

ABSTRACT
Dengue and Zika viral infections affect millions of people annually and can be complicated by shock and hemorrhage or neurological manifestations, respectively. Yet, a thorough understanding of the host response to these viruses is still lacking, in part because conventional approaches do not enable one to resolve heterogeneity in viral loads and cellular response between different individual cells. Here we present viscRNA-Seq (virus-inclusive single-cell RNA-Seq), an approach to probe the whole host transcriptome together with intracellular viral RNA at the single cell level that is applicable to both polyadenylated and non-polyadenylated viruses. We applied viscRNA-Seq to monitor dynamics of dengue and Zika virus infection in cultured cells and discovered a high heterogeneity of intracellular viral load which affects the virus-triggered host response. The single-cell distributions of both intracellular viral load and gene expression indicate that mean values measured in conventional bulk assays tend to over-represent highly infected cells. When these effects are accounted for, we discovered that several host factors show complex dynamics and a high degree of specificity for either virus. These host factors include both known and novel members of the endoplasmic reticulum translocon and proteins involved in signal peptide processing and intracellular membrane trafficking. We used loss-of-function and gain-of-function experiments to validate the viscRNA-Seq hits and discovered novel proviral and antiviral factors. Overall, viscRNA-Seq is a powerful approach to quantitatively assess the complex interplay between virus and host at the single cell level and at a genome-wide scale, and to elucidate the cellular pathways involved in viral infection.
INTRODUCTION
Flaviviruses, which include dengue (DENV) and Zika (ZIKV) viruses, infect several hundred million people annually and are associated with severe morbidity and mortality (Bhatt et al. 2013; Rasmussen et al. 2016; Guzman and Kouri 2003). Attempts to develop antiviral drugs that target viral proteins have been hampered in part by the high genetic diversity of these viruses. Since viruses usurp the cellular machinery at every stage of their life cycle, an alternative therapeutic strategy is to target host factors essential for viral replication (Bekerman and Einav 2015). To this end it is paramount to understand the interaction dynamics between viruses and the host, to identify pro- and antiviral host factors and to monitor their dynamics in the course of viral infection. The current model of flavivirus infection suggests that the virus enters its target cells via clathrin-mediated endocytosis, followed by RNA genome uncoating in the early endosomes and trafficking to ER-derived membranes for translation and viral RNA replication. Following assembly, viral particles are thought to bud into the ER lumen and are then released from the cell via the secretory pathway (Screaton et al. 2015). This pattern notwithstanding, an exhaustive list of host genes that interact, either directly or indirectly, with DENV or ZIKV is still lacking. Several high-throughput approaches have been applied to screen all 20,000 human genes for interactions with flaviviruses, including knockdown screens based on RNA interference (Sessions et al. 2009; Kwon et al. 2014; Le Sommer et al. 2012), knockout screens via haploid cell lines or CRISPR libraries (Marceau et al. 2016; Zhang et al. 2016), and bulk transcriptomics via microarrays or RNA-Seq (Sessions et al. 2013; Moreno-Altamirano et al. 2004; Fink et al. 2007; Conceição et al. 2010; Becerra et al. 2009; Liew and Chow 2006). While these approaches have provided important insights, our understanding of infection-triggered cellular responses is far from complete.
Knockdown, knockout, and population-level transcriptomics screens are extremely valuable tools but also share some limitations. First, because they are bulk assays, the heterogeneity of virus infection in single cells is obscured in the averaging process; differences in timing of virus entry and cell state across the culture and the fraction of uninfected cells are not accounted for. Second, because each population is a single data point and experiments cannot be repeated more than a handful of times, reproducibility and batch effects represent a challenge. Third, in knockout and knockdown screens the temporal aspect of infection is largely ignored, because successful knockdown can take days and recovery of the culture after infection in knockout screens lasts even longer. Fourth, because both knockdown and knockout can impair cellular viability and cannot probe essential genes, only a subset of genes can be probed by these techniques.
