

Abstract
Congenital diaphragmatic hernia (CDH) is one of the most common and lethal birth defects. Previous studies using exome sequencing support a significant contribution of coding de novo variants in complex CDH cases with additional anomalies and likely gene-disrupting (LGD) variants in isolated CDH cases. To further investigate the genetic architecture of CDH, we performed exome or genome sequencing in 283 proband-parent trios. Combined with data from previous studies, we analyzed a total of 357 trios, including 148 complex and 209 isolated cases. Complex and isolated cases both have a significant burden of deleterious de novo coding variants (1.7~fold, p= 1.2×10−5 for complex, 1.5~fold, p= 9.0×10−5 for isolated). Strikingly, in isolated CDH, almost all of the burden is carried by female cases (2.1~fold, p=0.004 for likely gene disrupting and 1.8~fold, p= 0.0008 for damaging missense variants); whereas in complex CDH, the burden is similar in females and males. Additionally, de novo LGD variants in complex cases are mostly enriched in genes highly expressed in developing diaphragm, but distributed in genes with a broad range of expression levels in isolated cases. Finally, we identified a new candidate risk gene MYRF (4 de novo variants, p-value=2×10−10), a transcription factor intolerant of mutations. Patients with MYRF mutations have additional anomalies including congenital heart disease and genitourinary defects, likely representing a novel syndrome.
1 Introduction
Congenital diaphragmatic hernia (CDH) is an anatomical defect of the diaphragm that leads to the protrusion of abdominal viscera into the thoracic cavity, compressing the lungs in utero and resulting in lung hypoplasia. CDH affects approximately 1 in 3000 live births and is often lethal1; 2. It can be isolated (50–60%) or associated with other birth defects and neurodevelopmental disorders 3; 4. Among these, cardiovascular malformations are the most common (~35% of CDH patients)5. Lung hypoplasia and the associated pulmonary hypertension are the main cause of the mortality and morbidity of CDH. Despite greatly improved survival rate with neonatal and surgical interventions, the overall mortality remains at ~30% 6–8.
The diaphragm develops between the fourth and tenth weeks of human gestation and in mice between embryonic day (E) 10.5 and E15.59. Both environmental and genetic factors have been implicated. The mesenchymal-derived pleuroperitoneal folds (PPFs) play a key role in diaphragm development, and mutations in PPF-derived muscle connective tissue fibroblasts can result in CDH 10. Most genes implicated in CDH have been identified through recurrent chromosomal anomalies and mutant mice11–17. The etiology is unclear for most CDH patients. The historical low reproductive fitness of CDH has limited the number of familial cases for genetic analysis. We and others have reported an enrichment of de novo deleterious genetic events in sporadic CDH patients18–20, especially LGD (likely gene disrupting) variants in complex cases.
To identify novel risk genes and compare the genetic architecture of complex and isolated cases, we performed whole exome sequencing (WES) in 79 proband-parent trios and whole genome sequencing (WGS) in 192 trios. Combined with previously published cases18; 19, we analyzed a total of 357 trios (Supplementary Table 1), including 148 complex and 209 isolated cases. We observed that there are different contribution from de novo variants in female and male CDH cases, and genes implicated by LGD variants in complex and isolated CDH cases have distinct expression patterns in early diaphragm development. Finally, we identified MYRF as a new candidate risk gene with de novo variants in four complex CDH patients.
Material and Methods
Patients
A total of 357 CDH patients and their unaffected parents were recruited for analysis in this study, including 74 trios from Boston Children’s Hospital (BCH) and Massachusetts General Hospital (MGH)18 (Boston Cohort) and 39 trios from a previous study 19 (Supplementary Table 1). Two hundred and eighty-three trios were recruited as part of the DHREAMS (Diaphragmatic Hernia Research & Exploration; Advancing Molecular Science) study (http://www.cdhgenetics.com/)20. Neonates, children and fetal cases with a diagnosis of diaphragm defects were eligible for DHREAMS. Clinical data were abstracted from the medical chart by study personnel at each of 16 clinical sites. Data on prenatal history, neonatal outcome, and longitudinal follow-up data including Bayley III and Vineland II developmental assessments and a parent interview about the patient's health since discharge at 2 years of age and/or 5 years of age were gathered in our birth cohort. A complete family history of diaphragm defects and major malformations was collected on all patients by a single genetic counsellor, and no patients had a family history of CDH.
