Allergy and Infectious Diseases (NIAID) grant R21AI156785 (to S.R.), by funding from the National Institute of General Medical Sciences of the National Institutes of Health grant R35GM143024 (to K.A) and by a Wisconsin Distinguished Graduate Fellowship Award from the University of Wisconsin-Madison and the William H. Peterson Fellowship Award from the University of Wisconsin-Madison Department of Bacteriology (to K.K.).
Footnotes
The only change was changing 10,000 to 10,073 in the abstract to specify the exact number of sequence motifs tested.
References