Here we report the development of viscRNA-Seq, an approach to sequence and quantify the whole transcriptome of single cells together with the viral RNA (vRNA) from the same cell. We applied this platform to DENV and ZIKV infections and investigated virus-host interactions in an unbiased, high-throughput manner, keeping information on cell-to-cell variability (i.e. cell state) and creating statistical power by the large number of single cell replicates while avoiding essential gene restrictions. By correlating gene expression with virus level in the same cell, we identified several cellular functions involved in flavivirus replication, including ER translocation, N-linked glycosylation and intracellular membrane trafficking. By comparing transcriptional dynamics in DENV versus ZIKV infected cells, we observed great variability in the specificity of these cellular factors for either virus, with a few genes including ID2 and HSPA5 playing opposite roles in the two infections. Using loss-of-function and gain-of-function screens we identified novel proviral (such as RPL31, TRAM1, and TMED2) and antiviral (ID2, CTNNB1) factors that are involved in mediating DENV infection. In summary, viscRNA-Seq sheds light on the temporal dynamics of virus-host interactions at the single cell level and represents an attractive platform for discovery of novel candidate targets for host-targeted antiviral strategies.
RESULTS
viscRNA-Seq recovers mRNA and viral RNA from single cells
To study the dynamical behaviour of human cells upon flavivirus infection, we developed an approach named viscRNA-Seq that captures and sequences the transcriptome of single cells (mRNA) together with the virus RNA (vRNA) from the same cell. This approach is modified from the commonly used Smart-seq2 for single cell RNA-Seq (Picelli et al. 2014). Briefly, single human cells are sorted into 384-well plates pre-filled with lysis buffer (Fig. 1C). In addition to ERCC (External RNA Controls Consortium) spike-in RNAs and the standard poly-T oligonucleotide (oligo-dT) that captures the host mRNA, the lysis buffer contains a DNA oligo that is reverse complementary to the positive-strand virus RNA (Fig. 1D). The addition of a virus-specific oligo overcomes limitations of other approaches and enables studying of viruses that are not polyadenylated (Russell, Trapnell, and Bloom 2017). Reverse transcription and template switching is then performed as in Smart-seq2, but with a 5’-blocked template-switching oligo (TSO) that greatly reduces the formation of artifact products (TSO concatemers). The cDNA is then amplified, quantified, and pre-screened for virus presence via a qPCR assay (Fig. 1E). Since many cells are not infected, this enables us to choose wells that contain both low and high vRNA levels and then to sequence their cDNA on an illumina NextSeq at a depth of ~ 400,000 reads per cell (Fig. 1F). This provides high coverage of transcriptome and allows high-quality quantitative detection of gene expression and intracellular viral load together with relatively large numbers of cells.

(A to F) Experimental design: (A) human hepatoma (Huh7) cells are infected with dengue or Zika virus at time 0 at multiplicity of infection (MOI) 0 (control), 1, or 10, then (B) harvested at different time points, (C) sorted and lysed into single wells. (D) Both mRNA and viral RNA (vRNA) are reverse transcribed and amplified from each cell, then (E) cells are screened for virus infection by qPCR. (F) Libraries are made and sequenced on an illumina NextSeq with a coverage of ~400,000 reads per cell. (G) The fraction of cells with more than 10 virus reads increases with MOI and time, saturating at 48 hours post infection. (H) Distributions of number of virus reads (left) and expression of an example stress response gene (right) inside single cells, showing the different dynamics of pathogen replication and host response. Whereas virus content can increase 1,000 fold and shows no saturation, expression of DDIT3/CHOP saturates after a 10 fold increase.
We applied viscRNA-Seq to an infection time course in cultured cells. We infected human hepatoma (Huh7) cells with DENV (serotype 2, strain 16681) at multiplicity of infection (MOI) of 1 and 10. In a separate experiment, Huh7 cells were infected with ZIKV (Puerto Rico strain, PRVABC59) at an MOI of 1. In both cases, uninfected cells from the same culture were used as controls (Fig. 1A). At four different times after infection – 4, 12, 24, and 48 hours – cells were harvested, sorted, and processed with viscRNA-Seq (Fig. 1B). Recovery of the ERCC spike-ins and number of expressed genes per cell confirmed that the libraries had high quality (Supplementary Fig. S1A-B). From each experimental condition, 380 cells were screened for virus ~100 of those were sequenced. In total ~7500 single cells were screened and ~2100 were sequenced (see Supplementary Table S1).