Patients without additional birth defects or neurodevelopmental disorder (NDD) at last contact were classified as isolated, and patients with the additional birth defects or NDD were classified as non-isolated (Details previously published19; 20). The diaphragm lesion was classified as left, right, bilateral or central. Pulmonary hypoplasia, cardiac displacement and intestinal herniation were considered to be part of the diaphragm defect sequence and were not considered to be an additional malformation. Subjects from BCH and MGH were described previously18. A blood, saliva, and/or skin/diaphragm tissue sample was collected from the affected patient and both parents. All participants provided informed consent/assent for participation in this study, which was approved by the institutional review boards of each participate study site.
Whole exome and whole genome sequencing
We included previously two sets of WES data for analysis18; 19. We performed whole exome sequencing (WES) at the University of Washington in 79 additional trios using genomic DNA largely from whole blood (73 trios, 93.4%), with a minority from saliva or tissues. DNA was processed with the Nimblegen SeqCap EZ Exome V2 exome capture reagent (Roche) and TruSeq DNA Sample Prep Kits (Illumina). Samples were multiplexed and sequenced with paired-end 75bp reads on Illumina HiSeq 2500 platform according to the manufacturer’s instructions (Illumina, Inc, San Diego, California, USA).
We sequenced another 192 trios at Baylor College of Medicine using whole genome sequencing (WGS) as part of NIH Gabriella Miller Kids First Pediatric Research Program. Among these, 27 trios that had no damaging de novo variants in previously published WES data were selected as “WES-negative” cases for WGS19. Genomic libraries were prepared by the Illumina TruSeq DNA PCR-Free Library Prep Kit. DNA was sheared into 350-bp average length using sonication on a Covaris LE220 instrument. The fragmented DNA was end-repaired, A-tailed and indexed using TruSeq Illumina adapters with overhang-T added to the DNA. The libraries were validated on a Bioanalyzer DNA High Sensitivity chip by size and quality, then pooled in equal quantities and sequenced as paired-end reads of 150-bp lengths on an Illumina HiSeq X platform.
Alignment and quality controls
Mapping, alignment, and variant calling were done according to the Broad Institute’s best practices using Burrows-Wheeler Aligner (bwa-mem, version 0.7.10)21 and Genome Analysis Toolkit (GATK;version3.3) (https://software.broadinstitute.org/gatk/best-practices/). Briefly, we mapped WES or reads to the reference genome (build GRCh37) using BWA-mem 22, mark PCR duplicates using Picard (v1.67), performed local realignment and quality recalibration using GATK 23. We jointly called variants in all WES samples using the GATK HaplotypeCaller. The output file was generated in the universal variant call format (VCF). We used the same procedure to analyze WGS samples.
Among new samples sequenced by WES, the mean depth of coverage is 59± 21 with 93±2.5% bases read with at least 15x in target regions. Among new samples sequenced by WGS, the mean depth of coverage is 39±2, with 99±0.25% bases read at least 15x (Supplementary Fig. 1).
We performed principal component analysis of common variants (allele frequency >5%) using Eigenstrat 24 to determine the population structure and ancestry of both cases and controls, with HapMap 3 sample collection data 25 as a reference.
Detection of de novo SNVs and indels
We used Plink26 (http://pngu.mgh.harvard.edu/purcell/plink/) to estimate Identity by Descent (IBD)27 to confirm the relatedness among familial trios. All trios were matched to parents-offspring with relatedness.