Intracellular viral load and gene expression are heterogeneous across cells
First, we focused on infection by DENV. As expected, qPCR measured an increase over time in the fraction of infected cells with both MOIs (Fig. 1G). Whereas most genes were rather homogeneously expressed, both intracellular viral load (number of vRNA reads per million transcripts) and expression of a subset of genes varied widely both across experimental conditions and cells from the same condition (Fig. 1H). Overall, between zero and a quarter of all reads from each cell is vRNA-derived, hence the dynamic range for intracellular viral load is extremely wide (5 decades). The distribution of both intracellular viral load and gene expression are rather symmetric in logarithmic space (Fig. 1H); as a consequence, mean expression as measured in a bulk assay is higher than the median and over-represents highly infected cells. The high coverage sequencing enables a quantitative measurement of the variation in the expression level of thousands of genes in each cell (Supplementary Fig. S1B). As a next step, we aimed at identifying which elements of this variation are induced by the infection.
Correlation between intracellular viral load and gene expression within single cells tracks infection-triggered host response
In a bulk assay each of the experimental conditions would be an average of all cells, making it difficult to extract clear statistical patterns. Leveraging both single-cell resolution and high throughput, we directly computed Spearman’s rank correlation coefficient between each gene expression and intracellular viral load across all cells. This metric does not require an explicit noise model for either expression or virus load and is therefore insensitive to outlier cells. As expected, most genes do not correlate with vRNA level and the distribution of their correlation coefficients decays rapidly away from zero (Fig. 2A). In panels 2B-D examples of strong anticorrelation, strong correlation, and absence of correlation are shown. Both the level of vRNA at which each gene starts to correlate and the slope of the response vary across genes and may reflect different infection stages (see below). Genes with extreme correlation consistently represent specific cellular functions. Most of the top correlated genes (Fig. 2A right inset) are involved in the ER unfolded protein response (UPR) (see e.g. DDIT3 in Fig. 3C), consistent with ER stress response triggered by flavivirus translation and RNA replication on ER-derived membranes (Medigeshi et al. 2007). Numerous strongly anticorrelated genes (Fig. 2A left inset) are components of actin and microtubules, indicating cytoskeleton breakdown (as an example, see ACTB in Fig. 2B). Molecular chaperones are found in both categories suggesting a more nuanced regulation.

(A) Distribution of Spearman correlation coefficients between dengue vRNA and mRNA from the same cell across all human genes. The insets list the top correlated (right) and anticorrelated (left) genes. Response to ER stress and apoptosis is activated as infection proceeds, whereas actin and microtubules pathways are downregulated. (B-E) Examples of correlation patterns observed across the transcriptome, as a scatter plot of gene expression versus vRNA content. Each dot is a single cell and the green shades indicate the density of cells. (B) anticorrelation at high vRNA content, (C) correlation at medium to high vRNA content, (D) no correlation, and (E) time-dependent correlation dynamics. (F) Expression versus vRNA content for gene COPE, as shown in panel E but splitting cells by time after infection. Correlation at each time is shown in the top left corner of each plot, and switches from strongly negative to strongly positive as infection proceeds. (G) Correlation between expression and dengue vRNA content switches from negative to positive (< −0.3 to > +0.3) for 6 genes (left panel) and in the opposite direction for 11 genes (right panel), highlighting potential multiple roles of these genes during dengue virus infection.

(A) Correlation between gene expression and vRNA during Dengue virus versus Zika virus infection. Each dot is a gene and the contour lines indicate the an estimate of the density of genes. Most genes do not correlate with either virus, but some genes correlate strongly with different degrees of virus specificities. Only cells with 500 or more virus reads per million transcripts are used for this analysis (see main text). (B-E) Examples of genes with different behaviour across the two viruses, as a scatter plot of gene expression versus vRNA content. Each dot is a single cell. Dengue plots are indicated by a D, Zika plots by a Z in the top left corner.
To understand whether correlated genes may represent pathways that are important for virus infection, we focused on the 1% most correlated subset of the transcriptome (correlation in excess of 0.3 in absolute value) and performed Gene Ontology (GO) enrichment analysis using the online service PANTHER (Mi et al. 2017). This statistical analysis confirms the qualitative picture emerging from the top correlates. At 4 hours post-infection upregulation of genes involved in translation and suppression of mRNA processing is demonstrated. At 48 hours post-infection there is upregulation of UPR, protein degradation via ERAD, and ER-to-Golgi anterograde transport via COPII-coated vesicles, and downregulation of cell cycle and cytoskeleton organization (supplementary tables ST1-ST4).
Several genes switch role during dengue infection
Naturally, cells that are infected for longer tend to harbor more vRNA. To disentangle the effect of time since infection from the vRNA level within each cell, we computed the same correlation coefficient within single time points. We discovered that most correlated genes exhibit either positive or negative correlation, but not both. This behaviour is expected for generic stress response genes; the sign of the differential expression is a hardwired component of their physiological function. However, a group of 17 “time-switcher” genes show both an anticorrelation of less than −0.3 and a correlation in excess of +0.3 at different time points post-infection, suggesting a more specific interaction with DENV. Of these, 6 genes transition from anticorrelation to correlation (e.g. COPE, Fig. 2E-F), 10 show the opposite trend, and a single gene (PFN1) follows a nonmonotonic pattern (Fig. 2G). Since 4 and not 2 time points were sampled, a consistent increase (or decrease) in correlation likely stems from a biological change rather than a technical noise. Of the six genes which switch from anti correlated to correlated, RPN1 and HM13 localize to the ER. RPN1 is a non-catalytic member of the oligosaccharide transfer (OST) complex, which is required for N-linked glycosylation of some ER proteins, whereas HM13 is a protease that cleaves the signal peptide after translocation into the ER. Both of these factors have been shown to be essential for DENV infection (Marceau et al. 2016). SQSTM1 is a scaffold protein involved in selective autophagy of polyubiquitinated substrates and has been shown to have a bimodal behaviour in DENV infection (Metz et al. 2015), whereas UBC is a major source of ubiquitin. Lastly, GORASP2 and COPE play a role in Golgi assembly and/or membrane trafficking. In particular COPE is a subunit of the coatomer complex (COPI) that mediates both intra-Golgi and Golgi-to-ER retrograde vesicle transport; another subunit of this complex, COPB1, has recently been shown to be essential for DENV (Iglesias et al. 2015). Interestingly, COPE also appears to be downregulated during early infection in “bystander” cells; i.e. cells that originate from an infected culture but are themselves not infected (Supplementary Fig. S4).
Host response differs for DENV and ZIKV
Next we sought to address the question of which elements of the host response are common between DENV and ZIKV, and therefore potentially common with other evolutionarily related viruses as well. To do so, we replicated the time course experiment with ZIKV at MOI 0 (control) and 1. Although Huh7 cells were also infected at an MOI of 10, cell death precluded sorting. Fig. 3A shows the correlations between gene expression (each dot is a human gene) and vRNA for both experiments and represents the two-dimensional equivalent of Fig. 2A. We discovered that the majority of genes are not correlated with either virus (contour lines indicate density of genes). Nevertheless, a clear pattern along the positive diagonal emerged with genes in this region, such as ATF3 (Fig. 3C) and ACTG1 (Fig. 3D), demonstrating a similar behaviour upon infection with either virus. A minority of genes are scattered away from the diagonal, indicating discordant