A variant that presents as a heterozygous genotype in the offspring and homozygous reference genotypes in both parents was considered to be a potential de novo variant. We used an established stringent filtering method to identify de novo variants as described previously 19; 28; 29. Briefly, we required the candidate variants have depth (minimum 5 alternate allele reads), alternate allele fraction (minimum 20%), Fisher Strand (FS) (maximum 25), Quality by depth (QD) (minimum 2), Phread-scaled genotype likelihood (PL) (minimum 60), population allele frequency(maximum 0.1% in ExAC), and parental read characteristics (minimum depth of 10 reference reads; alternate allele fraction less than 5%, minimum GQ of 30). Additionally, variants located in segmental duplication regions (maximum score 0.98) were excluded. All candidate de novo variants were manually inspected in the Integrated Genomics Viewer (IGV, http://software.broadinstitute.org/software/igv/). In addition, we validated all the de novo likely gene disrupting (LGD) (including frameshift, nonsense and splicing site) variants by dideoxynucleotide sequencing. Of 40 case variants that were submitted for validation by Sanger sequencing, all 40 were confirmed (precision =100%).
Among the 27 “WES-negative” cases, there were 12 de novo variants identified by WGS that were not detected by WES 19.
Annotation of variants
We used ANNOVAR30 to annotate variants and aggregate allele frequency and in silico functional predictions, then used average allele frequency in Exome Aggregation Consortium (ExAC) data to define rare variants (frequency < 1e-4). Rare de novo variants were classified as silent, missense, and likely-gene-disrupting (“LGD”, which includes stopgain, stoploss, canonical splicing site, or frameshift variants). In-frame insertions or deletions were not considered in the genetic analysis. We defined deleterious missense variants (“D-mis”) by CADD31 phred-scale score ≥25.
Statistical analysis
We performed statistical analyses using R package from the Comprehensive R Archive Network, and the denovolyzerR 32 package.
Global or gene set burden between case and mutation background rate
We calibrated the expected number of de novo variants in patients in each variant class in each gene based on the 3-nucleotide context-specific mutation rate estimated by Samocha et al.29; 32.
We used Poisson test to assess the significance of excess of observed de novo variants over expectation which was defined as enrichment rate (r). The positive predictive value (PPV) for de novo variants in each class was calculated as (r−1)/r. The Estimated number of true risk variants in each class is the number of observed variants (m) in cases multiplied by PPV: m * (r−1)/ r. The most severe predicted functional effect variants (LGD and D-mis) were used in further burden analyses based on the different phenotype, gender, gene set, and expression data.
Percent of CDH attributable to de novo variants
We calculated the percent of CDH patients with pathogenic variants in isolated and complex CDH groups, in male and female case groups, respectively. The fraction of individuals carrying at least one damaging de novo variant was determined, by subtracting the expected rate of damaging de novo variants per individual.
The formula is as follows:

where n1 is the total number of sub-group CDH patients with at least one de novo deleterious variant, r is the expected rate per healthy individual with at least one de novo deleterious variant, where the rate was estimated by 10,000 simulations of Poisson distribution of variants per person, and s1 is the total number of sub-group CDH patients.
Expression profile during diaphragm development
Mouse developing diaphragm (MDD) gene expression datasets from the pleuroperitoneal folds (PPFs)33 at embryonic day 11.5 (E11.5) were used in this study. High diaphragm expression is defined as the top quartile of probe sets based on RMA (Robust Multi-Array Average)-normalized expression levels of microarray data19.
Single genes with multiple de novo mutations
For MYRF, the number of observed deleterious de novo mutations was compared to the expected deleterious mutation background using a Poisson test. The p-value passed Bonferroni correction with all protein-coding genes annotated in CCDS34.