behaviour between DENV or ZIKV infection. For instance, ID2 expression decreses at high DENV level but increases at high ZIKV RNA level (Fig. 3B), while the opposite trend is observed with the chaperone HSPA5 (Fig. 3E and see below). A number of genes at the outskirts of the correlation plot are labeled and highlighted in red as they exhibit potentially interesting expression patterns upon infection: i.e. either an extremely strong correlation with both viruses or a high degree of virus specificity. These outliers include two subunits of the SEC61 complex (B, and G), several subunits of the TRAP complex and the OST, previously shown to be essential for DENV and/or WNV infection (Marceau et al. 2016; Zhang et al. 2016), and other genes that may be relevant to infection with either virus. To understand how these correlated genes share the heterogeneity of infected cells, we selected all genes with a correlation coefficient above 0.4 or below −0.4 and performed t-SNE dimensionality reduction (Maaten and Hinton 2008), coloring each cell by its intracellular viral load (Fig. 4A, left) or by time post-infection (right). Although uninfected cells form a mixed, heterogeneous cloud, infection pushes cells into more stereotypic states that are distinct for DENV and ZIKV infection (black arrows indicate average positions for cells at increasing intracellular viral load). Plotting gene expression dynamics on top of these visualizations enables to connect single cellular pathways to the global changes in cell state defined by virus infection (Fig. 4B). Remarkably, the temporal behaviour of a few genes is inconsistent with the global transcriptomics shifts: for instance the expression of HSPA5 increases until 24 hours after ZIKV infection, but is then sharply decreased at 48 hours post-infection and with higher intracellular viral load. To compare the temporal dynamics of gene expression during DENV and ZIKV infection, we identified “time switchers” for Zika infection (Fig. 4C). Although the number of genes that show both correlation and anti correlation is similar between the two viruses, the 11 Zika time switchers exhibit no correlation at 4 hours post-infection, followed by a non-monotonic behaviour as time passes. HSPA5 is included in this list, in agreement with its t-SNE visualization; this gene is therefore not only subject to opposite regulation in DENV versus ZIKV infection, but may play different roles at different times during the same infection. Among the other temporally regulated genes in ZIKV infection, is the circadian clock gene PER2 that resembles HSPA5 (Moni and Lio’ 2017)

(A) t-SNE dimensionality reduction using all genes that correlate with at least one virus (< −0.4 or > 0.4). Each dot is a cell and is colored by intracellular viral load (left panel) and time post-infection (right panel). Colors are shades of red for the dengue experiment, shades of blue for the Zika one. Arrows in the left panel indicate the average position of cells at increasing intracellular viral loads. (B) Expression of four example genes as in Fig. 3B-E on top of the t-SNE visualization. (C) Correlation between expression and Zika vRNA content switches from negative to positive (< −0.3 to > +0.3) for 1 gene (left panel) and in the opposite direction for 10 genes (right panel). Unlike in dengue virus infection (Fig. 2G), the temporal traces of Zika infection do not show a simple increase or decrease but rather complex dynamics.
Validation of proviral and antiviral host factors
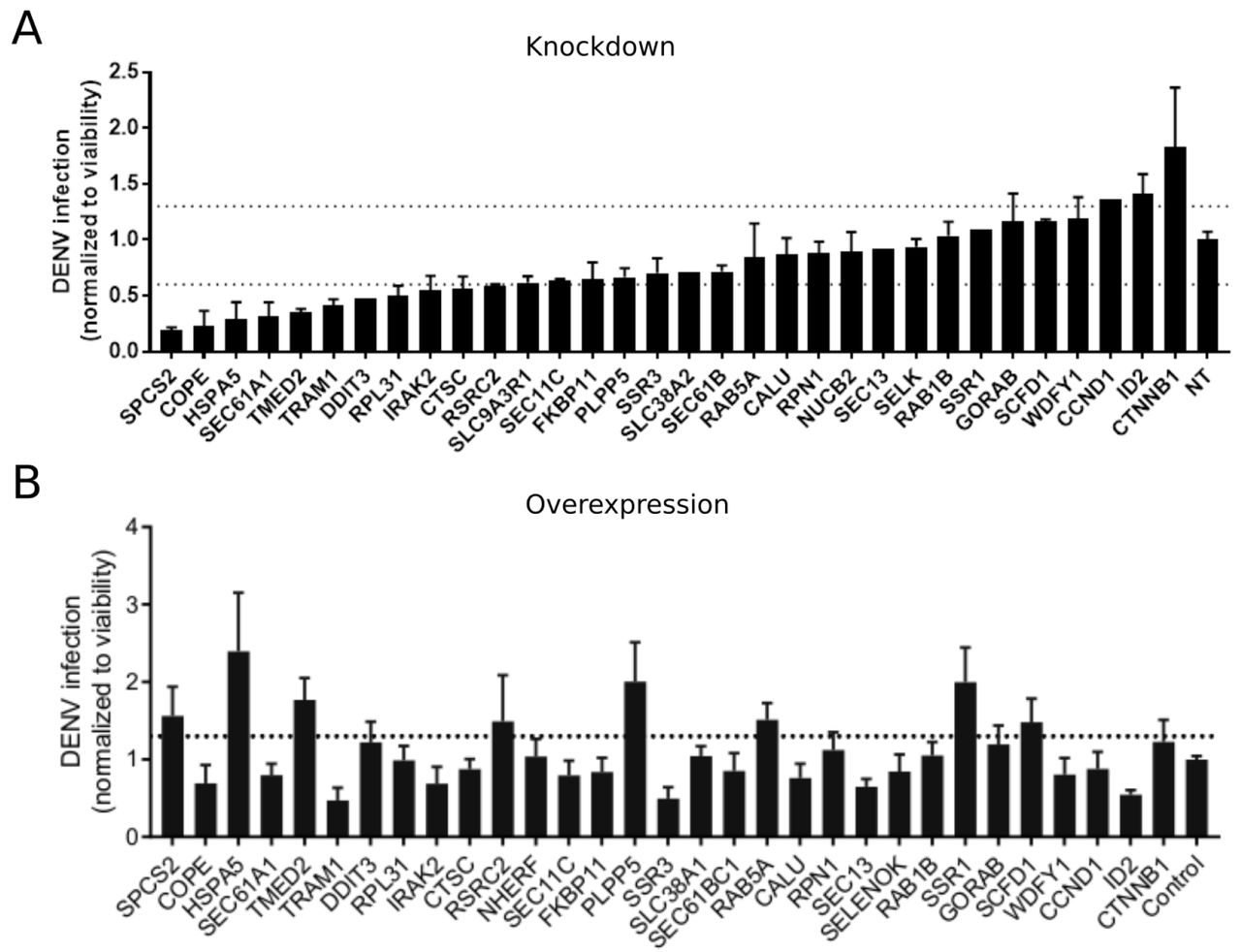
To probe the functional relevance of genes demonstrating interesting correlations with DENV viral load, we first conducted loss-of-function screens. We measured the effects of siRNA-mediated depletion of 32 individual genes in Huh7 cells on DENV infection as well as on cellular viability (Fig. 5A and Supplementary Fig. S5A). Using a cutoff of greater than 40% inhibition of viral infection as measured by luciferase assays normalized to cell viability in two independent screens, we identified multiple host factors essential for viral infection. These include a few components of the translocon previously shown to be essential for DENV: HM13 (Marceau et al. 2016) or WNV: SPCS2 (Zhang et al. 2016) as well as two novel components of the ER translocon: RPL31 and TRAM1 (Ng, Oresic, and Tortorella 2010). Depletion of two proteins involved in membrane trafficking, TMED2 (secretory pathway) and COPE (retrograde, Golgi to ER) as well as the ER-resident chaperone and ERAD protein HSPA5 and the multifunctional transcription factor in ER stress, DDIT3 also reduced DENV infection.

DENV infection relative to Non Targeting (NT) following siRNA-mediated knockdown (A) or overexpression of the indicated host factors (B) measured by luciferase assays at 48 hours post-infection and normalized to cell viability. Both data sets are pooled from 2 independent experiments each with triplicates. The dotted lines represent the cutoffs for positivity. Cellular viability measurements are shown in Supplementary Fig. S5.
In contrast, siRNA-mediated depletion of two genes that anticorrelate with intracellular virus load, ID2 and CTTNB1 (β-catenin), increased DENV infection, indicating that these proteins function as antiviral restriction factors, as previously reported in HIV (Kumar et al. 2008). Notably, ID2 and CTTNB1 are known interacting partners (Rockman et al. 2001), which may be acting via the interferon I pathway (Hillesheim et al. 2014). Suppression of another subset of overexpressed or underexpressed genes demonstrated no effect on DENV infection, suggesting that they were either non-essential or not restricting (possibly due to redundancy in host factors requirement) or that their siRNA-mediated depletion was ineffective.
To determine whether host factors found to be proviral are also rate limiting for infection, next we conducted a gain-of-function screen. Huh7 cells ectopically expressing 30 of the 32 individual gene products were infected with DENV. Using a cutoff of greater than 30% increase in viral infection normalized to cell viability in two independent screens, we identified HSPA5, TMED2, SPCS2, and DDIT3 as factors whose overexpression increased DENV infection (Fig. 5B and Supplementary Fig. S5B), indicating rate limitation associated with these important proviral factors. In contrast, ectopic expression of other proviral factors, such as COPE and TRAM1, decreased DENV infection, suggesting that DENV might be evolutionarily optimized for the natural expression level of these genes.