Results
Clinical data of the cohort
Patients were recruited from the multicenter, longitudinal DHREAMS study 35 and from the Boston Children’s Hospital/Massachusetts General Hospital. In the combined cohort, there were 210 (59%) male and 147 (41%) female CDH patients. The gender distribution with increase male prevalence (1.4:1) is consistent with published retrospective and prospective studies 36; 37. Among the 148 complex cases, the most frequent anomalies were congenital heart disease (41%), but neurodevelopmental delay, gastrointestinal, and other malformations were common (Table 1 and Supplementary Table 2). A total of 209 (59%) patients had isolated CDH without additional anomalies at last contact20. In the DHREAMS cohort (Online Methods) of 283 patients, 229 were part of the neonatal cohort (with 56% males), of which 152 had formal neurodevelopmental assessments at 2 years and/or 5 years. Nine (5.9%) patients evaluated had neurodevelopmental delay (NDD) with scores greater than 2 standard deviations below the mean (Supplementary Table 2).
Clinical and phenotypic summary of CDH patients (n=357)
Significant enrichment of coding de novo variants in both complex and isolated CDH
We identified 461 protein-coding de novo variants (Supplementary Table 3) (~1.29 per patient), including 190 damaging de novo variants in LGD and predicted deleterious missense variants (“D-mis” defined as CADD score ≥ 25, Supplementary Table 4). The overall de novo frequency in cases was 1.33 (255/192) in WGS and 1.25 (206/165) in WES. 41.2% (147/357) of probands carried at least one damaging de novo variant, including one de novo LGD in 8.4% (30/357), one de novo D-mis in 22.7% (81/357), and two or more damaging de novos in 10.1% (36/357).
We observed an overall enrichment of damaging de novo variants (fold enrichment (FE)=1.7, P=4.2×10−4 for LGD, and FE=1.5, P=3.2×10−6 for D-mis, respectively) in all CDH patients based on the expected mutation rate calibrated by the method described in Samocha et al.29; 32(Table 2, Online Methods). The positive predictive value (PPV) estimated from the enrichment rate for LGD and D-mis variants is 35%, which indicates about 67 damaging de novo variants contribute to CDH. The enrichment is still significant when stratifying complex and isolated CDH or by sex (Table 2). 22% of complex and 16% of isolated cases are explained by damaging de novo variants.
^LGD: likely-gene-disrupting, including frameshift, stopgain, stoploss, and splicing variants; *D-mis: missense predicted to be damaging by CADD phred score >= 25; ~Background expectation calibrated based on Samocha et al 2014 and Ware et al 201529; 32.
We then tested whether the burden of damaging de novo variants were concentrated in constrained genes (defined as ExAC 38 pLI≥0.5)38 across variant types and sub-phenotypes. Overall, the burden of LGD variants was concentrated in constrained genes for both complex and isolated cases. The burden of D-mis variants was concentrated in constrained genes for complex cases, whereas for isolated cases, the burden of D-mis variants was concentrated in other genes (pLI<0.5 or not available) (Supplementary Table 5 and 6). This suggests that de novo pathogenic variants in constrained genes are more likely to cause syndromic abnormalities while such variants in other genes are more likely to cause isolated cases. Since other genes are generally not dosage sensitive, the observed burden of D-mis in these genes suggests a role of dominant negative or gain of function in isolated CDH.
Different contribution of de novo variants to male and female CDH cases
Although CDH is more common in males, the enrichment of damaging de novo variants is higher in females than in males (FE=1.8 in female, FE=1.4 in male) (Table 2). We estimated that 27% of females can be explained by LGD or D-mis variants compared to 14% of males. In female cases, the enrichment rate of LGD or D-mis is comparable between complex and isolated cases (Supplementary Table 7). In contrast, in male cases, the enrichment rate is much higher in complex cases than isolated cases. In fact, there is essentially no enrichment of LGD or D-mis variants in male isolated cases (Figure 1a and Supplementary Table 7). Furthermore, in isolated female cases, LGD variants are mainly enriched in constrained genes (FE=3.3, P=0.001, Figure 1a), and D-mis variants were mainly in other genes (FE=2.2, P=0.0002) (Supplementary Table 8, Figure 1a). In complex CDH, the difference in enrichment rate of LGD and D-mis de novo variants in constrained genes between female and male cases is much smaller; and there is no significant enrichment of D-mis in other genes in either female or male cases (Supplementary Table 8, Figure 1b).

(a) Enrichment of LGD variants and D-mis in constrained or other genes in isolated female and male cases. Constrained genes with LGD variants and other genes with D-mis variants are mainly enriched in female isolated cases. There is no enrichment of damaging de novo variants in isolated male cases. (b) Enrichment of LGD and D-mis variants in constrained or other genes in complex female and male cases. Both LGD and D-mis de novo variants were mainly enriched in constrained genes in complex cases. P-values shown are from tests of enrichment analysis. Red dots represent female cases, blue dots represent male cases. Bars represent the 95% confidence intervals (CIs) of the point estimates. Constrained genes: genes with ExAC pLI≥0.5. Other genes: genes with pLI<0.5 or no pLI estimate from ExAC; D-mis are missense variants with CADD Phred score≥25.
Genes implicated by de novo LGD variants in complex and isolated CDH cases have distinct expression patterns in early diaphragm development
Genes associated with CDH are often expressed in pleuroperitoneal folds (PPF), an early structure critical in the developing diaphragm39,10. We analyzed the expression patterns of genes with LGD and D-mis variants using a mouse E11.5 PPF data set33. Isolated and complex cases have different patterns of LGD and missense variant burden. In complex cases, LGD de novo variants are dramatically enriched in genes in the top quartile of expression in developing diaphragm (E11.5) (FE=4.7, p=7×10−7) (Supplementary Table 9, Fig. 2). By contrast, in isolated cases, the burden of LGD de novo variants is distributed across genes with a broad range of expression in PPF (Supplementary Table 9 and 10, Fig. 2).

Enrichment rate of LGD de novo variants are shown in gene sets grouped by expression rank in E11.5 pleuroperitoneal folds (PPFs). In complex CDH cases, LGD de novo variants are dramatically enriched in the genes within the top quartile (0–25%) of expression in developing diaphragm (E11.5), and show no trend of enrichment in other quartiles. In isolated cases, LGD de novo variants have similar enrichment (~2x) across the 0–75% range of PPF gene expression. P values shown are from a test of enrichment. Bars represent the 95% CIs of the point estimates.
MYRF is a novel candidate risk gene of CDH
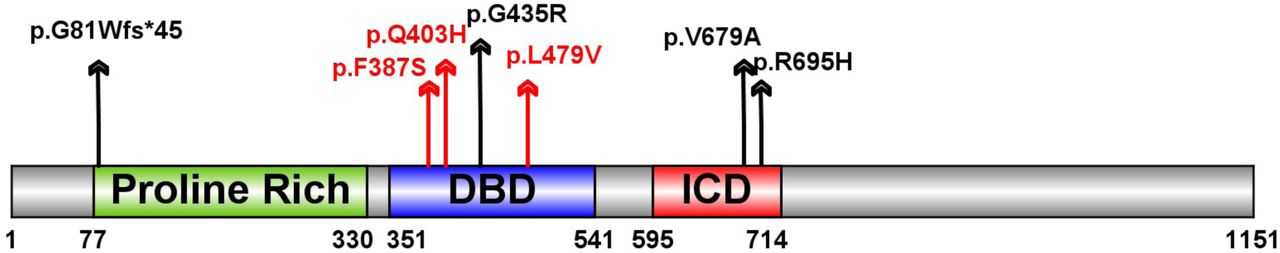
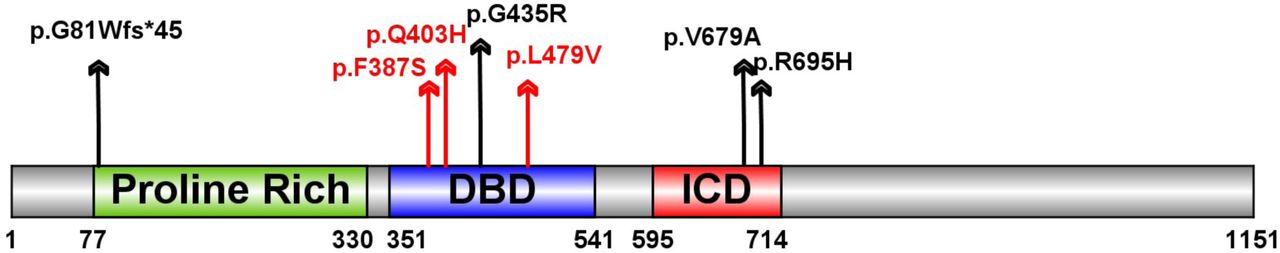
Two genes are observed with multiple damaging de novo variants. Wilms tumor 1 (WT1) has been previously implicated in CDH40 and has two D-mis variants. Myelin Regulatory Factor (MYRF), a transcription factor, has one de novo LGD and three D-mis variants (Fig. 3) in four complex CDH patients (p=2×10−10, based on comparison to expectation from background mutations 29; 32) (Table 3). A recent study of congenital heart disease (CHD) 41; 42 reported three additional damaging de novo missense variants (p.F387S, p.Q403H and p.L479V) in MYRF (Table 3, Fig 3a). All four CDH patients had CHD (Table 3). The CHD patient with the MYRF p.Q403H variant had hemidiaphragm eventration. Genitourinary