DISCUSSION
We have developed a new approach, designated viscRNA-Seq, to simultaneously quantify the whole transcriptome and intracellular viral load at the single cell level. This approach probes the natural gene expression dynamics of virus infections and is therefore complementary to knockout and knockdown genetic screens, which induce a controlled perturbation (Marceau et al. 2016; Zhang et al. 2016; Sessions et al. 2009; Kwon et al. 2014; Le Sommer et al. 2012). However, unlike those loss-of-function assays, viscRNA-Seq is able to fully discern cell-to-cell variation within a single experimental condition, is compatible with time-resolved sampling, and can be used to study essential genes. Although similar in spirit to a recent droplet-based approach that uses only a polyT capture oligonucleotide (Russell, Trapnell, and Bloom 2017), our approach can be easily adapted to any RNA virus, whether polyadenylated or not, by swapping one single oligonucleotide. Moreover, since RNA capture is highly efficient compared to droplet-based methods, an accurate quantification of both gene expression and viral RNA (vRNA) can be obtained with as little as 400,000 sequencing reads per cell. Since full-length transcripts are recovered as in the original Smart-seq2 (Picelli et al. 2014) and unlike in droplet-based protocols, viscRNA-Seq can be combined with enrichment PCRs before sequencing to focus on specific host or viral factors at a fraction of the sequencing cost.
We have applied this high-throughput technique to study the temporal infection dynamics of DENV and ZIKV, two major global health threats (Bhatt et al. 2013). Our first finding is that although the number of infected cells in the culture gradually increases with time as expected, there is a large heterogeneity across cells from the same Petri dish. Since flavivirus replication is not synchronized, such heterogeneity might reflect host-responses at different stages of viral life cycle. The single-cell distributions of both intracellular viral load and gene expression indicate that mean values measured via bulk assays tend to over-represent highly infected cells. Moreover, bulk transcriptomics studies cannot account for uninfected cells and are therefore limited to high MOI (Sessions et al. 2013); in contrast, we are able to study both high-MOl and low-MOl cultures equally well and to separate the effect of MOI from the actual infection state of each cell.
We then leveraged the statistical power of sequencing thousands of cells to correlate intracellular viral load with gene expression across the whole human transcriptome. The genes with the strongest positive correlation with both viruses are members of the unfolded protein response (UPR), particularly the PERK branch, including DDIT3, ATF3, and TRIB3. The strongest negative correlates with both viruses are components of the actin and microtubule networks (e.g. ACTB, ACTG1, TUBB1) as well as members of nucleotide biosynthesis, suggesting a disruption of both cytoskeleton and cellular metabolism. The URP response starts abruptly once 2,000 virus transcripts are present per million of total transcripts (i.e. when virus RNA comprises only 0.2% of the cellular mRNA); a threshold that is reached in most cells between 24 and 48 hours post-infection. Downregulation of cytoskeleton and metabolism, however, starts only at 10,000 virus transcripts per million of total transcripts; this higher threshold is reached in most cells at 48 hours post-infection. This delayed response may happen either because of direct cytopathic effects or as a consequence of the earlier UPR response. Interestingly, a recent transcriptomics study also found ER stress pathways to be differentially regulated during DENV infection (Sessions et al. 2013). However, because thousands of host genes were classified as differentially expressed in that study, this overlap may be in part coincidental due to the sheer number of “hits” reported by those authors. Indeed, the more quantitative statistics resulting from the large number of single cell replicates as opposed to bulk transcriptomics was a key factor that enabled us to narrow down the list of potentially relevant genes to a small number that could be subsequently validated.
A number of host genes correlate strongly with one virus but correlate less or do not correlate with levels of the other virus. Examples include subunits of several complexes involved in ER translocation and N-linked glycosylation (Supplementary Fig. S3): SEC61G, a subunit of the translocon; SSR3, a member of the TRAP complex; and OSTC, a subunit of the OST. Components of these three complexes were identified as essential host factors for DENV replication in a recent CRISPR-based knockout screen (Marceau et al. 2016). SEC11C, a subunit of the signal peptidase complex (SPCS), also behaves in this way, in agreement with the prior finding that this complex is essential for flavivirus infection (Zhang et al. 2016). Strikingly, we do not observe a dominant enrichment of interferon-related genes among the most strongly upregulated during flavivirus infection (Fink et al. 2007). This results may be caused by virus-induced blocking of the interferon-induced signaling cascade (Muñoz-Jordán et al. 2003); moreover, Huh7 cells are known to react more mildly than other culture systems to interferon stimulation (Guo, Zhu, and Seeger 2003).