anomalies were present in six of the seven patients, a female had a blind-ending vagina with no internal sex organs and five males had ambiguous genitalia or undescended testes. MYRF is a constrained gene intolerant of loss of function variants in the general populations (ExAC38 pLI=1). Although it has not previously been implicated in CDH or CHD, it is highly expressed in developing diaphragm and heart (ranked top 21% and 14% in mice E11.5 PPF 33 and E14.5 heart 43, respectively). Genital malformation may share developmental processes44 because PPF is physically connected dorsally to urogenital ridge.
Abbreviation: CDH (congenital diaphragmatic hernia); CHD (congenital heart disease); ASD (Atrial Septal Defect); VSD (Ventricular septal defect); TOF (Tetralogy of Fallot). The last three patients are ascertained by CHD as describe in Jin et al 2017 45
{kind=link}
{kind=link}
{kind=link}
Schematic of the MYRF protein with predicted sequence features, including N-terminal Proline Rich region, DNA-binding domain (DBD) and intramolecular chaperone domain (ICD). Variants identified in CDH indicated as black arrow, variants identified in congenital heart disease cases indicated with red arrows.
The three variants identified in CHD patients and p.G435R are located in the conserved DNA binding domain (DBD) of MYRF (Fig. 3), and could alter DNA binding46. The other two D-mis variants (p.V679R and p.R695H) are located in the intramolecular chaperone auto-processing domain (ICD) in a leucine zipper47. Mutations in the leucine zipper of the ICD domain may inhibit the trimerization of MYRF, resulting in the failure of formation of the N-terminal trimer47 which is important for the transcription factor function48. MYRF is thought to be an essential transcription factor for oligodendrocyte differentiation and myelination49. Conditional deletion of Myrf impaired motor learning50; 51 and the individual with the p.V679A variant we assessed at two years old had intellectual disability.
Discussions
CDH is slightly more common in males. For the first time, our study suggests that male and female CDH may have a different genetic architecture, especially among isolated CDH cases. Damaging de novo variants with large effect have a substantial contribution to isolated female cases but little contribution to isolated male cases. Given the higher frequency of males among isolated cases, a plausible explanation is that polygenic risk from inherited variants alone can cause isolated CDH in males, but due to a female protective effect52, additional highly penetrant de novo variants are often required to cause CDH in females to pass the threshold of liability. This is similar to what has been observed in autism which is also more common among males 53. Since there is a similar male/female ratio in overall cohort and our neonatal cohort (1.4:1), this difference is unlikely due to ascertainment or survival bias. The parental ages for male and female probands were similar and cannot account for the differences we observed in de novo variants.
Additionally, we found genes implicated in isolated and complex cases have distinct expression patterns in early development. In complex CDH, the burden of LGD and D-mis variants are concentrated in genes highly expressed in the FFP, an early embryonic diaphragm precursor, consistent with the pleiotropic effects of these genes on diaphragm and other organogenesis. By contrast, the burden of LGD variants in isolated cases is distributed across genes with a broader range of expression in PPF. Since the bulk expression data from PPFs is the sum of different cell types54, the lack of correlation of LGD enrichment and expression level in PPF suggests the possibility that a substantial portion of the implicated genes in isolated cases could be expressed only in sub-populations of cells in the PPF that are not relevant to organogenesis in other parts of the body. Single-cell mRNA-sequencing will be necessary to analyze gene expression pattern in specific cell types and further assess the cell type(s) responsible for isolated CDH.
Finally, MYRF is a novel candidate risk gene of CDH. The four CDH patients carrying damaging de novo variants in MYRF all have congenital heart defects, genitourinary anomalies including ambiguous genital, and this likely represents a novel syndrome. As we identify larger numbers of patients with mutations in genes associated with CDH, we will be able to better describe the spectrum of disease associated with the gene as well as the clinical outcomes including risk of pulmonary hypertension and respiratory complications which are life threatening concerns for CDH patients. Identification of additional high risk CDH genes should elucidate the developmental biology and provide targets for treatment and prevention.
Author Contributions
W.K.C and Y.S. conceived the study. F.A.H., J.M.W., and P.K.D. provided genomic data. H.Q., X.Z., Y.L., A.K., W.K.C., and Y.S. analyzed and interpreted the data. Y.L., X.Z., H.Q., W.K.C., and Y.S. wrote the manuscript. J.W., G.A., F.L., T.C., R.C., K.A., M.E.D., D.C., B.W.W., G.B.M., D.P., A.J.W., M.E., F.A.H., M.L., J.M.W., P.K.D. collected samples and clinical information. All authors contributed and discussed the results and critically reviewed the manuscript.
Competing Financial Interests statement
None declared
Acknowledgements
We would like to thank the patients and their families for their generous contribution. We are grateful for the technical assistance provided by Patricia Lanzano, Jiancheng Guo, and Liyong Deng from Columbia University, Jessica Kim at Boston Children’s Hospital, and Caroline Coletti and Pooja Bhayani at Massachusetts General Hospital. We thank our clinical coordinators across the DHREAMS centers: Trish Burns at Cincinnati Children's Hospital, Sheila Horak at Children's Hospital & Medical Center of Omaha, Brandy Gonzales at Oregon Health and Science University, Karen Lukas at St. Louis Children's Hospital, Jeannie Kreutzman at CS Mott Children's Hospital, Min Shi at Children's Hospital of Pittsburgh, Michelle Knezevich and Cheryl Kornberg at Medical College of Wisconsin. We thank University of Washington Center for Mendelian Genomics (UW-CMG) team for generating part of the WES data. The WES data generation at UW-CMG was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant HG006493 to Drs. Debbie Nickerson, Michael Bamshad, and Suzanne Leal. The whole genome sequencing data were generated by NIH Gabriella Miller Kids First Pediatric Research Program (X01HL132366). This work was supported by NIH grants R01HD057036 (L.Y., J.W., W.K.C.), R03HL138352 (A.K., W.K.C., Y.S.), R01GM120609 (H.Q., Y.S.), UL1 RR024156 (W.K.C.), and 1P01HD068250 (P.K.D, M.L., F.A.H., J.M.W., W.K.C.) Additional funding support was provided by grant from CHERUBS, a grant from the National Greek Orthodox Ladies Philoptochos Society, Inc. and generous donations from The Wheeler foundation, Vanech Family Foundation, Larsen Family, Wilke Family and many other families.
References