The expression of some host genes shows discordant correlation with DENV and ZIKV infection. Among the genes that are overexpressed during DENV infection but underexpressed during ZIKV infection are the molecular chaperone HSPA5 which has been shown to interact directly with the dengue E protein in liver cells (Jindadamrongwech, Thepparit, and Smith 2004), other members of the translocation machinery (SEC61B) or the TRAP complex (SSR1, SSR2), and ATF4, an ER-stress induced gene that interacts with DDIT3 and TRIB3. On the opposite end of the spectrum, genes that are underexpressed during DENV infection and overexpressed during ZIKV infection include the transcriptional regulator ID2. Both ID2 and cyclin D1 (CCND1), which is also strongly anticorrelated with DENV load, have been reported to be a target of β-catenin (Rockman et al. 2001; Shtutman et al. 1999).
A few host genes (17 for DENV, 11 for ZIKV) show a complex dependence on time and intracellular viral load; at early time points, gene expression correlates positively (or negatively) with viral load, but this behaviour is reversed at later time points. Among these genes are HM13, COPE, and SQSTM1 for DENV and HSPA5 for ZIKV. We speculate that these genes may play multiple roles during the virus replication cycle, acting as antiviral factors during certain phases of infection (e.g. cell entry) and as proviral during others (e.g. virion release). These genes may also represent virus triggered host-responses that were counteracted by viral proteins. Of these interesting hits, HM13 or signal peptide peptidase is involved in processing of signalling peptides after ER membrane translocation, a pathway that has been reported to be critical for several flaviviruses including dengue (Zhang et al. 2016). Furthermore SQSTM1, which is involved in selective autophagy, has been reported to affect DENV infection in a time-dependent manner, in agreement with our results (Metz et al. 2015), and to interact with the unrelated Chikunguya virus (Judith et al. 2013).
From the viscRNA-Seq screen we selected 32 candidate genes to determine whether they may play proviral or antiviral roles during DENV infection. The three genes HSPA5, SPCS2, and TMED2 showed clear proviral effects, reducing DENV replication upon knockdown and increasing it when ectopically expressed. The first two are known essential factors of DENV infection (Jindadamrongwech, Thepparit, and Smith 2004; Zhang et al. 2016), whereas TMED2, which is involved in coatomer complex (COPI) vesicle-mediated retrograde trafficking and trafficking from the golgi to the plasma membrane (Fiedler et al. 1996; Goldberg 2000), has not been reported before. Further in-depth studies are warranted to elucidate the role of this host factor in flavivirus infection. Unlike these proviral factors, the hits SSR3, COPE, and TRAM1 reduced viral replication under both knockdown and ectopic expression, although to different degrees depending on the direction of the perturbation. This suggests that DENV might be evolutionarily adapted to intermediate, wild type expression levels of these genes. Furthermore, it is striking that both TMED2 and COPE are involved in COPI-coated vesicle transport but produce opposite outcomes on DENV infection when ectopically expressed. Taken together with the time-switching correlation of COPE with intracellular DENV load, this result suggests a dual role for coatomer-coated vesicles during replication.
Overall, our study highlights the potential of single-cell level, high-throughput analyses to elucidate the interactions of human viruses with host cellular processes (Russell, Trapnell, and Bloom 2017). Combining temporal information, cell-to-cell variability, cross-virus comparison and high-quality expression data has allowed us to identify pathways that react similarly to infection by dengue and Zika viruses, such as the unfolded protein response, and others that are more virus-specific. Furthermore, our findings reveal two proteins involved in ER translocation as novel host factors essential for DENV infection. Lastly, these results indicate that coatomer-coated vesicle trafficking shows both complex temporal behaviour and includes a novel proviral factor, TMED2.
COMPETING INTERESTS
The authors declare no conflict of interests.
ACKNOWLEDGEMENTS
This work was supported by award 1U19 AMI10966201 from the National Institute of Allergy and Infectious Diseases (NIAID) to SE, 5T32AI007502 to EB, and grants from Stanford Bio-X and Stanford Institute for Immunity, Transplantation, and Infection. FZ is supported by a long-term EMBO fellowship (ALTF 269-2016). SYP was supported by the Child Health Research Institute, Lucile Packard Foundation for Children’s Health, as well as the Stanford Clinical and Translational Science Award (CTSA, grant UL1 TR000093).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}